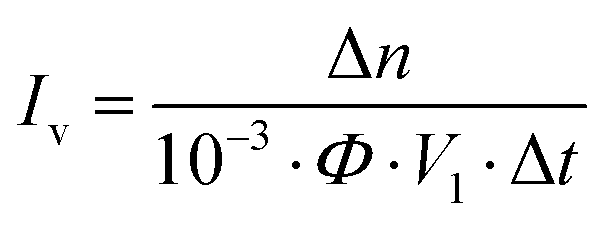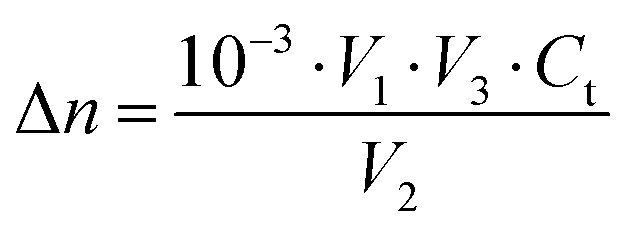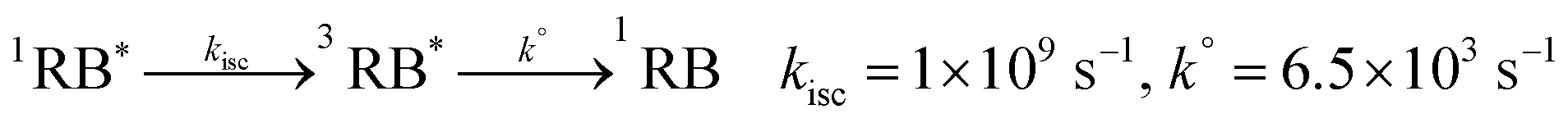Reaction of phenol with singlet oxygen†
Received
31st July 2018
, Accepted 21st November 2018
First published on 21st November 2018
Abstract
Photo-degradation of organic pollutants plays an important role in their removal from the environment. This study provides an experimental and theoretical account of the reaction of singlet oxygen O2(1Δg) with the biodegradable-resistant species of phenol in an aqueous medium. The experiments combine customised LED-photoreactors, high-performance liquid chromatography (HPLC), and electron paramagnetic resonance (EPR) imaging, employing rose bengal as a sensitiser. Guided by density functional theory (DFT) calculations at the M062X level, we report the mechanism of the reaction and its kinetic model. Addition of O2(1Δg) to the phenol molecule branches into two competitive 1,4-cycloaddition and ortho ene-type routes, yielding 2,3-dioxabicyclo[2.2.2]octa-5,7-dien-1-ol (i.e., 1,4-endoperoxide 1-hydroxy-2,5-cyclohexadiene) and 2-hydroperoxycyclohexa-3,5-dien-1-one, respectively. Unimolecular rearrangements of the 1,4-endoperoxide proceed in a facile exothermic reaction to form the only experimentally detected product, para-benzoquinone. EPR revealed the nature of the oxidation intermediates and corroborated the appearance of O2(1Δg) as the only active radical participating in the photosensitised reaction. Additional experiments excluded the formation of hydroxyl (HO˙), hydroperoxyl (HO2˙), and phenoxy intermediates. We detected for the first time the para-semibenzoquinone anion (PSBQ), supporting the reaction pathway leading to the formation of para-benzoquinone. Our experiments and the water-solvation model result in the overall reaction rates of kr-solvation = 1.21 × 104 M−1 s−1 and kr = 1.14 × 104 M−1 s−1, respectively. These results have practical application to quantify the degradation of phenol in wastewater treatment.
1. Introduction
Even though light-induced reactions remain one of the less explored areas of green chemistry, many researchers have attempted to use sunlight in wastewater-detoxification processes, particularly in those involving biodegradation-resistant materials such as phenol.1 An effective photosensitisation process relies on efficient light absorption. Unfortunately, the transparency of phenol mandates the use of an intermediate (dye) to absorb visible light in order to generate reactive oxygen species (ROS), mainly singlet oxygen O2(1Δg), to initiate the photo-degradation process.2–4
Dye-sensitised photo systems generate singlet oxygen O2(1Δg) efficiently,5,6 resulting in a wide range of applications of such processes in chemical,7 medical,8 biological,9 and wastewater10–12 treatments. Singlet oxygen O2(1Δg) represents one of the most reactive ROS, residing 94 kJ mol−1 (ΔGH0298) above the ground state triplet oxygen (O2(3Σ−g)).13,14 One of the prominent effects of singlet oxygen stems from its role in chain initiation and propagation15–17 with aromatic organic compounds such as phenol. Reactions of O2(1Δg) include [π2+π2] 1,2-cycloaddition to isolated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, [π2+π4] 1,4-cycloaddition to conjugated double bonds either in alkenes or aromatics, and the ene (Schenck) reaction normally involving the transfer of an allylic hydrogen from the carbon atom to the oxygen molecule.18,19
C bonds, [π2+π4] 1,4-cycloaddition to conjugated double bonds either in alkenes or aromatics, and the ene (Schenck) reaction normally involving the transfer of an allylic hydrogen from the carbon atom to the oxygen molecule.18,19
Removing phenol and its derivatives, by reacting them with the ROS such as singlet oxygen, represents an attractive pathway for decreasing the toxicity of contaminated water. Phenols pollute surface water through industrial effluents in areas such as refineries (6–500 mg L−1) and coal processing (9–6800 mg L−1) plants, severely affecting aquatic life, even at concentrations as low as 1 ppb.4,20 On the other hand, the steady-state concentration of singlet oxygen that exists in natural water varies within a range of (6–28) × 10−14 M.21 In natural waters, singlet oxygen forms with a quantum yield of 1 to 3%,22–24 indicating its likelihood to coexist with phenol.
Previous investigations on the photooxidation of phenol with singlet oxygen reported different products, such as para-benzoquinone, hydroquinone, and hydroxybiphenyls.10–12,25 However, the underlining reaction mechanism remains poorly understood. Both the product distribution and photochemical efficiency strongly depend, in most cases, on the concentration of oxygen, a selected sensitiser and the pH of the reaction. For instance, alkaline conditions prompt phenol to deprotonate into phenoxide ion (aka phenolate, PhO−, pKa = 9.8) leading to the formation of 2-hydroxy-1,4-benzoquinone, 2-hydroxyhydroquinone, 2,5-dihydroxy-1,4-benzoquinone and others, rather than the usual para-benzoquinone.4 Briviba et al.12 and Pizzocaro et al.25 demonstrated that reaction of O2(1Δg) with the phenol molecule leads selectively to the formation of para-benzoquinone. The absence of catechol and ortho-benzoquinone suggested the formation of a 1,4-endoperoxide (2,3-dioxabicyclo[2.2.2]octa-5,7-dien-1-ol) as an initial intermediate via a 1,4-cycloaddition mechanism, with subsequent steps proposed to involve unimolecular elimination of water. Other experimental studies attributed the formation of para-benzoquinone-type derivatives to the abstraction of phenolic hydrogen by O2(1Δg), followed by reactions with the ground-state triplet oxygen (O2(3Σ−g)).26,27 Hence, the overall oxidation pathways of phenol by singlet oxygen still remain largely speculative. Moreover, experiments appear insufficient for conclusive mechanistic and kinetic insights because of uncertainties relating to the very short lifetime of singlet molecular oxygen (∼10−4 s)14 and the possible parallel reactions involving triplet oxygen O2(3Σ−g), and hydroxyl radical (HO˙).28 Consequently, experimental investigation of the reaction of O2(1Δg) with phenol must be coupled with a sophisticated theoretical analysis.
To this end, the present contribution develops an experimental apparatus to react phenol with O2(1Δg) in a semi-batch reactor with known initial concentrations of phenol and O2(1Δg) produced continuously in situ from bubbled O2(3Σ−g). We identify and quantify the intermediate and product species as a function of time using UV-vis and high-performance liquid chromatography (HPLC). Reported findings map out the governing mechanism for the reaction of phenol with singlet oxygen, where the appearance of radical species is confirmed by electron paramagnetic resonance (EPR) measurements. These results, supplemented by the quantum chemical calculations, afford formulation of a kinetic model that offers practical applications to water treatment.
2. Materials and methodology
2.1. Materials
We obtained all chemicals with the highest commercially available purity. The sensitiser (rose bengal, 85%, Acros Organics), reactant (phenol, >99%, Sigma Aldrich) and expected products (1,4-benzoquinone, 99%; hydroquinone, 98%; catechol, >99%) were sourced from Sigma Aldrich, as were scavengers of the singlet oxygen (sodium azide, >99.99%; glutathione, >98%; DL-dithiothreitol, >99%; all Sigma Aldrich) and scavengers of hydroxyl radical (tert-butyl alcohol, >99%; sodium formate, 99.998%). 2-Propanol (>99%) was from Ajax Finechem. TEMP (2,2,6,6-tetramethyl-4-piperidone 99.9%), which functions as an EPR spin trap for singlet O2, and TEMPO (2,2,6,6-tetramethyl-4-piperidone-N-oxyl, 97%), a standard product of the reaction of TEMP with singlet oxygen, were from Merck. Actinometry reagents (iron chloride, >99%; potassium oxalate monohydrate, 98.5%; sodium acetate anhydrous, 99%; and 1,10-phenanthroline monohydrate, 99.5%) were from Chem Supply, and iron sulfate heptahydrate and sulfuric acid (98%) were from Fisher Scientific. All aqueous solutions were prepared for EPR, UV-vis, and HPLC standards and mobile phases using ultrapure water (resistivity >18 MΩ cm), tapped from a Sartorius Arium Pro UV/DI water purification system.
