Excellent benzocoumarin-based ratiometric two-photon fluorescent probe for H2O2 detection†
Received
27th September 2018
, Accepted 20th November 2018
First published on 21st November 2018
Abstract
The level of hydrogen peroxide (H2O2) plays an essential role in regulating biological processes. The in vivo or in vitro detection of H2O2 in deep tissues by utilizing two-photon (TP) fluorescent probes can significantly alleviate the detection damage inflicted onto living organisms as well as facilitate high-resolution imaging when compared with one-photon (OP) fluorescent probes. However, few TP fluorescent probes possess both high fluorescence efficiency and easily distinguishable spectra for measuring H2O2. Therefore, an in-depth understanding of the relationship between the electronic structure and TP fluorescent properties and fabricating probes with excellent performance are still challenging. Consequently, we designed a series of benzocoumarin-based ratiometric TP fluorescent probes and corresponding product molecules for H2O2 detection. Thereafter, we theoretically evaluated the TP recognition performance of these compounds and studied the relationship between their molecular structure and TP performance by means of time-dependent density functional theory and quadratic response theory. Moreover, we determined their spectral properties and fluorescence efficiencies. Fortunately, in this study, we were able to propose an excellent TP probe BC-3 and the corresponding product molecule DCCA-3, which exhibit large TPA cross-sections in the NIR region (3420 GM/988 nm; 316 GM/939 nm) and large Stokes (116 nm; 60 nm) and emission (225 nm) shifts. Therefore, this probe enables the simultaneous NIR and TP imaging of H2O2, which is a unique ability and has never been previously reported. Moreover, we comprehensively investigated the effect of the benzene-fused position in the coumarin backbone on the transition dipole moment and nonradiative decay channels, explaining the fluorescence near-quenching mechanism of benzo[f]coumarin derivative DCCA-4 for the first time.
1. Introduction
Hydrogen peroxide (H2O2), a major member of the reactive oxygen species (ROS), plays an essential role in regulating biological processes.1 However, the aberrant production of H2O2 can give rise to many diseases such as cancer,2 neurodegenerative disorders,3 and cardiovascular diseases.4 Therefore, it is necessary to efficiently monitor the level of H2O2 in living cells. In the last few years, many techniques for H2O2 detection have been developed, but most of them are harmful to living cells, which is not suitable for in vivo monitoring.5 Fluorescent probes have become powerful tools for the detection of H2O2 because of their high sensitivity and real-time imaging ability.6 Until now, most of them have been designed based on one-photon (OP) excitation with relatively short excitation wavelengths, which may lead to photodamage and photobleaching. However, two-photon (TP) fluorescent probes can alleviate these limitations and have promising potential due to their advantages such as localized excitation, deeper penetration depth, and lower tissue auto-fluorescence.7 However, applications of TP fluorescent probes are relatively scarce due to their small cross-section, low fluorescent efficiency, and incomplete internal mechanism when compared with OP fluorescent probes.8 Therefore, research involving TP fluorescent probes with high efficiency and large cross-section is extremely urgent for applications in monitoring the level of H2O2.
In the last few years, most of the TP fluorescent probes for H2O2 detection have possessed the turn-on or turn-off mechanism, which can be easily interfered.8a,c,d,f,g However, ratiometric fluorescent probes can simultaneously maintain a record of the fluorescence intensities and ratio at two wavelengths, which facilitates reducing the influence of environmental factors on the detection ability.9 The large emission separation between the probe and its product and large Stokes shift are two particular indexes for ratiometric fluorescent probes, but it is very difficult to achieve the target.9 Recently, Lin et al. designed a ratiometric fluorescent probe BC based on oxonium as the recognition group by Baeyer–Villiger oxidative rearrangement reaction to obtain the product as the coumarin derivative DCCA.10 As elucidated in the experimental results, this TP fluorescent probe BC exhibits high sensitivity, excellent selectivity, good water solubility, near-infrared (NIR) emission, and large emission shift.10 However, there is still a considerable avenue for further improvement in the small excitation and emission wavelengths, Stokes shift, and TPA cross-section of the product (coumarin derivative DCCA) for facile detection after reaction with H2O2. The imperfect photophysical performance of the product compound can be a limitation for monitoring the level of H2O2. Therefore, it is one of the main purposes of this research to provide design strategies to enable both TP probes and corresponding product molecules with large TPA cross-sections and easily resolved fluorescence spectra.
To the best of our knowledge, benzocoumarins with larger electronic conjugation can alleviate these abovementioned limitations and can prove to be promising candidates for TP fluorescent probes. Recently, Cho et al. have proposed that benzo[h]coumarin derivatives possess large TPA cross-sections in the long-wavelength light region.11 Kim, Lee, and Ahn et al. have reported that both TPA cross-sections and Stokes shifts for benzo[g]coumarin derivatives are larger than those for the corresponding coumarin derivatives.12 Moreover, the group of Kyo Han Ahn and Daniel T. Gryko systemically researched the optical properties of π-expanded coumarins.13 It has been shown that benzocoumarins with appropriate substituents can be excited at longer wavelengths and may have higher fluorescence quantum yields than the corresponding coumarin derivatives. Moreover, many benzocoumarin derivatives have been used for biological imaging and in vivo detection due to their desirable biocompatibility, low toxicity, and high photostability.11,12 Interestingly, when compared with benzo[g]coumarin derivatives, the corresponding benzo[f]coumarin derivatives have very weak emissions in polar media, but the internal mechanism for this phenomenon is unclear.13 These studies indicate that benzocoumarins may prove to be promising candidates for use as TP fluorescent probes, and the benzene-fused position plays an important role in determining the molecular optical properties.
Based on the above studies, we designed a series of probe molecules (BC-2, BC-3, and BC-4) with oxonium-based site and benzocoumarin derivatives as the product molecules (DCCA-2, DCCA-3, and DCCA-4) in order to obtain excellent ratiometric TP fluorescent probes with large cross-sections before and after their reaction with H2O2. The structures of these compounds are shown in Fig. 1. DCCA-2, DCCA-3, and DCCA-4 are benzo[g]coumarin, benzo[h]coumarin, and benzo[f]coumarin derivatives, respectively. We calculated the TPA cross-sections and fluorescence quantum yields to evaluate the photophysical properties of these compounds. We demonstrated the effect of the benzene-fused position on the molecular optical properties and explained the fluorescence near-quenching mechanism of the benzo[f]coumarin derivative DCCA-4 by analyzing the difference between the isomeric benzocoumarin derivatives in the excited-state decay channels and relevant transition dipole moments. It is expected that this theoretical investigation can provide a new insight into the design of ratiometric TP fluorescent probes with high efficiency and large cross-sections for the in vivo monitoring of the level of H2O2.
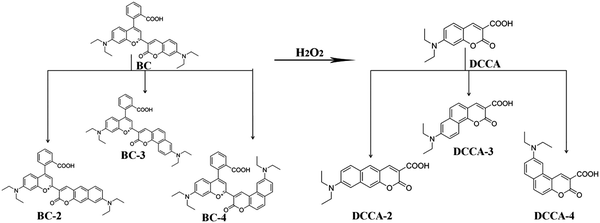 |
| | Fig. 1 Structures and corresponding names of the investigated molecules. | |
2. Computational details
The ground-state geometry optimizations and vibrational frequency calculations of all the studied compounds were performed by using density functional theory (DFT), employing the B3LYP functional and 6-31G(d) basis.14 The same level of calculations for the first excited state was obtained by using time-dependent density functional theory (TD-DFT), and no imaginary frequency was found. On the basis of structural stability, we have evaluated the electron distribution and frontier molecular orbital (FMO) levels. Thereafter, the absorption and emission spectra are obtained by the TD-DFT//B3LYP/6-311+G(d) method. The results obtained from the B3LYP theory are in good agreement with the experimental results, as listed in Tables S1 and S2 (ESI†). To evaluate the solvent effect, a polarizable continuum model (PCM)15 was used. These geometric and electronic structure calculations were performed by using the Gaussian 09 program.16 The electron transition characteristics were analyzed by the Multiwfn program.17 The radiative and nonradiative rates were examined by the MOMAP program,18 which takes into account the normal mode displacements and Duschinsky rotation effect using the DUSHIN program.19 Moreover, the two-photon absorption (TPA) properties including the TPA cross-sections (σTPA) and corresponding TPA wavelengths (λT) in the NIR spectral region were calculated with the help of the quadratic response theory20 at the TD-DFT//B3LYP/6-311+G(d) level using the DALTON program.21
3. Results and discussion
3.1 Molecular design
In order to investigate how the benzene-fused position influences the molecular optical properties, we designed a series of probe molecules (BC-2, BC-3, and BC-4) and the corresponding product molecules (DCCA-2, DCCA-3, and DCCA-4), as shown in Fig. 1. According to experimental studies, the ratiometric fluorescent probe molecule BC with oxonium as the recognition group can react with H2O2 to yield the coumarin derivative DCCA because of the Baeyer–Villiger oxidative rearrangement reaction.10 This is the key factor for the above reaction in which the C atom of the reactive site (Fig. S7: yellow atom, ESI†) possesses more positive charge than the neighboring atoms, which supports the nucleophilic addition of H2O2 to the specific reactive site (C atom) in the first step of the reaction mechanism.10 By means of the natural band orbital (NBO) analysis (Table S7, ESI†), all the designed probe molecules (BC-2–4) have the abovementioned characteristics that can maintain the same recognition response mechanism as the experimental molecule. Besides the above compounds, the position isomerization of their donor substituents was also considered (DCCA-2a, DCCA-2b, DCCA-2c, DCCA-3a, DCCA-3b, DCCA-3c, DCCA-4a, DCCA-4b, and DCCA-4c) (Fig. S1, ESI†) in order to obtain the optimal molecular configuration with TP fluorescent property before and after reactions with H2O2. As shown in Fig. S1, the planar rigidity for the DCCA-2, DCCA-3, and DCCA-4 molecules are better than those for the other isomers. The study of different resonance behaviors of all the designed compounds suggest that DCCA-Xa, DCCA-Xb, and DCCA-Xc (X = 1, 2, 3) isomers have unfavorable electron conjugation for electron polarization, and therefore, this would be detrimental to their nonlinear optical properties and fluorescent emissions (Fig. S2). Hence, we will only consider the optical properties of the cationic probe molecules (BC-2, BC-3, and BC-4) and neutral product molecules (DCCA-2, DCCA-3, and DCCA-4) in the following discussion.
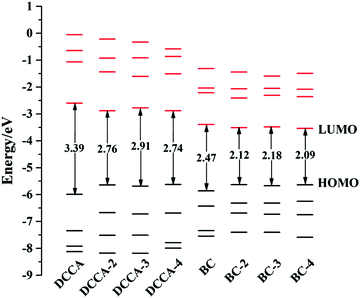
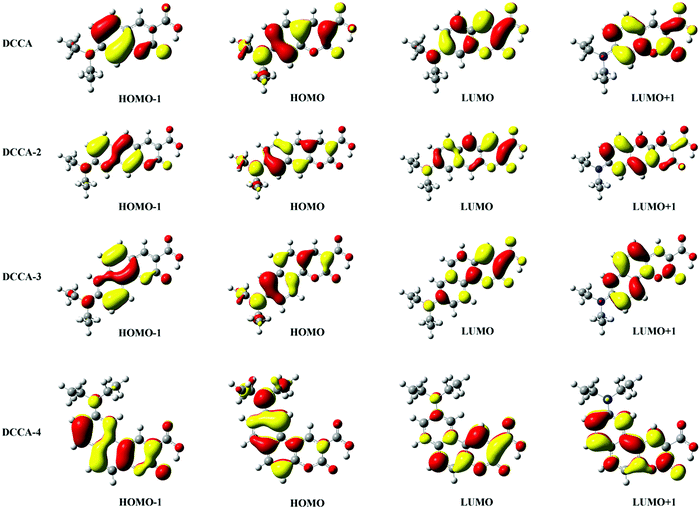
3.2 Optical properties and electronic structures
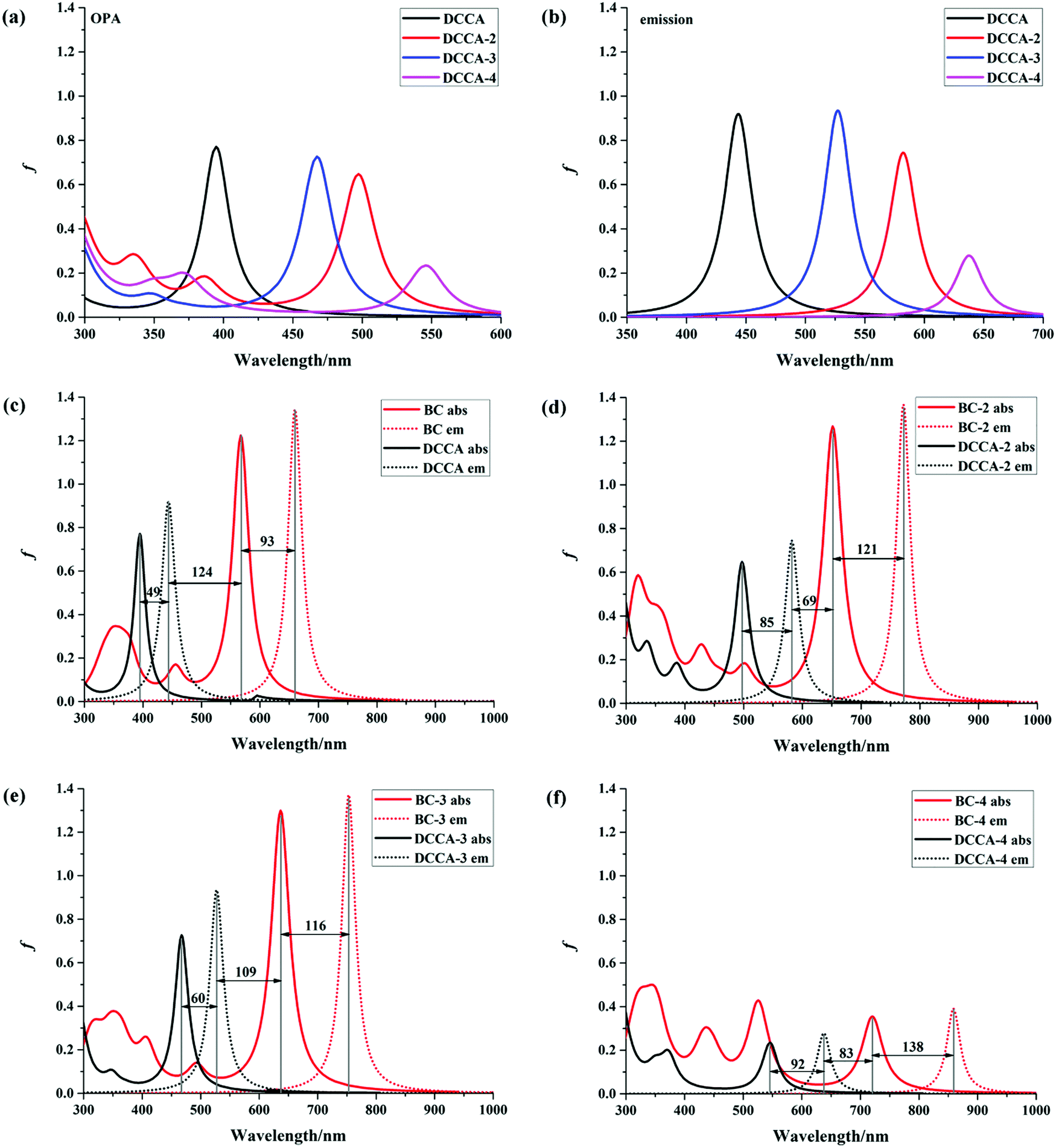
The absorption and emission spectra are very useful to obtain a comprehensive insight into the optical properties of a compound. The one-photon absorption (OPA) and emission spectra are shown in Fig. 2, and the corresponding calculated values are listed in Table 1 and Tables S3, S4 (ESI†). The calculated maximum OPA (395 nm) and emission (444 nm) peaks for the DCCA molecule and the emission spectral separation (217 nm; Fig. 2c) between the probe molecule (BC) and product molecule (DCCA) are in good agreement with the experimental values (411, 472, and 221 nm).10 As shown in Fig. 2a and b, the OPA spectra of the benzene-fused molecules DCCA-2–DCCA-4 are red-shifted to the blue-green light region and the fluorescent spectra for benzene-fused molecules red-shift to