2.2. Photooxidation of phenol with singlet oxygen
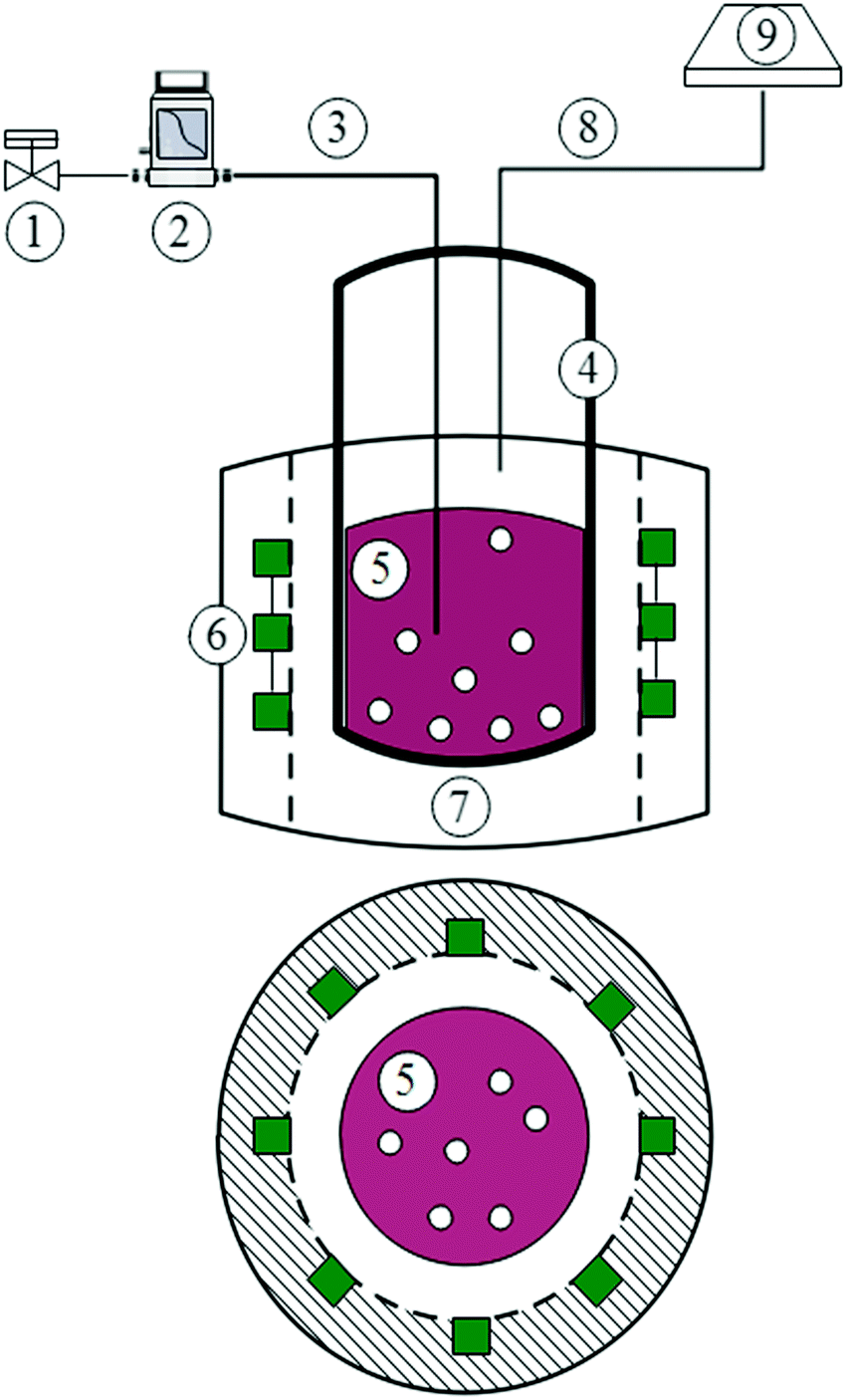
Generally, polychromatic (lamp)29 and monochromatic (laser)30 lights generate the electromagnetic energy in the UV and visible range for photoreactions. The selection criteria depend on the environmental conditions and the required efficiency. For example, laser sources illuminate living cells in biological systems, while monochromatic light such as UV radiation covers larger areas; e.g., in wastewater treatment.29 Examples of monochromatic lamps include light-emitting diodes (LEDs), xenon arcs, and medium-pressure Hg lamps selected by their relative photon intensity and efficiency. Among them, LEDs exhibit the most effective operation, with the lighting efficiency of photo-oxygenated system attaining 70%.31 Hence, the present study deployed LEDs as a light source. Fig. 1 illustrates the photoreactor. We employed 24 green LEDs (4.5 × 10 × 500 mm), operated between 7 and 24 V. The LEDs form a 10 cm-long illuminating zone, assembled from eight strips, each consisting of three units. We uniformly mounted the strips onto the inside of a white PVC tube casing that surrounds the reactor. The dead-zone thickness between the LED units in a single strip amounted to 0.68 cm, based on a 120° beam angle, prompting us to centre the quartz reactor 1 cm away from the PVC holder.
 |
| | Fig. 1 Schematic diagram of photochemical reactor shown in side and cross-sections. 1: O2/He (3%) valve, 2: mass flow controller, 3: inlet gas, 4: pyrex reactor, 5: phenol dyed solution (pH = 6), 6: LED strip (7 V), 7: PVC holder, 8: off-gas outlet, 9: exhaust. | |
Fig. S1 in ESI† shows that, rose bengal (RB) displays a strong absorption in the green band with a maximum around 510 nm and molar absorption coefficient as high as 105 M−1 cm−1.32,33 RB constitutes an efficient photosensitiser with a quantum yield of (0.76–0.85).34 To ensure a clean and pure source of singlet oxygen, the energy difference between the sensitiser and its first triplet excited state should be equal or greater than the singlet oxygen energy level of about 94 kJ mol−1 (i.e. 0.98 eV) above the ground-state triplet oxygen.35 The energy difference between RB singlet ground and its first triplet excited state corresponds to 166 kJ mol−1 (1.72 eV).36 Another reason for using RB lies in its stability over the time scale of our experiments (i.e., around 1 h). We conducted the bleaching test of RB by illuminating a 35 μM of the sensitiser-buffered solution for 0–24 h, followed by testing the absorbance (abs) in a range of 200–800 nm using a UV-vis spectrophotometer. The bleaching rate of rose bengal (rB), calculated from the rate of change of absorbance ((abs − abs°) × t (s)−1), amounts to 3.5 × 10−15 M s−1.
We prepared buffered solutions (pH = 6 or pD = 6.29, confirmed by a Hach pH-meter) of 1 mM phenol and 35 μM rose bengal (5, Fig. 1) freshly in D2O or H2O in a temperature-controlled laboratory maintained at 25 °C. The illumination experiments lasted 1 to 50 min and involved 5 mL aliquots, sourced from the stock solution and charged into the photoreactor. During the photo-induced reaction, a calibrated mass flow controller (Brooks 4800 series) fed a 3% O2 (in He balance) continuously at a constant rate of 500 mL min−1. The helium carrier gas (instead of the generally applied N2 medium) reduced the intermolecular interaction between the excited-state molecules and the inert gas. Okamoto et al.37 reported no effect of superficial gas velocity (i.e., no mass transfer limitation) on the phenol reaction rate at above 0.35 cm s−1, prompting us to adopt 0.44 cm s−1 as the superficial gas velocity in our experiments. We estimated the concentration of oxygen in the aqueous phase to reach 3.9 × 10−5 M based on Henry's law (PO2 = KH˙[O2], where KH = 769.23 atm M−1)38 and 3% concentration of O2 in He carrier gas (P = 1 atm).
2.3. Analytical techniques
A Shimadzu HPLC instrument, equipped with a C18 analytical column (4.6 mm diameter and 150 mm length) and controlled by data acquisition software, served to quantify the conversion of phenol and the yields of the reaction products from the LED photoreactor. The analytical method involved injections of samples of 10 μL in volume and maintaining the oven temperature at 30 °C. The mobile phase comprised a solution of 0.01 mol L−1 phosphate buffer (pH 4.5), methanol, and tetrahydrofuran (THF) in the ratio of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5, v/v, flowing at a rate of 1 mL min−1, under low-pressure conditions. The analyte elutes into a UV-vis detector operating at 254 nm using a reversed phase elution mode. Under these conditions, phenol eluted at 25.5 min and para-benzoquinone at 4 min (see Fig. S2, ESI†). Furthermore, we obtained the absorption spectra of RB (Fig. S1, ESI†), phenol (not shown), hydroquinone (Fig. S3, ESI†), para-benzoquinones (Fig. S3, ESI†), and complex of ferrous ion and 1,10-phenanthroline (actinometry experiment, Fig. S4, ESI†) in a quartz cell (1 cm pathlength) using Agilent Carry 5000 UV-vis-NIR spectrophotometer operated at a resolution of 1 nm. Based on the spectra in Fig. S1, S3 and S4 (ESI†), we were able to confirm 254 nm as an optimum wavelength for HPLC analyses.
:5:5, v/v, flowing at a rate of 1 mL min−1, under low-pressure conditions. The analyte elutes into a UV-vis detector operating at 254 nm using a reversed phase elution mode. Under these conditions, phenol eluted at 25.5 min and para-benzoquinone at 4 min (see Fig. S2, ESI†). Furthermore, we obtained the absorption spectra of RB (Fig. S1, ESI†), phenol (not shown), hydroquinone (Fig. S3, ESI†), para-benzoquinones (Fig. S3, ESI†), and complex of ferrous ion and 1,10-phenanthroline (actinometry experiment, Fig. S4, ESI†) in a quartz cell (1 cm pathlength) using Agilent Carry 5000 UV-vis-NIR spectrophotometer operated at a resolution of 1 nm. Based on the spectra in Fig. S1, S3 and S4 (ESI†), we were able to confirm 254 nm as an optimum wavelength for HPLC analyses.