the yellow-red region when compared with those of the experimental DCCA molecule, which contributes toward facilitating spectral recognition and reducing photodamage. As shown in Tables S3 and S4 (ESI†), the first excited states (S1) with the (π, π*) electron configuration corresponding to the maximum OP absorption and emission peaks for all the molecules are constructed by the highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) and LUMO → HOMO transitions, respectively. The FMO energy levels and contour surfaces of the FMOs of the investigated molecules in the ground state were examined and plotted in Fig. 3, 4 and Fig. S3 (ESI†). Evidently, the electron cloud on both HOMO and LUMO of the benzocoumarin derivatives are more polarized when compared with the coumarin derivative DCCA, which indicates that benzene-fused molecules experience larger intramolecular charge transfer (ICT) than the experimental molecule upon excitation. When compared with the experimental DCCA molecule, the HOMO energy levels of benzocoumarin derivatives dramatically increase and the LUMO energy levels decrease, which leads to a decrease in the HOMO–LUMO energy gaps (ΔEH–L) and red-shifts in the spectra. With regard to isomeric benzocoumarin derivatives, the FMO levels of DCCA-3 are more stabilized and the LUMO energy level of DCCA-3 is marginally higher than those of DCCA-2 and DCCA-4. In addition, the absorption and emission spectra for all the investigated coumarin derivatives showed a gradual red-shifted characteristic with an increase in the solvent polarity in the order of gas < toluene < CHCl3 < MeOH < DMSO, as shown in Fig. S4 (ESI†). The fluorescent spectra show a more obvious shift than the OPA spectra with an increase in the polarity of the solvents, indicating that the solvent effect has a significant influence on the emitting electronic state rather than on the Franck–Condon state. However, the oscillator strengths (f) of all the benzocoumarin derivatives are smaller than those of DCCA, which lie in the order of DCCA (0.77) > DCCA-3 (0.72) > DCCA-2 (0.64) > DCCA-4 (0.23) (Table 1 and Fig. 2a). To explain this phenomenon, the OPA transition probability δOPA was further investigated. It is well known that the oscillator strength (f) is related to δOPA, which is proportional to the excitation energy (ω0f) and square of the ground-to-excited-state transition moment (![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) 0f). The expression for OPA transition probability is given as follows:22
0f). The expression for OPA transition probability is given as follows:22| | 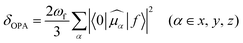 | (1) |
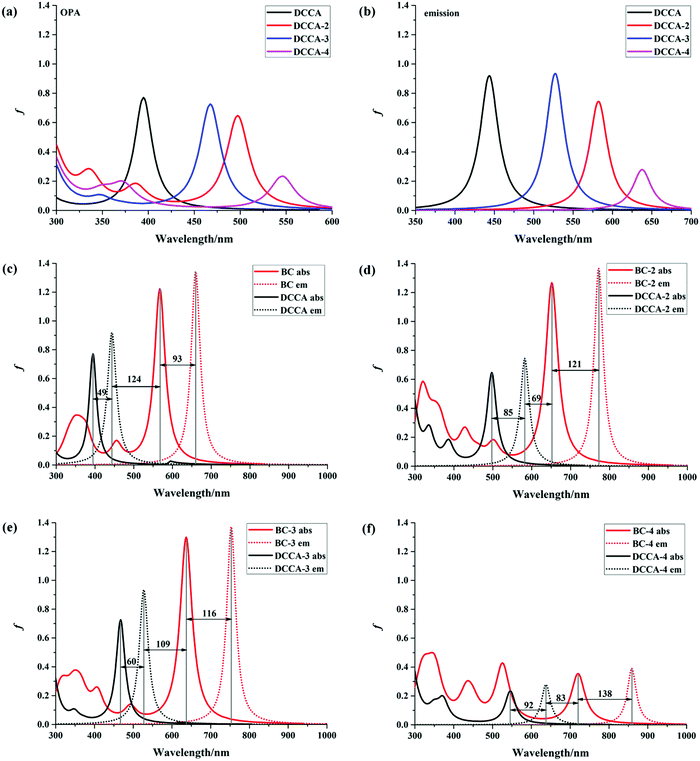 |
| | Fig. 2 OPA and fluorescence spectra of all the studied molecules in water. | |
Table 1 Calculated OPA parameters including the maximum absorption peaks (λabs), vertical excitation energies (ω0f), oscillator strengths (f), transition moments (μ0f), and corresponding transition characteristic of the investigated coumarin derivatives by B3LYP/6-311+G(d) method in a water solvent. The absorption wavelengths (λexp) are obtained from the experimental results
| Molecule |
λ
exp/nm |
λ
0abs/nm |
ω
0f/eV |
f
|
μ
0f/D |
Transition nature |
|
DCCA
|
411 |
394.63 |
3.14 |
0.77 |
3.15 |
S0 → S1 |
H → L |
(98.11%) |
|
DCCA-2
|
|
497.23 |
2.49 |
0.64 |
3.23 |
S0 → S1 |
H → L |
(96.18%) |
|
DCCA-3
|
|
467.36 |
2.65 |
0.72 |
3.33 |
S0 → S1 |
H → L |
(97.90%) |
|
DCCA-4
|
|
545.79 |
2.27 |
0.23 |
2.02 |
S0 → S1 |
H → L |
(99.17%) |
 |
| | Fig. 3 Calculated FMO levels for the studied compounds in the ground state. | |
 |
| | Fig. 4 Contour surfaces of the FMOs for the coumarin derivatives (DCCA, DCCA-2, DCCA-3, and DCCA-4) in the ground state. | |
It is obvious that the transition moment increases in the order of DCCA-4 (2.02 D) < DCCA (3.15 D) < DCCA-2 (3.23 D) < DCCA-3 (3.33 D); however, the excitation energy decreased in the order of DCCA (3.14 eV) > DCCA-3 (2.65 eV) > DCCA-2 (2.49 eV) > DCCA-4 (2.27 eV) (see Table 1). The relatively small excitation energy and transition moment for DCCA-4 are derived from the small HOMO–LUMO energy gap and short charge transfer (CT) distance (Fig. 5), which results in its small oscillator strength (OPA transition probability). However, the OPA intensity of the benzene-fused DCCA-3 and DCCA-2 molecules can be comparable to the experimental DCCA molecule because of their more dominant transition moments. In conclusion, it is an effective approach to red-shift the OP absorption and emission spectra via an increase in the phenyl group, but the benzene-fused position is very important for the OPA intensity.
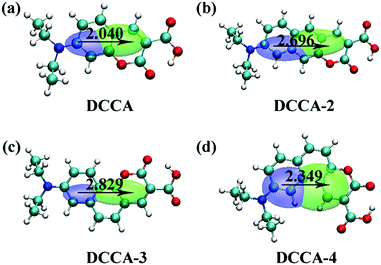 |
| | Fig. 5 Contour map of the electron density difference between the ground state S0 and the first excited state S1 for coumarin derivatives (DCCA, DCCA-2, DCCA-3, and DCCA-4). In the diagram, green (blue) indicates an increase (decrease) in the electron density. The CT distances for DCCA, DCCA-2, DCCA-3, and DCCA-4 are 2.040 Å, 2.696 Å, 2.829 Å, and 2.349 Å, respectively. | |
For fluorescent probe molecules, the OPA and fluorescent spectra of all the benzene-fused probes are located in the red and NIR regions, as shown in Fig. 2c–f. Moreover, benzene-fused molecules not only maintain large emission separations between the probe and product molecules but also exhibit large Stokes shifts, which is desirable for forming strong signal contrast before and after recognizing H2O2 and to avoid confusion and interference. For example, the emission shift between probe BC-3 and the corresponding DCCA-3 product molecule is up to 225 nm, and the Stokes shift is 116 (60) nm for the BC-3 molecule (DCCA-3), as shown in Fig. 2e. This large emission separation is related to the electron-withdrawing ability of the electron acceptor. After reaction with H2O2, the electron-acceptor group becomes neutral in the product from electropositive in the probe, resulting in the decreased ability of attracting electrons and a dramatic increase in the energy gap. As shown in Fig. 3, the LUMO energy levels of the product molecules drastically increase when compared with those of the probe molecules, which leads to the distinctly increased ΔEH–L value of the product molecules. Obviously, the Stokes shifts of the benzene-fused DCCA-2 (85 nm) and DCCA-4 (92 nm) are much larger than that of DCCA (49 nm) (Fig. 2c, d, and f). The relatively larger Stokes shifts for benzocoumarin derivatives are due to their corresponding smaller energy gaps, ΔEH–L. Evidently, the absorption and emission spectra of BC-2 and its product DCCA-2 are located in different visible light regions (red, green, and yellow), large oscillator strength, large emission shift (190 nm), and large Stokes shifts (121 nm for BC-2 and 85 nm for DCCA-2), which demonstrates that the BC-2 molecule is also a potential fluorescent labeling material. However, BC-4 molecule may not be suitable as a fluorescent probe because of its small oscillator strength (f) in the visible region, which indicates low fluorescence efficiency.
3.3 Fluorescence quantum yield and radiative and nonradiative decay rates
For fluorescence detection and imaging, fluorescence quantum yield is an important index to determine fluorescence efficiency. According to Kasha's rule, the high singlet excited states Sn initially decay to the first singlet excited state S1, and these decay processes are so fast that their energy consumption can be negligible. Therefore, we only consider the decay processes of the first singlet excited state S1 and the fluorescence quantum yield mainly depends on the competition between the radiative and nonradiative decay rates from S1 to the ground state S0. The radiative rate (kr) is mainly controlled by the electric transition dipole moment (μeg) and adiabatic excitation energy (Ead).23 As listed in Table 2, the DCCA molecule possesses the largest kr value (2.76 × 108 s−1) due to its large Ead (2.99 eV) and μeg (8.90 D) values. The transition dipole moment (μS1–S0) between the S0 and S1 states is proportional to the HOMO–LUMO orbital overlap:24| | | μS1–S0 = |〈ΦS1||ΦS0〉| ≈ |〈L||H〉 | (2) |
where is the dipole operator, ΦS1/S0 is the electronic wavefunction of the S1/S0 state, and L/H denotes the LUMO/HOMO orbital, which is the main electronic configuration of the S1 and S0 states for all the studied compounds. As shown in Fig. 4, the large π-conjugation and electronic delocalization contribute toward the HOMO–LUMO orbital overlap; therefore, the μeg values for benzene-fused DCCA-2 (9.02 D) and DCCA-3 (9.75 D) are larger than that for DCCA (8.90 D), as listed in Table 2. However, the small μeg value of DCCA-4 (6.10 D) can be attributed to the short distance in the electric transition, resulting in the kr value of DCCA-4 to be not as large as the others. Moreover, the small excitation energies of benzene-fused molecules is detrimental to radiative decay, particularly for DCCA-4 (Evt = 2.08 eV and kr = 4.93 × 107 s−1). This suggests that it is very important to maintain large π-conjugation and excitation energy values for obtaining a high radiative rate in the design of new benzene-fused compounds.
Table 2 Fluorescence properties, including the vertical excitation energies (Evt), adiabatic energies difference between S1 and S0 (Ead), electric transition dipole moments (μeg), radiative rates (kr), internal conversion rates (kic), and fluorescence quantum yields (Φ), are calculated by using the B3LYP/6-31G(d) method in a water solvent
| Molecules |
E
vt/eV |
E
ad/eV |
μ
eg/D |
k
r/s−1 |
K
ic/s−1 |
Φ
|
|
DCCA
|
2.91 |
2.99 |
8.90 |
2.76 × 108 |
2.65 × 108 |
0.51 |
|
DCCA-2
|
2.22 |
2.34 |
9.02 |
1.31 × 108 |
9.28 × 108 |
0.12 |
|
DCCA-3
|
2.47 |
2.55 |
9.75 |
2.13 × 108 |
3.67 × 108 |
0.37 |
|
DCCA-4
|
2.08 |
2.21 |
6.10 |
4.93 × 107 |
5.56 × 108 |
0.08 |
The nonradiative rate is another key parameter for determining the fluorescence quantum yield. The internal conversion (IC) decay is related to the geometric distortions between the two electronic states and adiabatic excitation energy.25 Reorganization energy is a helpful measure for the determining the extent of geometry relaxation, which facilitates the determination of electron-vibration coupling between the two electronic states and determining the IC decay.26 The reorganization energies and corresponding crucial vibrational frequencies are shown in Fig. 6. From this figure, the following can be concluded. (1) There is one high-frequency mode (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond stretching mode) at about 1720 cm−1 with large reorganization energy in all the compounds. (2) The low-frequency modes (<500 cm−1) are suppressed for DCCA-3 when compared with DCCA. The total reorganization energy is 683 cm−1 for DCCA and 612 cm−1 for DCCA-3; the contribution of the low-frequency (<500 cm−1) modes to the total reorganization energy is more than 15% in DCCA, while it is roughly 8% in DCCA-3. There are only two molecule scissor bending modes (667 and 727 cm−1) with large reorganization energies (71 and 49 cm−1) in DCCA-3. (3) In the low-frequency region, the side-chain twisting motion at 127 cm−1 possesses a notably large reorganization energy value (103 cm−1) for DCCA-2. (4) Generally, the reorganization energy for DCCA-4 (maximum: 162 cm−1) is much larger than that for DCCA (maximum: 114 cm−1), as shown in Fig. 6a and d. In addition, to comprehensively examine the vibration differences in the low-frequency-regime (<500 cm−1), we investigated the Huang–Rhys (HR) factors for all the investigated compounds (Fig. 7), which characterizes the changes in the vibrational quanta when transitioning from one electronic state to another.23 Evidently, the low-frequency side-chain twisting and molecule scissor bending motions are suppressed for DCCA-3 when compared with the other molecules. There is a notable side-chain twisting motion (at 127 cm−1) with a large HR factor (0.81) for DCCA-2. Apparently, the HR factors for DCCA-3 (maximum: 0.11) and DCCA-4 (maximum: 0.17) are much smaller than that for DCCA-2 (maximum: 0.81). Therefore, DCCA-2 exhibits a larger nonradiative rate when compared with its isomeric counterparts, i.e., DCCA-3 and DCCA-4.