An electron paramagnetic resonance (EPR) spectrometer served to quantitate the concentration of spin-trapped singlet oxygen, as well as to monitor in situ the appearance of radical species. Chemical spin traps represent highly reactive species that capture singlet oxygen by forming a stable compound (endoperoxide), detectable by the EPR spectrometry. We used the most common singlet oxygen spin trap, 2,2,6,6-tetramethyl-4-piperidone (TEMP), because it reacts rapidly and selectively with O2(1Δg) to form 2,2,6,6-tetramethyl-4-piperidone-N-oxyl (TEMPO), a distinct and stable endoperoxide, with no side products. EPR spectroscopy readily detects TEMPO.39 TEMP demonstrates high solubility in water and transparency to a range of excitation wavelengths.40,41 We did not select other common spin traps (tetrapotassium rubrene-2,3,8,9-tetracarboxylate (RTC) and disodium 9,10-anthracenedipro panoate (ADP)) because of their reduced transparency.42
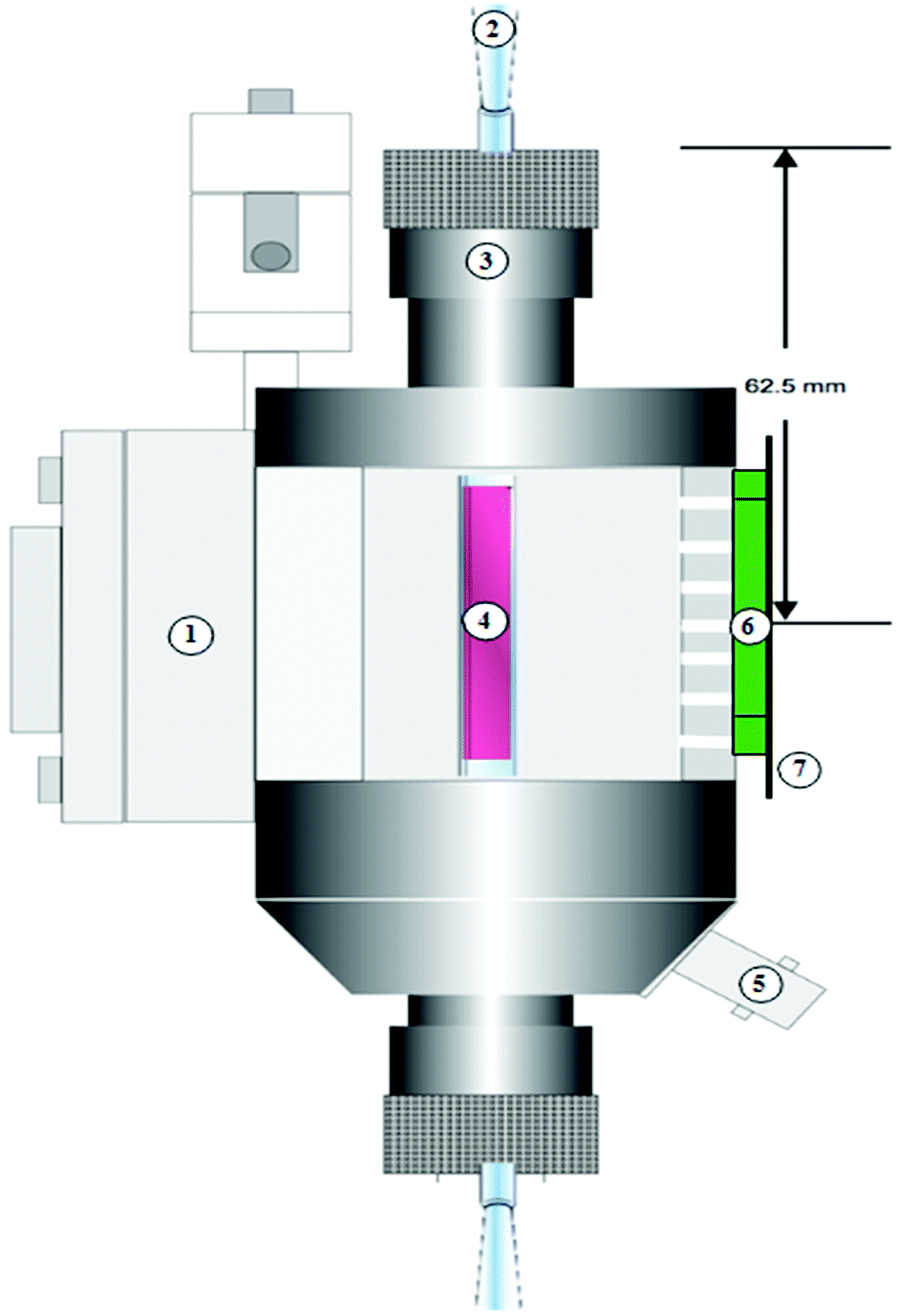
The EMXPlus 10/2.7 EPR spectrometer, fitted with an ER 4102ST general-purpose rectangular X-band cavity and cooling side plates, averaged 5 scans at a resonant frequency of 9.76 GHz. We placed the samples in a 10.5 mm flat cell (WG-812-Q), and bubbled a mixture of 3% oxygen in He carrier gas for a least 20 min. Fig. 2 illustrates an LED light, fixed on a wooden holder to prevent metallic interference with the magnetic field, that irradiated the sample in situ (i.e., inside the EPR resonator cavity). The spectrometer functioned at microwave power of 20 mW, modulation frequency of 100 kHz, modulation amplitude of 1 G, sweep width of 100 G, and with the time constant set to 0.01 ms, conversion time to 50 ms, sweep time to 100 s, receiver gain to 2 × 105, number of data points to 10000, and the centre field to 3487.00 G.
 |
| | Fig. 2
In situ EPR photoreactor schematic; 1: EPR resonator, 2: inlet to the quartz flat cell (Wilmad WG-812-Q), 3: collet nut, 4: dyed sample, 5: modulation cable, 6: green LED (24 V), 7: wooden support. | |
2.4. Estimated LED light intensity
A potassium ferrioxalate actinometer43 was used to obtain light intensity based on the Hatchard–Parker approach for reactors in Fig. 1 and 2.44,45 This method provides reliable results between 253 and 577 nm, based on established quantum yields for potassium ferrioxalate.43 ESI† provides a more detailed description of the experimental procedure; Fig. S4 (ESI†) illustrates the measurement of the absorbance of the ferrioxalate solution by UV-vis spectroscopy.
Eqn (1)–(3) yield the number of absorbed photons:
| |  | (1) |
| |  | (2) |
| |  | (3) |
where Δ
n denotes change of the photo-generated ferrous iron (M),
Φ the ferrioxalate quantum yield at the operated wavelength (0.86, 510 nm
43), Δ
t irradiation time (s),
V1 irradiated volume (mL), and
V2 the volume taken from the irradiated samples (mL),
V3 the diluted volume (mL),
Ct the concentration of ferrous iron after dilution (M), abs. the absorbance at 510 nm which has been corrected by the background of a blank sample,
l equals cm (the light path of the quartz cell) and
ε represents the molar absorptivity in M
−1 cm
−1 and the slope of the standard curve (
ε = 11
073, Fig. S4, ESI
†). Our measurement of
ε concurs well with the recently reported values of 11
050 and 11
100 M
−1 cm
−1.
43
After acquiring Δn/Δt = 5.96 × 10−8 mol min−1 (the slope of the plot in Fig. 3) for the photoreactor (Fig. 1, LED operated at 7 V), the light intensity = 1.157 × 10−9 E L−1 s−1. On the other hand, for the in situ EPR reactor (Fig. 2), the light intensity amounts to 32.75 × 10−5 E L−1 s−1, reflecting the higher voltage of 24 V of the LED light.
 |
| | Fig. 3 Photogenerated ferrous ions (Fe2+) using 24 green LEDs at 0, 15, 30, and 50 min irradiation times. | |
2.5. Computational details
The Gaussian 09 suite of programs46 was used for all energy calculations (Δ‡H0298, ΔrH0298, Δ‡G0298, and ΔrG0298) and structural optimisations. Possible biradical character of the phenol + O2(1Δg) system required determining the spin state of intermediate and transition structures along the entire potential energy surface. Severe spin contaminations required energy refinements based on the approximated spin-projection scheme (AP) pioneered by the Yamagushi group,47 in which the located transition structures and intermediates converge on a pure low-spin solution with zero-spin densities. Structural optimisations that involve fixing the frontier molecular orbitals (i.e., the broken symmetry solution) allow assessing a plausible spin contamination by examining a value of the total spin angular momentum operator S2.48 A value of this operator of near unity for high spin configuration indicates a significant spin contamination. We found that expected values of the spin contamination operator of all of the present intermediates and transition structures clustered close to zero. This means that the phenol + O2(1Δg) system exists in a genuine singlet state, eliminating the need for further energy refinement. However, the energy of the O2(1Δg) suffers from contamination by the triplet state, as evident from the value of the spin contamination operator S2 of approximately 1. As such, we applied the AP approach to refine the energy of the singlet oxygen molecule.
Cremer and co-workers49 showed that unrestricted density functional methods (UDFT) perform well in comparison to multireference methods for deriving energies and geometries of singlet biradicals. Employing hybrid DFT functionals further enhances the accuracy of the UDFT in describing biradical systems.50 Accordingly, we deployed the hybrid DFT functional of M062X51 with the extended 6-311+G(3df,2p)52 basis set. The M062X functional has been parameterised to provide accurate and cost-effective performance for general application in organic chemistry.53–55 We modelled reactions in the aqueous phase by deploying the polarisable continuum model (PCM) with a dielectric constant (εc) equal to 78.39 at 298.15 K. Attempts to investigate the catalytic effect of discrete water molecules, rather than applying PCM, did not yield viable pathways. All transition structures contain one and only one imaginary frequency. We have confirmed the identity of each transition structure by performing intrinsic reaction coordinate (IRC) calculation. In the next section, we quote changes in enthalpy and Gibbs energy calculated at 298.15 K. Finally, the KiSThelP code56 was used for computation of the reaction rate constants in the temperature range of 300–400 K, as included in Table 1.
Table 1 Arrhenius rate parameters fitted in the temperature range of 300–400 K and 1 M for the phenol reaction channels with singlet oxygen; Asolvation is cm3 molecule−1 s−1 or s−1 for unimolecular reaction and Ea-solvation in kJ mol−1
| Reaction |
A
solvation (s−1, cm3 molecule−1 s−1) |
E
a-solvation (kJ mol−1) |
k (M−1 s−1, s−1) |
| 300–400 K |
300–400 K |
298.15 K |
| Phenol + O2(1Δg) → M1 |
8.24 × 10−15 |
15 |
1.21 × 104 |
| Phenol + O2(1Δg) → M2 |
1.03 × 10−14 |
41 |
4.07 × 10−1 |
| Phenol + O2(1Δg) → M3 |
9.75 × 10−15 |
37 |
1.93 × 100 |
| Phenol + O2(1Δg) → M4 |
1.76 × 10−15 |
111 |
3.78 × 10−14 |
| Phenol + O2(1Δg) → M5 |
1.37 × 10−13 |
116 |
3.91 × 10−13 |
|
M6 → M7 |
1.57 × 108 |
57 |
1.62 × 10−2 |
|
M7 → M8 |
1.31 × 1021 |
134 |
2.91 × 10−3 |
|
M8 → para-BQ + H2O |
2.07 × 1021 |
130 |
3.47 × 10−2 |
3. Results and discussion
3.1. Generation of singlet oxygen
Eqn (4)–(9) describe Type II dye-photoreaction mechanism for rose bengal.57 Starting from a sensitiser molecule in its ground state, 1RB absorbs green photon to form a singlet excited dianion (1RB*, eqn (4)) with singlet state energy of ES = 213 kJ mol−1.58 Subsequently, the 1RB* spin-flips to the triplet state (3RB*, ET = 166 kJ mol−1, reaction (6))59 through intersystem crossing (ISC). 3RB* may deactivate by phosphorescence or non-radiative processes60 to 1RB with a rate constant of k° = 6.5 × 103 s−1,57 and 1RB bleaches with the calculated rate of rB = 3.5 × 10−15 M s−1; see Section 2.2. While the photosensitiser molecule relaxes to its ground state, it transfers the excitation energy to a molecule of triplet oxygen 3O2, forming singlet state oxygen 1O2 (reaction (7), kt = 1.2 × 109 M−1 s−157). The vibronic coupling with water quenches singlet oxygen with a pseudo-first-order rate constant of kd = 5 × 105 s−1.61 We do not account for the radiative decay of singlet oxygen (kp, reaction (9)) as this pathway makes negligible contribution to the decay process in solutions.62 The product of hν in reaction (4) represents the light intensity absorbed by RB, calculated earlier using the actinometry technique, and 1RBb denotes the rose bengal bleaching product.| |  | (4) |
| |  | (5) |
| |  | (6) |
| |  | (7) |
| |  | (8) |
| |  | (9) |
The lifetime of singlet oxygen changes dramatically in different solvents63 depending on the energy-transfer efficiency from electronic to intramolecular vibrational states. The closer the vibrational mode of the solvent to that of singlet oxygen, the higher the effectiveness of the deactivating process. For instance, O–D and O–H bonds absorb photons with energy corresponding to 2550 and 3500 cm−1, respectively, whereas singlet oxygen emits photons characterised by energy matching 3286 cm−1; hence, it quenches far faster in water than deuterium oxide (heavy water).64 Snowden65 estimated the natural lifetime of singlet oxygen to be thirteen times higher in D2O than in H2O.
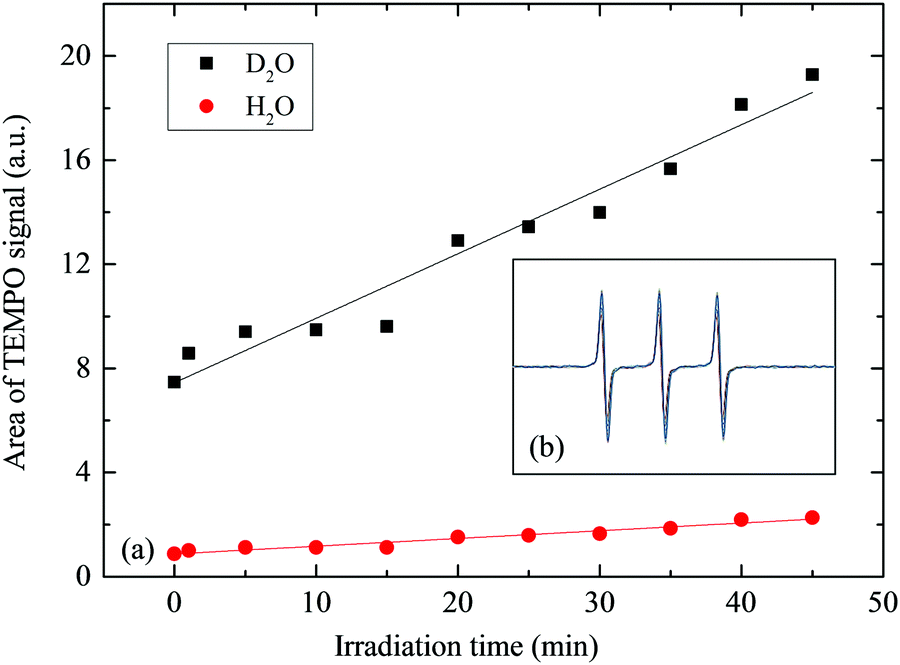
The strictly hindered amine (TEMP) reacts selectively with singlet oxygen to form nitroxide radical (TEMPO) that displays a characteristic EPR signal.66,67 In Fig. 4, we compare the TEMPO signals in 50% (MeOH, D2O) and in 50% (MeOH, H2O) by double-integrating the spectral areas. The yield of TEMPO is on average 8.45 times higher in D2O than H2O, corroborating a longer half-life of O2(1Δg) in D2O. Fig. 4b depicts the in situ spectral profiles of TEMPO after the reaction of TEMP with the photogenerated singlet oxygen, as recorded for the 35 μM RB sensitiser present in 50% (by volume) solution of MeOH in D2O. The signal increased steadily over the analysis time interval of 35 min. Fig. 5 contrasts the experimental and modelled profiles of concentration of singlet oxygen in water, the former obtained from the EPR spin-trap technique and the latter from the solution of the coupled ordinary differential equations that describe the kinetics of the process, as coded in the POLYMATH software (Table S2, ESI†). The measured trend correlates well with the kinetic approximation, indicating an expected concentration of singlet oxygen between (3–4) × 10−6 M in response to 3 h illumination by 24 V LED light.
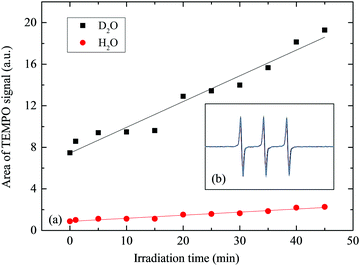 |
| | Fig. 4 Double integrated area of the time course TEMPO signals in H2O (red circles) and D2O (black squares) (a), TEMPO spectra recorded by in situ EPR after illuminating TEMP sample in dyed D2O solution at pD = 6.35, g = 2.00062, and centre field = 3487.00 G (b). | |
 |
| | Fig. 5 EPR concentration profile of singlet oxygen generated in H2O using 24 V LED. The dash-dotted line represents the corresponding values obtained from POLYMATH kinetic solution detailed in Table S1 (ESI†). | |
3.2. Reaction of singlet oxygen with phenol
The photoreaction of phenol with singlet oxygen follows eqn (10) and (11). Eqn (10) illustrates the physical quenching (spin orbital coupling), with no oxygen consumption (kq = 8 × 106 M−1 s−1),68 whereas, eqn (11) describes the overall formation of para-benzoquinone (BQ), written here as a global step and broken down into elementary steps in the next section.| |  | (10) |
| |  | (11) |
3.2.1. Mechanistic and kinetic analysis.
As pictured in Fig. 6, the reaction of phenol with O2(1Δg) branches into five initial channels with Gibbs free energies of activation spanning a relatively wide range between 59 kJ mol−1 and 168 kJ mol−1 (Fig. S5 in ESI† depicts geometries of the located transition structures of all considered reactions). Fig. 6 portrays two [π2+π4] 1,4-cycloaddition reactions producing intermediates M1 and M2 (1,4-peroxide structures). The concerted 1,4-cycloaddition of O2(1Δg) at ipso C(OH) and a para positions requires a 17–22 kJ mol−1 lower energy barrier than corresponding addition at ortho and meta sites. This observation could easily be rationalised based on the charge distribution in the phenol molecule. The hydroxyl group donates charge to ortho and para carbon atoms, leaving C(OH) as an electron-deficient site. Calculated Mulliken charges at para C, meta (C), ortho (C) and ipso C sites amount to −0.194 eV, −0.133 eV, −0.143 eV, 0.354 eV, respectively. It follows that the para carbon appears relatively more susceptible to O2(1Δg) 1,4-cycloaddition than the ortho C atom, and consistently we find a lower energy barrier for the formation of M1 in comparison to the emergence of M2 (59 kJ mol−1versus 81 kJ mol−1).
 |
| | Fig. 6 Potential energy map for the initial channels in the reaction of O2(1Δg) with phenol. Values in bold and italic denote Gibbs energy of reaction and activation (Δ‡G0298 and ΔrG0298) calculated at 298.15 K, respectively in kJ mol−1. The TS1 (green) line represents the preferred pathway, subsequently illustrated in Fig. 7. | |
Two ene-type reactions lead to the formation of the two hydroperoxide ketones M3 and M4. Transition structures TS3 and TS4 are concerted abstractions of the phenolic H simultaneously with the addition of the O2(1Δg) at an ortho or para positions, respectively. TS3 incurs a significantly lower energy barrier than TS4 (76 kJ mol−1versus 168 kJ mol−1) and contrasts with direct H-abstraction reaction, which is associated with a sizable barrier 165 kJ mol−1.
Table 1 lists the reaction rate constants for the five initial channels fitted in the low to intermediate temperature window of 300–400 K. Inspection of values in Table 1 reveals that, the formation of M1 intermediate holds importance throughout the considered temperature range with a rather negligible contribution from the other four channels.