O double bond stretching mode) at about 1720 cm−1 with large reorganization energy in all the compounds. (2) The low-frequency modes (<500 cm−1) are suppressed for DCCA-3 when compared with DCCA. The total reorganization energy is 683 cm−1 for DCCA and 612 cm−1 for DCCA-3; the contribution of the low-frequency (<500 cm−1) modes to the total reorganization energy is more than 15% in DCCA, while it is roughly 8% in DCCA-3. There are only two molecule scissor bending modes (667 and 727 cm−1) with large reorganization energies (71 and 49 cm−1) in DCCA-3. (3) In the low-frequency region, the side-chain twisting motion at 127 cm−1 possesses a notably large reorganization energy value (103 cm−1) for DCCA-2. (4) Generally, the reorganization energy for DCCA-4 (maximum: 162 cm−1) is much larger than that for DCCA (maximum: 114 cm−1), as shown in Fig. 6a and d. In addition, to comprehensively examine the vibration differences in the low-frequency-regime (<500 cm−1), we investigated the Huang–Rhys (HR) factors for all the investigated compounds (Fig. 7), which characterizes the changes in the vibrational quanta when transitioning from one electronic state to another.23 Evidently, the low-frequency side-chain twisting and molecule scissor bending motions are suppressed for DCCA-3 when compared with the other molecules. There is a notable side-chain twisting motion (at 127 cm−1) with a large HR factor (0.81) for DCCA-2. Apparently, the HR factors for DCCA-3 (maximum: 0.11) and DCCA-4 (maximum: 0.17) are much smaller than that for DCCA-2 (maximum: 0.81). Therefore, DCCA-2 exhibits a larger nonradiative rate when compared with its isomeric counterparts, i.e., DCCA-3 and DCCA-4.
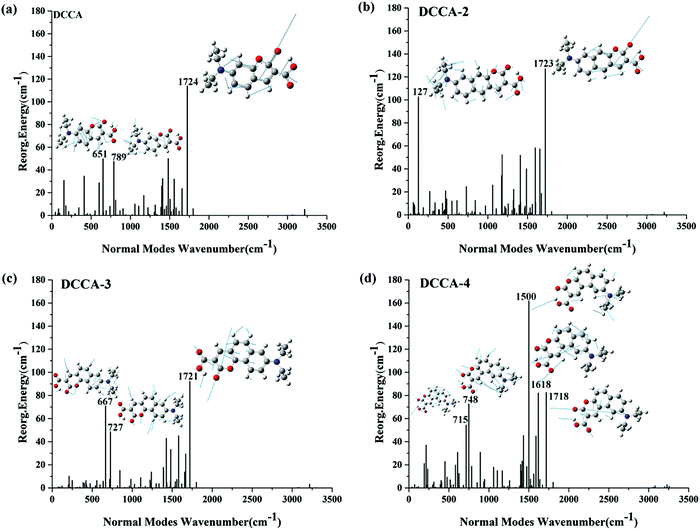 |
| | Fig. 6 Reorganization energies and crucial displacement vectors for the normal modes with large reorganization energies for DCCA, DCCA-2, DCCA-3, and DCCA-4. | |
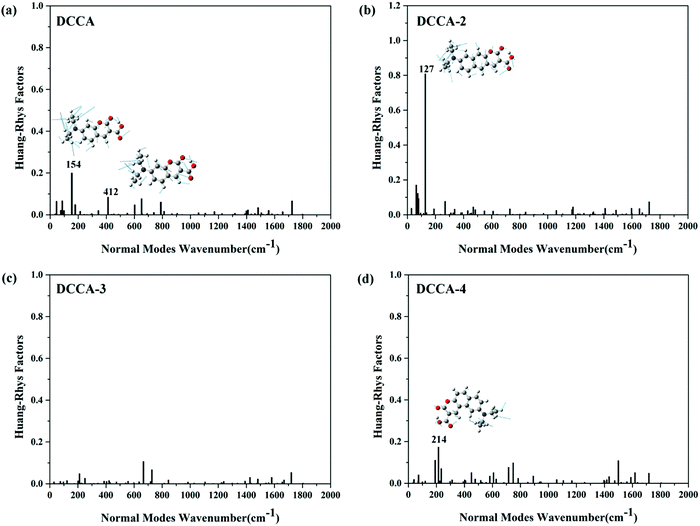 |
| | Fig. 7 HR and crucial displacement vectors for the normal modes with large HR in the low-frequency regime (<500 cm−1) for DCCA, DCCA-2, DCCA-3, and DCCA-4. | |
Therefore, we can conclude that the relatively large quantum yields (0.51 and 0.37) of DCCA and DCCA-3 are due to the large adiabatic excitation energy and transition dipole moments contributing toward the radiative decay. For the designed DCCA-3 compound, the low-frequency motion of the molecule scissor bending and side-chains twisting are hindered; therefore, their contributions toward IC decay are suppressed, leading to a large quantum yield. Although DCCA-2 also possesses a large transition dipole moment (9.02 D), the side-chain twisting mode at 127 cm−1 with a large HR factor and reorganization energy are conducive to IC decay; therefore, the fluorescence quantum yield for DCCA-2 (0.12) is smaller than that for DCCA-3 (0.37). However, DCCA-4 with a small transition dipole moment (6.10 D) and large reorganization energy leads to an extremely small quantum yield (0.08), resulting in fluorescence near-quenching. Therefore, the benzene-fused position plays an important role in the electronic transition character and molecular geometric relaxation. Benzocoumarin derivative DCCA-3 exhibits excellent luminous performance.
3.4 TPA property
As mentioned above, one of the main objectives of the present study is to design a fluorescent probe with large TPA cross-sections. The TPA spectra of all the studied compounds are shown in Fig. 8 and the corresponding calculated values are listed in Table 3, which are in good agreement with the experimental values.10 Evidently, the maximum TPA cross-section (σTmax) of the BC probe molecules series are reasonably large (>2000 GM); this value for BC-3 can go up to 3420 GM in the NIR region (988 nm), which can be attributed to the large conjugated bridge structure contributing toward ICT. The σTmax value of BC-2 is 2740 GM at 1004 nm, which can be attributed to the electronic transition from HOMO−1 to LUMO, exhibiting a combination of local excitation (LE) and CT characteristic (Fig. S3, ESI†). However, the maximum TPA band in the NIR region (2260 GM/867 nm) for BC-4 can be attributed to the electronic excitation S0 → S4, which can be described as the transition configuration from HOMO → LUMO+1 with an obvious CT characteristic, as shown in Fig. S3 (ESI†). Moreover, there is another absorption band (562 GM) at 1055 nm for BC-4, which can be attributed to the electronic transition from HOMO−1 to LUMO. Apparently, the large extent of π-conjugation is conducive toward the increase in the σTmax value and the red-shift in the spectra. The maximum TPA cross-sections for benzene-fused DCCA-2 (282 GM/1000 nm) and DCCA-3 (316 GM/939 nm) product molecules are more than three times as large as that for DCCA (90.6 GM/792 nm). It is noteworthy that the TPA action cross-section (ΦσTmax) for DCCA-3 is up to 117 GM, while that for the experimental DCCA molecule is only 46 GM (<50 GM). However, the σTmax value for DCCA-4 (118.6 GM) is rather small as compared to that for DCCA-3 (316 GM).