By applying the Curtin–Hammond principle,69 the ratio of para–ipso and meta–ortho products ([Ppara]/[Pmeta]) is dictated by the free energy gap (ΔGG0P–M298) between the para and meta transition states (TS1 and TS2) rather than the stabilities of the two intermediates (M1 and M2); i.e., ΔGG0P–M298 = 21 kJ mol−1. Our ratio of [Ppara]/[Pmeta] at 298.15 K corresponds to ∼9000:1. This finding indicates an expected dominance of the para isomer and a negligible abundance of the meta isomer, excluding the meta-channel from further mechanistic analysis. The prominence of the channel leading to the formation of M1 concurs with the experimental detection of the 1,4-peroxide structures during UV radiation of several phenolic compounds.7 Consistently with the results of the present calculations, literature suggests no ene-type reaction at the ortho position as a main reaction channel in the system of phenol and O2(1Δg). In contrast, we predict that the commonly discussed route affording the M4 intermediate constitutes an unlikely pathway (cf.Table 1).
The formation of phenoxy radical via direct abstraction could be ruled out in view of the high Gibbs free energy barrier embedded in TS5. Moreover, our in situ EPR results revealed no formation of phenoxy radicals. This absence could potentially be explained easily through the ionisation of the hydroxyl group (i.e., deprotonation) as in eqn (15), where Ka represents the acidity constant of the phenol group –OH. We calculated the deprotonation degree of the phenolic –OH group at pH = 6 as α = 1.6 × 10−4. Therefore, the αkPO in eqn (14) remains negligible with αkPO− ≈ 0.70
| |  | (12) |
| | 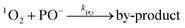 | (13) |
| |  | (14) |
| |  | (15) |
Next, we turn our attention to the selective formation of
para-benzoquinone from reaction of phenol and O
2(
1Δ
g). Any major product flux toward
para-benzoquinone follows the two major initial pathways, producing
M1 and
M3 intermediates. Herein, we show that an unimolecular isomerisation of the
M1 moiety produces
para-benzoquinone
via a facile mechanism.
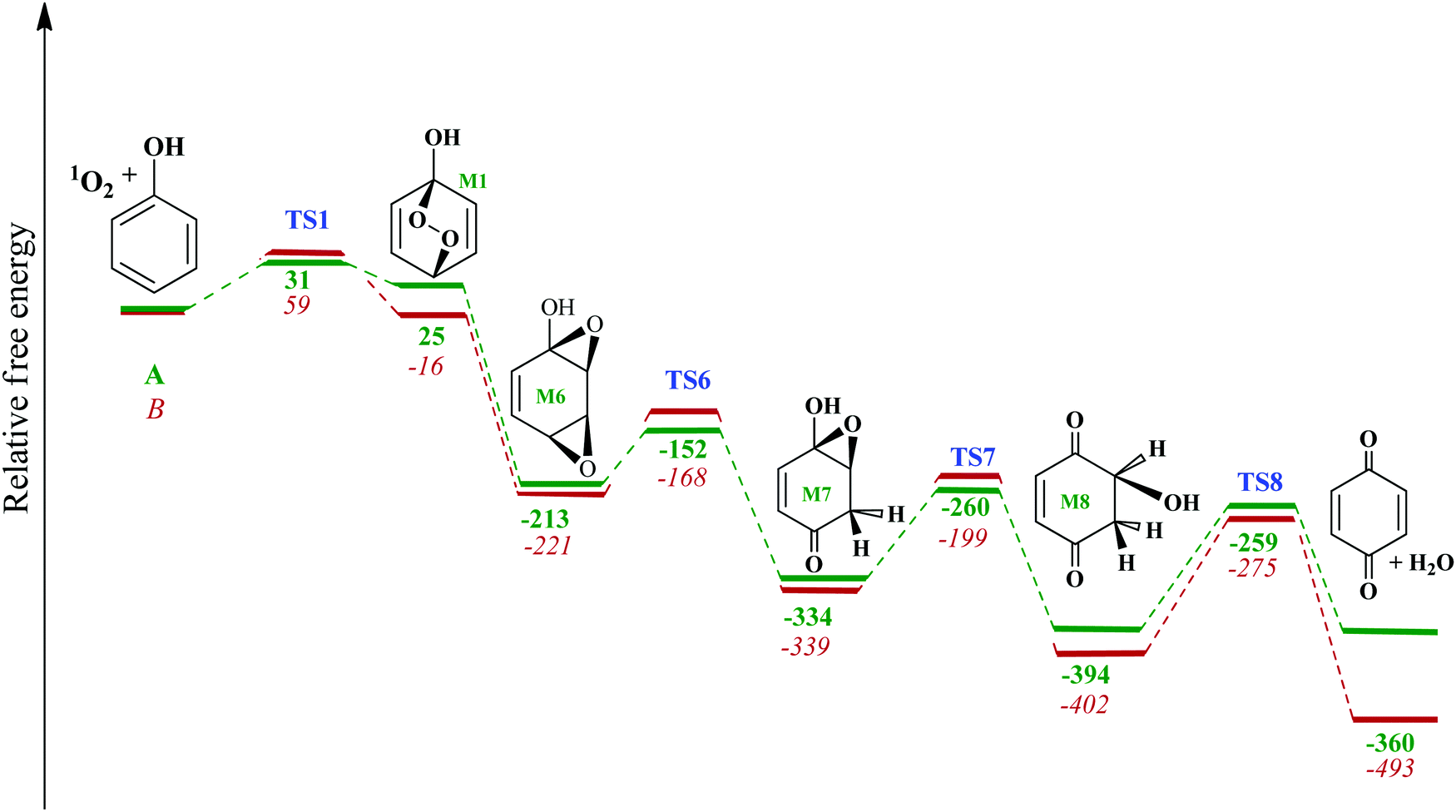
Fig. 7 depicts the potential energy surface for the formation of
para-benzoquinone from oxidation of phenol by singlet oxygen. The
M1 intermediate incurs endothermicity of 25 kJ mol
−1 (with exergicity of 16 kJ mol
−1) relative to the separated reactants, and it could further stabilise into the three-member-ring structure
M6 without passing through an intrinsic reaction barrier. A 1,2-hydrogen transfer
via TS6 proceeds with an enthalpic barrier of 53 kJ mol
−1 and yields the
M7 intermediate. In the subsequent step, intramolecular hydrogen transfer from the hydroxyl group through TS7 forms the significantly de-energised
M8 intermediate through an enthalpic barrier of 140 kJ mol
−1. Thus, the
M7 →
M8 step represents one of the “bottlenecks” of the mechanism shown in
Fig. 7. Finally, elimination of water from intermediate
M8 yields
para-benzoquinone. The enthalpy of the final products falls 360 kJ mol
−1 below the separated reactants.
 |
| | Fig. 7 Enthalpy (bold non-italic green) and Gibbs free energy (italic red) map for the formation of para-BQ from the reaction of singlet oxygen with phenol using M062X (solvation) model. All values are in kJ mol−1 at 298.15 K. | |
3.2.2. Formation of product species.
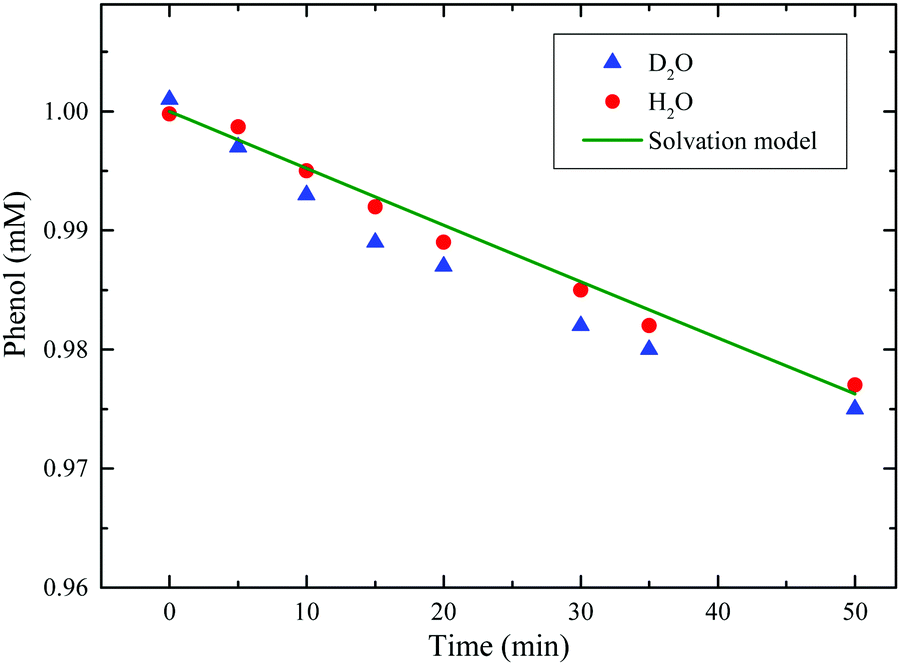
HPLC data provides major insights into the O2(1Δg) interaction with phenol. The photolysis of phenol-RB buffered solution ultimately yielded para-benzoquinone with no other products detected (Fig. 8). This high selectivity towards the formation of para-adduct product was also observed in the case of singlet-oxygen reaction with salicylic acid.71 In Fig. 9, singlet oxygen degrades phenol with a second-order rate constant of kr = 1.14 × 104 M−1 s−1 in H2O. In addition, in deuterium oxide, phenol reacts by approximately 7.5-fold faster than in water, supporting the involvement of singlet oxygen. We detected no trace of para-benzoquinone when executing the photoreaction of phenol in the absence of rose bengal or oxygen.