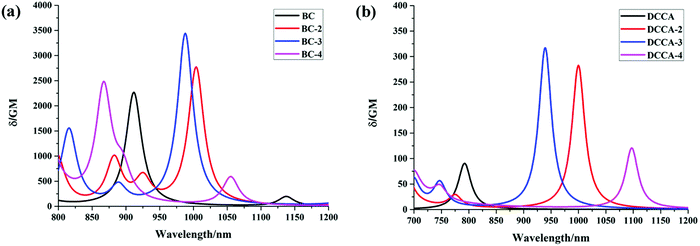 |
| | Fig. 8 TPA spectra of the studied molecules. | |
Table 3 Calculated TPA properties including the maximum TPA cross-section (σTmax), corresponding TPA wavelength (λTmax), and transition nature of all the investigated molecules by means of the quadratic response theory method
| Molecules |
λ
Tmax/nma |
σ
Tmax/GMa |
λ
Tmax/nmb |
σ
Tmax/GMb |
Transition natureb |
|
Experimental data.10
Results were calculated at the B3LYP/6-311+G(d) level of theory using DALTON programs in a water solvent.
|
|
BC
|
|
|
911.65 |
2260 |
S0 → S2 |
H−1 → L |
92.24% |
|
|
|
1137.48 |
187.8 |
S0 → S1 |
H → L |
98.81% |
|
BC-2
|
|
|
1003.92 |
2740 |
S0 → S2 |
H−1 → L |
89.79% |
|
|
|
925.26 |
486 |
S0 → S3 |
H−2 → L |
90.69% |
|
|
|
882.46 |
906 |
S0 → S4 |
H → L+1 |
90.58% |
|
BC-3
|
|
|
987.93 |
3420 |
S0 → S2 |
H−1 → L |
93.26% |
|
|
|
888.78 |
356 |
S0 → S3 |
H−2 → L |
96.17% |
|
|
|
815.69 |
1502 |
S0 → S4 |
H → L+1 |
67.57% |
|
BC-4
|
|
|
867.03 |
2260 |
S0 → S4 |
H → L+1 |
95.79% |
|
|
|
1055.19 |
562 |
S0 → S2 |
H−1 → L |
96.57% |
|
|
|
895.20 |
568 |
S0 → S3 |
H−2 → L |
96.46% |
|
DCCA
|
760 |
164.7 |
792.24 |
90.6 |
S0 → S1 |
H → L |
98.11% |
|
DCCA-2
|
|
|
999.88 |
282 |
S0 → S1 |
H → L |
96.18% |
|
|
|
774.91 |
23.8 |
S0 → S2 |
H−1 → L |
88.42% |
|
DCCA-3
|
|
|
939.28 |
316 |
S0 → S1 |
H → L |
97.90% |
|
|
|
746.90 |
47 |
S0 → S2 |
H−1 → L |
78.70% |
|
DCCA-4
|
|
|
1097.21 |
118.6 |
S0 → S1 |
H → L |
99.17% |
|
|
|
746.90 |
26 |
S0 → S2 |
H−1 → L |
94.10% |
In order to further explain the effect of benzene-fused positions on the TPA of coumarin derivatives, we calculated and analyzed the TPA cross-sections for the series of DCCA compounds through a two-state model, where the TP transition probability (δ) can be expressed as follows:27
| | 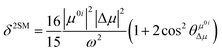 | (3) |
where
μ0i is the transition dipole moment from the ground to the
ith excited state and Δ
μ is the difference between the excited and ground-state dipole moments. Further,
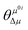
refers to the angle between
μ0i and Δ
μ. Furthermore,
ω is the excitation energy. These values are listed in
Table 4. Evidently, the regularity of
δ2SM values calculated by using the two-state model is in good agreement with those calculated by using the response theory for the studied molecules. In all the benzocoumarin derivatives, an increase in the physical parameters such as the excitation energy (
ω) and dipole moment difference (Δ
μ) can facilitate an increase in the
δ values as compared to those for
DCCA. As shown in
Fig. 9 and
Table 4, the Δ
μ values for benzene-fused
DCCA-2,
DCCA-3, and
DCCA-4 compounds are much larger than that for
DCCA, which can be attributed to the relatively larger excited-state dipole moments
μ1 for the former compounds. Moreover, the
δ2SM value for
DCCA-4 (10727.05) is much smaller than that for
DCCA-3 (16274.94) because the
μ0i value for
DCCA-4 is the smallest in all the investigated compounds due to the geometric characteristic and short CT distance (
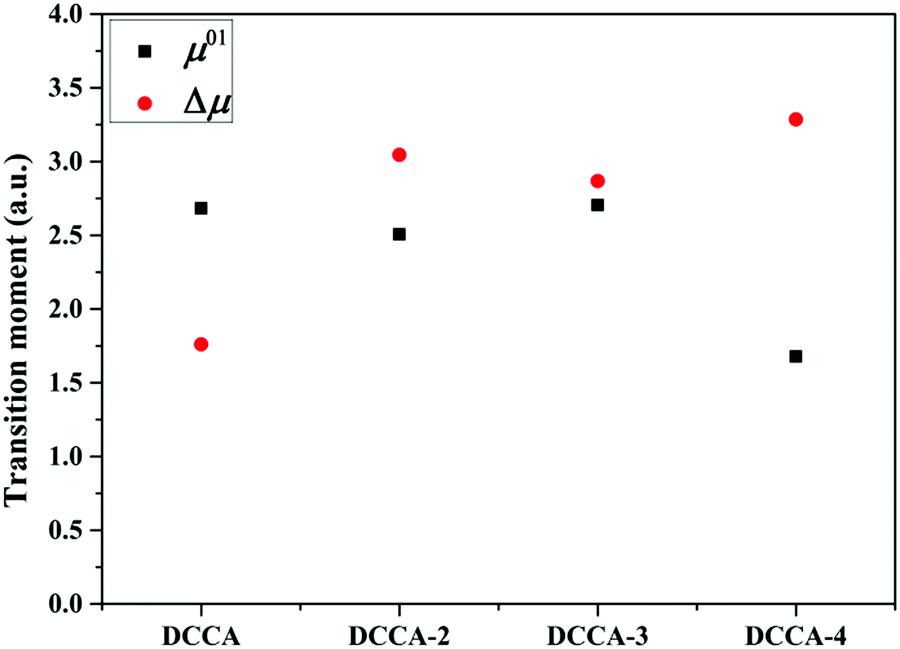
Fig. 5). Therefore, it is evident that the benzene-fused position plays an important role in determining the excitation energy and transition dipole moment, and it also provides an effective way to increase the TPA cross-section through an increase in the phenyl group in a suitable position in the coumarin backbone.