 |
| | Fig. 8 Concentration profiles for the formation of para-benzoquinone via the water-solvation model (green line) and the published experimental measurements of Briviba et al.12 (blue triangles, methylene blue) versus our present results (red circles, rose bengal). | |
 |
| | Fig. 9 Concentration profiles for phenol depletion via our experimental results in H2O (red circles) and D2O (blue triangles), versus water-solvation model (green line). The overall mechanism for the water solvation model is discussed in Section 3.2.1 and cast into a set of ordinary differential equations that are listed in the ESI.† | |
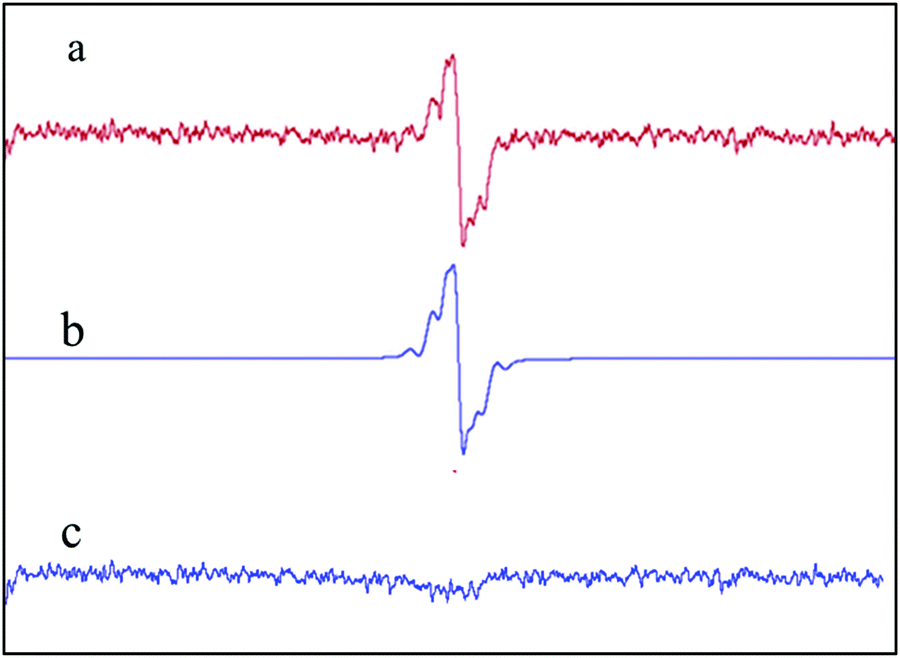
Illumination of phenol in RB-buffered solution (50% MOH, pH = 6.5) gives rise to the spectrum illustrated in Fig. 10, showing a well-defined 1:4:6:4:1 stick configuration that represents the interaction of the radical with four identical protons. The five superhyperfine lines in Fig. 11 show splitting of para-semibenzoquinone anion (PSBQ) signal.72–74 In addition to the PSBQ signal, we observed the appearance of a semi-reduced anion spectrum of RB (i.e., the spectrum of an RB trianion radical), as shown in Fig. 12, and described in eqn (16) and (17). The pattern of RB trianion radical illustrates an unpaired electron interacting with two equivalent protons (known as position 1 and 8 in the RB structure). Even though benzoquinone, as reported by Foote et al.,75 does not interact with singlet oxygen, it quenches the triplet state of rose bengal. Fig. 13 graphs the spin counts profiles of both signals.
| | 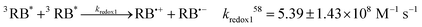 | (16) |
| | 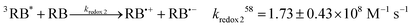 | (17) |
| | RB˙− + BQ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) RB + BQ˙− RB + BQ˙− | (18) |
| | | BQ˙− + RB˙+ → RB + BQ | (19) |
In several mechanisms suggested for the photosensitised reaction of phenol, researchers argued the effect of hydroxyl radical (HO˙) on the product formation and selectivity. Reaction of phenol with hydroxyl radical proceeds
via ortho,
meta, and
para routes.
76 Even though the
meta-product was not detected in some scenarios, the
ortho analogue was always present. For example, when HO˙ is generated using Fe(
II)–EDTA/H
2O
2, hydroquinone (
para) and catechol (
ortho) are produced in equivalent quantities without appearance of the
meta-product.
77 We tested the effect of radical scavengers for both singlet oxygen and hydroxyl radical (O
2(
1Δ
g), HO˙) in order to verify their existence in our system. As noted in
Table 2, the addition of the HO˙ scavengers (
tert-butyl alcohol, 2-propanol,
78 and sodium formate
79,80) seemed to have no obvious effect on the benzoquinone production. In contrast, O
2(
1Δ
g) scavengers (sodium azide,
61,81 dithiothreitol,
82 and glutathione
83) suppress the formation of
para-benzoquinone (
e.g., sodium azide <0.01,
Table 2). The failure of the HO˙ scavengers to block the formation of benzoquinone demonstrates the absence of HO˙ radical in this reaction, whereas a significant effect of O
2(
1Δ
g) scavengers further supports the involvement of singlet oxygen O
2(
1Δ
g). Moreover, the presence of only the
para-product in the reaction of phenol with O
2(
1Δ
g) indicates no involvement of HO˙ in the oxidation process.
 |
| | Fig. 10
In situ EPR spectrum of 0.5 mM para-benzoquinone and 35 μM RB in 50% buffered methanol solution (pH = 6.5) after illumination for 1 min. | |
 |
| | Fig. 11 Experimental (a), simulated (b), residual (c) and time-course (d) EPR spectra obtained upon continuous in situ irradiation of 35 μM RB and 1 mM phenol in 50% buffered methanol solution (pH = 6.5) for 3 h. Simulation parameters: RB trianion; g = 2.00439, hyperfine splitting HFS = 1.14224 ± 0.00005, para-semibenzoquinone anion; g = 2.00446, HFS = 2.36955 ± 0.00005. | |
 |
| | Fig. 12 EPR signal of (a) RB trianion radical formed after illuminating a sample containing 1 mM phenol, 35 μM rose bengal in 50% methanol solution, at pH = 6 for 1 min, (b) simulated EPR spectrum and (c) the subtract. Simulation parameters: g = 2.00439, HFS = 1.14224 ± 0.00005. | |
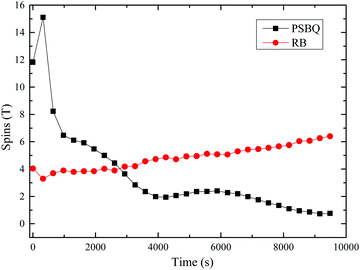 |
| | Fig. 13 Spins-course EPR spectra obtained upon continuous irradiation in situ of 35 μM RB and 1 mM phenol in 50% buffered methanol solution (pH = 6.5) for 2.6 h. Simulation parameters: RB trianion (red circles); g = 2.00439, HFS = 1.14224 ± 0.00005, para-semibenzoquinone anion (PSBQ, black squares); g = 2.00446, HFS = 2.36955 ± 0.00005. | |
Table 2 Effect of O2(1Δg) and HO˙ scavengers on the formation of benzoquinone after 30 min of illumination (pH = 6, H2O) at 298.15 K
| Scavengers |
Benzoquinone (14 μM) |
| O2(1Δg) scavengers |
Sodium azide (10 mM) |
<0.01 |
| Dithiothreitol (10 mM) |
1.05 ± 0.05 |
| Glutathione (10 mM) |
1.43 ± 0.17 |
|
|
| HO˙ scavengers |
Sodium formate (10 mM) |
13.55 ± 0.51 |
|
tert-Butyl alchohol (100 mM) |
13.02 ± 0.32 |
| 2-Propanol (100 mM) |
14.30 ± 0.28 |
Applying the values of the kinetic parameters for solution from Table 2 and converting the units to L and mol, our calculated rate constant of photooxidation of phenol with singlet delta oxygen at 298.15 K is kr-solvation = 1.21 × 104 M−1 s−1, which is consistent with the present experimental measurement of kr = 1.14 × 104 M−1 s−1 as well as the literature value of kr = 2.4 × 104 M−1 s−112 in methanol solution.84 We solved the system of ordinary differential equations (ODEs) for our solvation model (see Table S2 in ESI†) using POLYMATH85 software. The initial concentrations corresponded to 35 μM and 1 mM for rose-bengal sensitiser and phenol, respectively.
In the kinetic modelling, we apply a batch reactor model with a steady-state concentration of singlet oxygen and a sensitiser quantum yield of 26.6 × 10−6 M−1 and 0.76, with the kinetic expressions derived from the solvation model. As illustrated in Fig. 8 and 9, the results of the model compare well with the experimental measurements. In Fig. 8, we also include the experimental measurements of Briviba et al.,12 demonstrating close agreement. The small difference reflects the lower quantum yield of methylene-blue dye used by Briviba et al.12 The vibronic coupling of singlet oxygen with water or the physical quenching of singlet delta oxygen62 with phenol may also explain the small discrepancy.
4. Conclusions
This contribution has addressed the mechanistic and kinetic aspects of the photo-induced reaction of phenol with singlet oxygen, coupling the experimental and theoretical results for the first time. Singlet oxygen reaction with phenol proved to have an extreme selectivity toward the para position. The suggested mechanism proceeds via the 1,4-endoperoxide species (M1) intermediate to yield para-benzoquinone, the only product detected. Herein, we show that the unimolecular isomerisation of the M1 moiety produces para-benzoquinone via a facile mechanism. Our mechanistic maps are supported by the in situ detection of para-semibenzoquinone anion (PSBQ) in the EPR resonator and by the results of singlet-oxygen spin trapping and experiments involving O2(1Δg) and HO˙ scavengers. The addition of the HO˙ scavengers has no obvious effect on the benzoquinone production. In contrast, O2(1Δg) scavengers suppress the formation of benzoquinone (e.g., sodium azide <0.01). Moreover, phenol disappears more rapidly in deuterium oxide compared to normal water (by approximately 7.5 times), supporting the involvement of singlet oxygen. Our experimental and modelling results should apply to decomposition of other biologically active phenolic entities, typically occurring in aqueous media.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We gratefully acknowledge grants of computing time from the National Computational Infrastructure (NCI) and from the Pawsey Supercomputing Centre, Australia as well as the financial supports from the Australian Research Council (ARC). J. N. thanks Murdoch University for the award of a MUSS postgraduate scholarship. Our thanks are due to Dr Juita and Dr Marc Hampton for their valuable help.
References
- Y. Zhou, J. Jiang, Y. Gao, S.-Y. Pang, Y. Yang, J. Ma, J. Gu, J. Li, Z. Wang, L.-H. Wang, L.-P. Yuan and Y. Yang, Activation of peroxymonosulfate by phenols: important role of quinone intermediates and involvement of singlet oxygen, Water Res., 2017, 125, 209–218 CrossRef CAS PubMed.
- B. Koehler, F. Barsotti, M. Minella, T. Landelius, C. Minero, L. J. Tranvik and D. Vione, Simulation of photoreactive transients and of photochemical transformation of organic pollutants in sunlit boreal lakes across 14 degrees of latitude: a photochemical mapping of Sweden, Water Res., 2018, 129, 94–104 CrossRef CAS PubMed.
- D. Vione, D. Fabbri, M. Minella and S. Canonica, Effects of the antioxidant moieties of dissolved organic matter on triplet-sensitized phototransformation processes: implications for the photochemical modeling of sulfadiazine, Water Res., 2018, 128, 38–48 CrossRef CAS PubMed.
- N. A. Garcia, New trends in photobiology: singlet-molecular-oxygen-mediated photodegradation of aquatic phenolic pollutants. A kinetic and mechanistic overview, J. Photochem. Photobiol., B, 1994, 22, 185–196 CrossRef CAS.
- J. Al-Nu'airat, M. Altarawneh, I. Oluwoye, X. Gao and B. Z. Dlugogorski, Role of Singlet Oxygen in Combustion Initiation of Aromatic Fuels, Energy Fuels, 2018 DOI:10.1021/acs.energyfuels.8b02312.
-
M. Wainwright, Photosensitisers in Biomedicine, John Wiley & Sons, 2009 Search PubMed.
- S. Takizawa, R. Aboshi and S. Murata, Photooxidation of 1, 5-dihydroxynaphthalene with iridium complexes as singlet oxygen sensitizers, Photochem. Photobiol. Sci., 2011, 10, 895–903 RSC.
- S. Bhatnagar, A. Kumar and S. C. Ameta, Photooxidation of Some Pharmaceutical Drugs by Singlet Molecular Oxygen, Asian J. Pharm. Biol. Res., 2011, 1, 210–217 Search PubMed.
- K. R. Weishaupt, C. J. Gomer and T. J. Dougherty, Identification of singlet oxygen as the cytotoxic agent in photo-inactivation of a murine tumor, Cancer Res., 1976, 36, 2326–2329 CAS.
- C. Li and M. Z. Hoffman, Oxidation of Phenol by Singlet Oxygen Photosensitized by the Tris(2,2‘-bipyridine)ruthenium(II) Ion, J. Phys. Chem. A, 2000, 104, 5998–6002 CrossRef CAS.
- C. Pizzocaro, M. Bolte, H. Sun and M. Z. Hoffman, Metal complex sensitized photooxidation of phenol in aqueous solution, New J. Chem., 1994, 18, 737–743 CAS.
- K. Briviba, T. P. A. Devasagayam, H. Sies and S. Steenken, Selective para-hydroxylation of phenol and aniline by singlet molecular oxygen, Chem. Res. Toxicol., 1993, 6, 548–553 Search PubMed.
- A. U. Khan, Singlet molecular oxygen from superoxide anion and sensitized fluorescence of organic molecules, Science, 1970, 168, 476–477 CrossRef CAS PubMed.
- E. L. Clennan and A. Pace, Advances in singlet oxygen chemistry, Tetrahedron, 2005, 61, 6665–6691 CrossRef CAS.
- A. M. Starik and N. S. Titova, Possibility of initiation of combustion of CH4–O2 (Air) mixtures with laser-induced excitation of O2 molecules, Combust., Explos. Shock Waves, 2004, 40, 499–510 CrossRef.
- A. M. Starik, P. S. Kuleshov, A. S. Sharipov and N. S. Titova, Kinetics of ignition and combustion in the Al–CH4–O2 system, Energy Fuels, 2014, 28, 6579–6588 CrossRef CAS.
- A. Starikovskiy and N. Aleksandrov, Plasma-assisted ignition and combustion, Prog. Energy Combust. Sci., 2013, 39, 61–110 CrossRef CAS.
- A. Maranzana, C. Canepa, G. Ghigo and G. Tonachini, Theoretical study on the reactivity and regioselectivity of the ene reaction of 1Δg O2 with α,β-unsaturated carbonyl compounds, Eur. J. Org. Chem., 2005, 3643–3649 CrossRef CAS.
-
A. G. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Handbook of Organic Photochemistry and Photobiology, CRC Press, 2012, vol. 1 Search PubMed.
- S. Mohammadi, A. Kargari, H. Sanaeepur, K. Abbassian, A. Najafi and E. Mofarrah, Phenol removal from industrial wastewaters: a short review, Desalin. Water Treat., 2015, 53, 2215–2234 CrossRef CAS.
-
I. J. Tinsley, Chemical Concepts in Pollutant Behavior, John Wiley & Sons, 2004 Search PubMed.
- R. G. Zepp, N. L. Wolfe, G. L. Baughman and R. C. Hollis, Singlet oxygen in natural waters, Nature, 1977, 267, 421–423 CrossRef CAS.
- W. R. Haag and J. Hoigne, Singlet oxygen in surface waters. 3. Photochemical formation and steady-state concentrations in various types of waters, Environ. Sci. Technol., 1986, 20, 341–348 CrossRef CAS PubMed.
- F. H. Frimmel, H. Bauer, J. Putzien, P. Murasecco and A. M. Braun, Laser flash photolysis of dissolved aquatic humic material and the sensitized production of singlet oxygen, Environ. Sci. Technol., 1987, 21, 541–545 CrossRef CAS PubMed.
- C. Pizzocaro, M. Bolte and M. Z. Hoffman, Cr(bpy)33+ -sensitized photo-oxidation of phenol in aqueous solution, J. Photochem. Photobiol., C, 1992, 68, 115–119 CrossRef CAS.
- B. Heyne, S. Kohnen, D. Brault, A. Mouithys-Mickalad, F. Tfibel, P. Hans, M.-P. Fontaine-Aupart and M. Hoebeke, Investigation of singlet oxygen reactivity towards propofol, Photochem. Photobiol. Sci., 2003, 2, 939–945 RSC.
- B. Heyne, D. Brault, M.-P. Fontaine-Aupart, S. Kohnen, F. Tfibel, A. Mouithys-Mickalad, G. Deby-Dupont, P. Hans and M. Hoebeke, Reactivity towards singlet oxygen of propofol inside liposomes and neuronal cells, Biochim. Biophys. Acta, Gen. Subj., 2005, 1724, 100–107 CrossRef CAS PubMed.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake and S. A. Rusak,
et al., Methods for reactive oxygen species (ROS) detection in aqueous environments, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS.
- N. De la Cruz, J. Giménez, S. Esplugas, D. Grandjean, L. F. De Alencastro and C. Pulgarin, Degradation of 32 emergent contaminants by UV and neutral photo-fenton in domestic wastewater effluent previously treated by activated sludge, Water Res., 2012, 46, 1947–1957 CrossRef CAS PubMed.
- I. E. Kochevar and R. W. Redmond, [2] Photosensitized production of singlet oxygen, Methods Enzymol., 2000, 319, 20–28 CAS.
- K. N. Loponov, J. Lopes, M. Barlog, E. V. Astrova, A. V. Malkov and A. A. Lapkin, Optimization of a scalable photochemical reactor for reactions with singlet oxygen, Org. Process Res. Dev., 2014, 18, 1443–1454 CrossRef CAS.
- T. Sarna, J. Zajac, M. K. Bowman and T. G. Truscott, Photoinduced electron transfer reactions of rose bengal and selected electron donors, J. Photochem. Photobiol., C, 1991, 60, 295–310 CrossRef CAS.
- S. M. Linden and D. C. Neckers, Type I and type II sensitizers based on rose bengal onium salts, Photochem. Photobiol., 1988, 47, 543–550 CrossRef CAS PubMed.
- P. Esser, B. Pohlmann and H.-D. Scharf, The photochemical synthesis of fine chemicals with sunlight, Angew. Chem., Int. Ed. Engl., 1994, 33, 2009–2023 CrossRef.
- F. Nifiatis, J. C. Athas, K. Don Dasitha Gunaratne, Y. Gurung, K. Mae Monette and P. J. Shivokevich, Substituent effects of porphyrin on singlet oxygen generation quantum yields, Open Spectrosc. J., 2011, 5, 1 CrossRef CAS.
- S. Fukuzumi, S. Fujita, T. Suenobu, H. Yamada, H. Imahori, Y. Araki and O. Ito, Electron transfer properties of singlet oxygen and promoting effects of scandium ion, J. Phys. Chem. A, 2002, 106, 1241–1247 CrossRef CAS.
- K.-I. Okamoto, F. Hondo, A. Itaya and S. Kusabayashi, Kinetics of dye-sensitized photo-degradation of aqueous phenol, J. Chem. Eng. Jpn., 1982, 15, 368–375 CrossRef CAS.
-
D. Green and R. Perry, Perry's Chemical Engineers’ Handbook, McGraw Hill Professional, 8th edn, 2007 Search PubMed.
- S. K. Han, T.-M. Hwang, Y. Yoon and J.-W. Kang, Evidence of singlet oxygen and hydroxyl radical formation in aqueous goethite suspension using spin-trapping electron paramagnetic resonance (EPR), Chemosphere, 2011, 84, 1095–1101 CrossRef CAS PubMed.
- H. Qi, X. Dong, Y. Zhao, N. Li, H. Fu, D. Feng, L. Liu and C. Yu, ROS production in homogenate from the body wall of sea cucumber Stichopus japonicus under UVA irradiation: ESR spin-trapping study, Food Chem., 2016, 192, 358–362 CrossRef CAS PubMed.
- E. Yaghini, K. F. Pirker, C. W. Kay, A. M. Seifalian and A. J. MacRobert, Quantification of reactive oxygen species generation by photoexcitation of pegylated quantum dots, Small, 2014, 10, 5106–5115 CrossRef CAS PubMed.