Table 4 Simulated physical parameters (excited energy ω, transition dipole moment μ0i from the ground to the ith excited state, ground-state dipole moment μ0, excited-state dipole moment μ1, difference between the excited and ground-state dipole moments Δμ, and the angle between the two-state dipole moment vectors 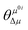 ) related to the TPA spectra by the two-state model and B3LYP/6-311+G(d) method using Gaussian software, and the numbers in the parentheses refer to the response theory values calculated by the DALTON software
) related to the TPA spectra by the two-state model and B3LYP/6-311+G(d) method using Gaussian software, and the numbers in the parentheses refer to the response theory values calculated by the DALTON software
|
|
i
|
ω
|
μ
0i
|
μ
0
|
μ
1
|
Δμ |
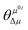
|
δ
2SM
|
|
|
DCCA
|
1 |
0.13 |
2.68 |
4.91 |
6.65 |
1.76 |
8.92 |
4466.20 |
(3603.82) |
|
DCCA-2
|
1 |
0.10 |
2.51 |
5.36 |
8.36 |
3.04 |
164.32 |
17100.06 |
(15566.16) |
|
DCCA-3
|
1 |
0.11 |
2.70 |
5.15 |
8.01 |
2.87 |
174.53 |
16274.94 |
(14116.65) |
|
DCCA-4
|
1 |
0.09 |
1.68 |
4.15 |
7.30 |
3.29 |
6.33 |
10727.05 |
(7136.297) |
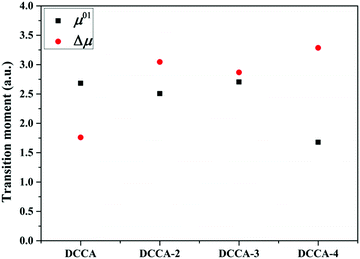 |
| | Fig. 9 Transition dipole moment and dipole moment difference of the investigated compounds. | |
4. Conclusions
In this paper, we successfully designed an excellent ratiometric TP BC-3 fluorescent probe and the corresponding DCCA-3 product molecule, which possess large TPA cross-sections in the NIR region (3420 GM/988 nm; 316 GM/939 nm) and large Stokes (116 nm; 60 nm) and emission (225 nm) shifts. These abovementioned characteristics contribute toward facilitating spectral recognition and reducing photodamage. It is noteworthy that the effective TPA cross-section (Φσ) for the benzocoumarin derivative DCCA-3 is up to 117 GM, while this value for the experimental DCCA molecule is only 46 GM. Moreover, the significant influence of the benzene-fused position in the coumarin backbone on the transition dipole moment and nonradiative decay channels was comprehensively investigated by the TP transition assignment and normal mode analyses. The crucial characteristic frequency regions for the different benzene-fused styles and the resulting different electron-vibration coupling effects were discussed. In the three isomeric benzocoumarin derivatives, DCCA-3 possesses the largest TP cross-section and fluorescence quantum yield due to large transition dipole moment and small nonradiative decay rate. The nonradiative decay rate is larger for DCCA-2 because of the drastic side-chain twisting modes in the low-frequency-regime, resulting in the relatively smaller fluorescence quantum yield for DCCA-2 (0.12) than that for DCCA-3 (0.37). For DCCA-4, the short CT distance and large geometric relaxation lead to its extremely weak fluorescence. Summarily, the appropriate benzene-fused expansion of the coumarin backbone is an effective way to increase the absorption and emissive wavelength, TPA cross-section, and fluorescence efficiency. In this paper, we comprehensively discuss the effect of benzene-fused position on a molecule's optical properties and explained the fluorescence near-quenching mechanism for benzo[f]coumarin derivative DCCA-4 for the first time. Moreover, we provided a worthwhile idea for the design and application of ratiometric TP fluorescent probes with high efficiency and large cross-sections for the in vivo monitoring of the level of H2O2.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Natural Science Foundation of China (No. 21473071, 21173099, 20973078 and 20673045) and the Major State Basis Research Development Program (2013CB 834801).
Notes and references
-
(a) R. S. Balaban, S. Nemoto and T. Finkel, Cell, 2005, 120(4), 483–495 CrossRef CAS PubMed;
(b) D. Chen, K. P. Taylor, Q. Hall and J. M. Kaplan, Genetics, 2016, 204(3), 1151–1159 CrossRef CAS PubMed.
- T. Finkel, M. Serrano and M. A. Blasco, Nature, 2007, 448(7155), 767–774 CrossRef CAS PubMed.
- M. T. Lin and M. F. Beal, Nature, 2006, 443(7113), 787–795 CrossRef CAS PubMed.
- A. M. Shah, Heart, 2004, 90(5), 486–487 CrossRef CAS PubMed.
-
(a) M. Tarvin, B. McCord, K. Mount, K. Sherlach and M. L. Miller, J. Chromatogr. A, 2010, 1217(48), 7564–7572 CrossRef CAS PubMed;
(b) P. Gimeno, C. Bousquet, N. Lassu, A. F. Maggio, C. Civade, C. Brenier and L. Lempereur, J. Pharm. Biomed. Anal., 2015, 107, 386–393 CrossRef CAS PubMed;
(c) E. Y. Jomma, N. Bao and S.-N. Ding, Anal. Methods, 2017, 9(23), 3513–3518 RSC.
-
(a) H. Maeda, Y. Fukuyasu, S. Yoshida, M. Fukuda, K. Saeki, H. Matsuno, Y. Yamauchi, K. Yoshida, K. Hirata and K. Miyamoto, Angew. Chem., Int. Ed. Engl., 2004, 43(18), 2389–2391 CrossRef CAS PubMed;
(b) A. R. Lippert, G. C. V. De Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44(9), 793–804 CrossRef CAS PubMed;
(c) G. Li, D. Zhu, Q. Liu, L. Xue and H. Jiang, Org. Lett., 2013, 15(4), 924–927 CrossRef CAS PubMed;
(d) J. Xu, Y. Zhang, H. Yu, X. Gao and S. Shao, Anal. Chem., 2016, 88(2), 1455–1461 CrossRef CAS PubMed;
(e) X. Liu, B. Hu, R. Cheng, F. Kong, X. Pan, K. Xu and B. Tang, Chem. Commun., 2016, 52(40), 6693–6696 RSC.
-
(a) R. M. Williams, W. R. Zipfel and W. W. Webb, Curr. Opin. Chem. Biol., 2001, 5(5), 603–608 CrossRef CAS PubMed;
(b) F. Helmchen and W. Denk, Nat. Methods, 2005, 2(12), 932–940 CrossRef CAS PubMed;
(c) W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21(11), 1369–1377 CrossRef CAS PubMed.
-
(a) E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127(47), 16652–16659 CrossRef CAS PubMed;
(b) J. Jing and J.-L. Zhang, Chem. Sci., 2013, 4(7), 2947 RSC;
(c) K. M. Zhang, W. Dou, P. X. Li, R. Shen, J. X. Ru, W. Liu, Y. M. Cui, C. Y. Chen, W. S. Liu and D. C. Bai, Biosens. Bioelectron., 2015, 64, 542–546 CrossRef CAS PubMed;
(d) M. Ren, B. Deng, J. Y. Wang, X. Kong, Z. R. Liu, K. Zhou, L. He and W. Lin, Biosens. Bioelectron., 2016, 79, 237–243 CrossRef CAS PubMed;
(e) Q. Ma, X. li, J. Zhang, X. Zhu, L. Zhou and H. Liu, Anal. Methods, 2017, 9(31), 4558–4565 RSC;
(f) H. Li, Q. Yao, J. Fan, J. Du, J. Wang and X. Peng, Biosens. Bioelectron., 2017, 94, 536–543 CrossRef CAS PubMed;
(g) K. Naidu Bobba, M. Won, I. Shim, N. Velusamy, Z. Yang, J. Qu, J. S. Kim and S. Bhuniya, Chem. Commun., 2017, 53(81), 11213–11216 RSC.
-
(a) M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44(13), 4185–4191 RSC;
(b) D. Andina, J. C. Leroux and P. Luciani, Chemistry, 2017, 23(55), 13549–13573 CrossRef CAS PubMed.
- B. Dong, X. Song, X. Kong, C. Wang, Y. Tang, Y. Liu and W. Lin, Adv. Mater., 2016, 28(39), 8755–8759 CrossRef CAS PubMed.
-
(a) H. M. Kim, P. R. Yang, M. S. Seo, J. S. Yi, J. H. Hong, S. J. Jeon, Y. G. Ko, K. J. Lee and B. R. Cho, J. Org. Chem., 2007, 72(6), 2088–2096 CrossRef CAS PubMed;
(b) J. H. Son, C. S. Lim, J. H. Han, I. A. Danish, H. M. Kim and B. R. Cho, J. Org. Chem., 2011, 76(19), 8113–8116 CrossRef CAS PubMed.
-
(a) I. Kim, D. Kim, S. Sambasivan and K. H. Ahn, Asian J. Org. Chem., 2012, 1(1), 60–64 CrossRef CAS;
(b) D. Kim, S. Singha, T. Wang, E. Seo, J. H. Lee, S. J. Lee, K. H. Kim and K. H. Ahn, Chem. Commun., 2012, 48(82), 10243–10245 RSC;
(c) D. Kim, S. Sambasivan, H. Nam, K. H. Kim, J. Y. Kim, T. Joo, K. H. Lee, K. T. Kim and K. H. Ahn, Chem. Commun., 2012, 48(54), 6833–6835 RSC;
(d) Y. W. Jun, S. Sarkar, S. Singha, Y. J. Reo, H. R. Kim, J. J. Kim, Y. T. Chang and K. H. Ahn, Chem. Commun., 2017, 53(78), 10800–10803 RSC;
(e) S. Sarkar, M. Santra, S. Singha, Y. W. Jun, Y. J. Reo, H. R. Kim and K. H. Ahn, J. Mater. Chem. B, 2018, 6(27), 4446–4452 RSC.
-
(a) D. Kim, Q. P. Xuan, H. Moon, Y. W. Jun and K. H. Ahn, Asian J. Org. Chem., 2014, 3(10), 1089–1096 CrossRef CAS;
(b) M. Tasior, D. Kim, S. Singha, M. Krzeszewski, K. H. Ahn and D. T. Gryko, J. Mater. Chem. C, 2015, 3(7), 1421–1446 RSC.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113(17), 2019–2039 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105(8), 2999–3093 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria;M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada;M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., P. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers;K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand;K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski;R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian,Inc., Wallingford, CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
-
Z. Shuai, Q. Peng, Y. Niu and H. Geng, MOMAP, Revision 0.3.001, Beijing, China, 2016, MOMAP: a free and open-source molecular materials property prediction package; avaliable online http://www.shuaigroup.net.
-
(a) P. Weber and J. R. Reimers, J. Phys. Chem. A, 1999, 103(48), 9830–9841 CrossRef CAS;
(b) Z. L. Cai and J. R. Reimers, J. Phys. Chem. A, 2000, 104(36), 8389–8408 CrossRef CAS.
- P. Sałek, O. Vahtras, T. Helgaker and H. Ågren, J. Chem. Phys., 2002, 117(21), 9630–9645 CrossRef.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekstrom, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernandez, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hattig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenaes, S. Host, I. M. Hoyvik, M. F. Iozzi, B. Jansik, H. J. Jensen, D. Jonsson, P. Jorgensen, J. Kauczor, S. Kirpekar, T. Kjaergaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnaes, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Salek, C. C. Samson, A. S. de Meras, T. Saue, S. P. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thogersen, O. Vahtras, M. A. Watson, D. J. Wilson, M. Ziolkowski and H. Agren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(3), 269–284 CAS.
- R. C. Hilborn, Am. J. Phys., 1982, 50, 982–986 CrossRef CAS.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129(30), 9333–9339 CrossRef CAS PubMed.
- X. K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J. L. Bredas, Adv. Mater., 2017, 29, 46 Search PubMed.
-
(a) S. H. Lin, J. Chem. Phys., 1966, 44(10), 3759–3767 CrossRef;
(b) Y. Niu, Q. Peng and Z. Shuai, Sci. China, Ser. B: Chem., 2008, 51(12), 1153–1158 CrossRef CAS.
- Q. Peng, Y. Niu, Q. Shi, X. Gao and Z. Shuai, J. Chem. Theory Comput., 2013, 9(2), 1132–1143 CrossRef CAS PubMed.
-
(a) N. A. Murugan, R. Zalesny, J. Kongsted, A. Nordberg and H. Agren, Chem. Commun., 2014, 50(79), 11694–11697 RSC;
(b) M. M. Alam, M. Chattopadhyaya and S. Chakrabarti, Phys. Chem. Chem. Phys., 2012, 14(3), 1156–1565 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06050a |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a








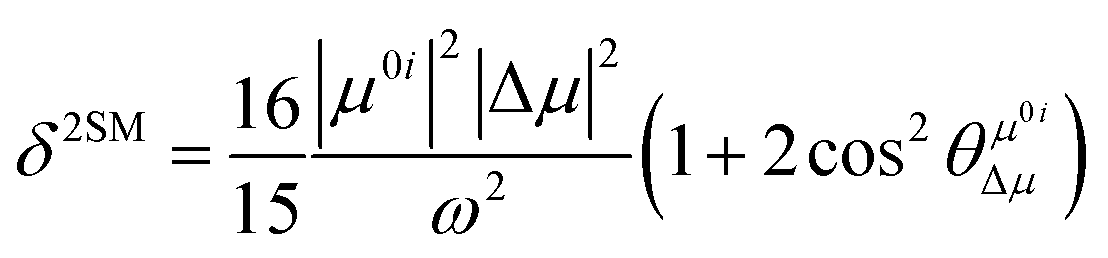
 refers to the angle between μ0i and Δμ. Furthermore, ω is the excitation energy. These values are listed in Table 4. Evidently, the regularity of δ2SM values calculated by using the two-state model is in good agreement with those calculated by using the response theory for the studied molecules. In all the benzocoumarin derivatives, an increase in the physical parameters such as the excitation energy (ω) and dipole moment difference (Δμ) can facilitate an increase in the δ values as compared to those for DCCA. As shown in Fig. 9 and Table 4, the Δμ values for benzene-fused DCCA-2, DCCA-3, and DCCA-4 compounds are much larger than that for DCCA, which can be attributed to the relatively larger excited-state dipole moments μ1 for the former compounds. Moreover, the δ2SM value for DCCA-4 (10727.05) is much smaller than that for DCCA-3 (16274.94) because the μ0i value for DCCA-4 is the smallest in all the investigated compounds due to the geometric characteristic and short CT distance (Fig. 5). Therefore, it is evident that the benzene-fused position plays an important role in determining the excitation energy and transition dipole moment, and it also provides an effective way to increase the TPA cross-section through an increase in the phenyl group in a suitable position in the coumarin backbone.
refers to the angle between μ0i and Δμ. Furthermore, ω is the excitation energy. These values are listed in Table 4. Evidently, the regularity of δ2SM values calculated by using the two-state model is in good agreement with those calculated by using the response theory for the studied molecules. In all the benzocoumarin derivatives, an increase in the physical parameters such as the excitation energy (ω) and dipole moment difference (Δμ) can facilitate an increase in the δ values as compared to those for DCCA. As shown in Fig. 9 and Table 4, the Δμ values for benzene-fused DCCA-2, DCCA-3, and DCCA-4 compounds are much larger than that for DCCA, which can be attributed to the relatively larger excited-state dipole moments μ1 for the former compounds. Moreover, the δ2SM value for DCCA-4 (10727.05) is much smaller than that for DCCA-3 (16274.94) because the μ0i value for DCCA-4 is the smallest in all the investigated compounds due to the geometric characteristic and short CT distance (Fig. 5). Therefore, it is evident that the benzene-fused position plays an important role in determining the excitation energy and transition dipole moment, and it also provides an effective way to increase the TPA cross-section through an increase in the phenyl group in a suitable position in the coumarin backbone.
 ) related to the TPA spectra by the two-state model and B3LYP/6-311+G(d) method using Gaussian software, and the numbers in the parentheses refer to the response theory values calculated by the DALTON software
) related to the TPA spectra by the two-state model and B3LYP/6-311+G(d) method using Gaussian software, and the numbers in the parentheses refer to the response theory values calculated by the DALTON software