- V. Nardello, D. Brault, P. Chavalle and J.-M. Aubry, Measurement of photogenerated singlet oxygen (1O2(1Δg)) in aqueous solution by specific chemical trapping with sodium 1,3-cyclohexadiene-1,4-diethanoate, J. Photochem. Photobiol., B, 1997, 39, 146–155 CrossRef CAS.
-
P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, John Wiley & Sons, 2009 Search PubMed.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS.
- C. A. Parker, Proc. R. Soc. London, Ser. A, 1953, 220, 104–116 CrossRef.
-
M. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Peterssonet al., Gaussian 09, Revision A. 02, Gaussian, Inc., Wallingford CT, 2009, p. 200 Search PubMed.
- S. Yamanaka, T. Kawakami, H. Nagao and K. Yamaguchi, Effective exchange integrals for open-shell species by density functional methods, Chem. Phys. Lett., 1994, 231, 25–33 CrossRef CAS.
- M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- J. Gräfenstein, E. Kraka, M. Filatov and D. Cremer, Can unrestricted density-functional theory describe open shell singlet biradicals?, Int. J. Mol. Sci., 2002, 3, 360–394 CrossRef.
- T. Saito, S. Nishihara, Y. Kataoka, Y. Nakanishi, T. Matsui, Y. Kitagawa, T. Kawakami, M. Okumura and K. Yamaguchi, Transition state optimization based on approximate spin-projection (AP) method, Chem. Phys. Lett., 2009, 483, 168–171 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. A. Montgomery Jr, J. W. Ochterski and G. A. Petersson, A complete basis set model chemistry. IV. An improved atomic pair natural orbital method, J. Chem. Phys., 1994, 101, 5900–5909 CrossRef.
- J. Al-Nu’airat, M. K. Altarawneh, X. Gao, P. R. Westmoreland and B. Z. Dlugogorski, Reaction of aniline with singlet oxygen (O21Δg), J. Phys. Chem. A, 2017, 121, 3199–3206 CrossRef PubMed.
- N. Zeinali, M. Altarawneh, D. Li, J. Al-Nu’airat and B. Z. Dlugogorski, New Mechanistic Insights: Why do plants produce isoprene?, ACS Omega, 2016, 1, 220–225 CrossRef CAS.
- J. Al-Nu’airat, B. Z. Dlugogorski, I. Oluwoye, G. Xiangpeng and M. K. Altarawneh, Effect of Fe2O3 nanoparticles on
combustion of coal surrogate (anisole): enhanced ignition and formation of persistent free radicals, Proc. Combust. Inst., 2018 DOI:10.1016/j.proci.2018.06.081.
- S. Canneaux, F. Bohr and E. Henon, KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
-
K. Gollnick, Type II photooxygenation reactions in solution, Advances in Photochemistry, 2007, ch. 1, vol. 6, DOI:10.1002/9780470133361.
- L. Ludvikova, P. Fris, D. Heger, P. Sebej, J. Wirz and P. Klan, Photochemistry of rose bengal in water and acetonitrile: a comprehensive kinetic analysis, Phys. Chem. Chem. Phys., 2016, 18, 16266–16273 RSC.
- E. Chesneau and D. C. Neckers, Electron transfer sensitized photobleaching of rose bengal induced by triplet benzophenones, J. Photochem. Photobiol., C, 1988, 42, 269–281 CrossRef CAS.
- X.-F. Zhang, I. Zhang and L. Liu, Photophysics of halogenated fluoresceins: involvement of both intramolecular electron transfer and heavy atom effect in the deactivation of excited states, Photochem. Photobiol., 2010, 86, 492–498 CrossRef CAS PubMed.
- N. Hasty, P. B. Merkel, P. Radlick and D. R. Kearns, Role of azide in singlet oxygen reactions: reaction of azide with singlet oxygen, Tetrahedron Lett., 1972, 13, 49–52 CrossRef.
- C. Schweitzer and R. Schmidt, Physical mechanisms of generation and deactivation of singlet oxygen, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed.
- B. F. Minaev, Spin-orbit coupling mechanism of singlet oxygen 1Δg quenching by solvent vibrations, Chem. Phys., 2017, 483, 84–95 CrossRef.
-
E. Boix-Garriga, B. Rodriguez-Amigo, O. Planas and S. Nonell, Properties of Singlet Oxygen, 2016, ch. 2, pp. 23–46 Search PubMed.
-
M. A. J. Rodgers and T. Snowden, Lifetime of 1O2 in liquid water as determined by time-resolved infrared luminescence measurements, 1982 Search PubMed.
- G. Nardi, I. Manet, S. Monti, M. A. Miranda and V. Lhiaubet-Vallet, Scope and limitations of the TEMPO/EPR method for singlet oxygen detection: the misleading role of electron transfer, Free Radical Biol. Med., 2014, 77, 64–70 CrossRef CAS PubMed.
- L.-Y. Zang, Z. Zhang and H. P. Misra, EPR Studies of trapped singlet oxygen (lO2) generated during photoirradiation of hypocrellin A, Photochem. Photobiol., 1990, 52, 677–683 CrossRef CAS PubMed.
- P. R. Erickson, N. Walpen, J. J. Guerard, S. N. Eustis, J. S. Arey and K. McNeill, Controlling factors in the rates of oxidation of anilines and phenols by triplet methylene blue in aqueous solution, J. Phys. Chem. A, 2015, 119, 3233–3243 CrossRef CAS PubMed.
- J. I. Seeman, Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett Winstein-Holness kinetics, Chem. Rev., 1983, 83, 83–134 CrossRef CAS.
- F. E. Scully and J. Hoigné, Rate constants for reactions of singlet oxygen with phenols and other compounds in water, Chemosphere, 1987, 16, 681–694 CrossRef CAS.
- J. B. Feix and B. Kalyanaraman, Production of singlet oxygen-derived hydroxyl radical adducts during merocyanine-540-mediated photosensitization: analysis by ESR-spin trapping and HPLC with electrochemical detection, Arch. Biochem. Biophys., 1991, 291, 43–51 CrossRef CAS PubMed.
- I. H. Leaver, Semiquinone radical intermediates in the eosin-sensitized photooxidation of phenols, Aust. J. Chem., 1971, 24, 891–894 CrossRef CAS.
- I. H. Leaver, An ESR study of radical intermediates in the photoreduction of xanthene dyes, Aust. J. Chem., 1971, 24, 753–763 CrossRef CAS.
- C. Lambert, T. Sarna and T. G. Truscott, Rose bengal radicals and their reactivity, J. Chem. Soc., Faraday Trans., 1990, 86, 3879–3882 RSC.
- C. S. Foote, R. W. Denny, L. Weaver, Y. Chang and J. Peters, Quenching of singlet oxygen, Ann. N. Y. Acad. Sci., 1970, 171, 139–148 CrossRef CAS.
- N. V. Raghavan and S. Steenken, Electrophilic reaction of the hydroxyl radical with phenol. Determination of the distribution of isomeric dihydroxycyclohexadienyl radicals, J. Am. Chem. Soc., 1980, 102, 3495–3499 CrossRef CAS.
- C. P. Moorhouse, B. Halliwell, M. Grootveld and J. M. Gutteridge, Cobalt (II) ion as a promoter of hydroxyl radical and possible ‘crypto-hydroxyl’radical formation under physiological conditions. Differential effects of hydroxyl radical scavengers, Biochim. Biophys. Acta, Gen. Subj., 1985, 843, 261–268 CrossRef CAS.
- S. M. Khopde, K. I. Priyadarsini, P. Venkatesan and M. N. A. Rao, Free radical scavenging ability and antioxidant efficiency of curcumin and its substituted analogue, Biophys. Chem., 1999, 80, 85–91 CrossRef CAS.
- A. Henglein and C. Kormann, Scavenging of OH radicals produced in the sonolysis of water, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1985, 48, 251–258 CrossRef CAS.
- K. Makino, M. M. Mossoba and P. Riesz, Chemical effects of ultrasound on aqueous solutions. Evidence for hydroxyl and hydrogen free radicals (OH and H) by spin trapping, J. Am. Chem. Soc., 1982, 104, 3537–3539 CrossRef CAS.
- M. Y. Li, C. S. Cline, E. B. Koker, H. H. Carmichael, C. F. Chignell and P. Bilski, Quenching of singlet molecular oxygen (1O2) by azide anion in solvent mixtures, Photochem. Photobiol., 2001, 74, 760–764 CrossRef CAS PubMed.
- T. P. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, Activity of thiols as singlet molecular oxygen quenchers, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS.
- M. V. M. Lafleur, J. J. Hoorweg, H. Joenje, E. J. Westmijze and J. Retel, The ambivalent role of glutathione in the protection of DNA against singlet oxygen, Free Radical Res., 1994, 21, 9–17 CrossRef CAS PubMed.
- N. Miyoshi and G. Tomita, Fluorescein-photosensitized Furan Oxidation in Methanolic and Reversed Micellar Solutions, Part II Kinetic Analysis, Z. Naturforsch., B: J. Chem. Sci., 1980, 35, 107–111 Search PubMed.
- POLYMATH is copyrighted by M. Shacham, M.B. Cutlip and M. Elly, http://www.polymath-software.com/, accessed Feb 23, 2018.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Bleaching test of rose bengal in buffered solution (pH = 6), Fig. S2. HPLC spectrum of (35 μM RB, 1 mM phenol) illuminated sample in buffered solution, Fig. S3. UV-vis absorption spectra for benzoquinone and hydroquinone, Fig. S4. Calibration curve for ferrioxalate actinometer, Fig. S5. Optimised structures of transition states of phenol oxidation by singlet oxygen, Table S1. POLYMATH85 ordinary differential equations (ODEs) and parameters for the estimated singlet oxygen concentration profile, Table S2. POLYMATH85 ordinary differential equations (ODEs) and parameters for the solvation model, and detailed description of the actinometry experiments. See DOI: 10.1039/c8cp04852e |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Bogdan Z.
Dlugogorski
a,
Bogdan Z.
Dlugogorski