
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New rhenium-tricarbonyl complexes bearing halogen-substituted bidentate ligands: structural, computational and Hirshfeld surfaces studies†‡
Reza
Kia
 *a,
Soheil
Mahmoudi
a and
Paul R.
Raithby
b
*a,
Soheil
Mahmoudi
a and
Paul R.
Raithby
b
aChemistry Department, Sharif University of Technology, P.O. Box 11155-3516, Tehran, Iran. E-mail: rkia@sharif.edu; zsrkk@yahoo.com; Tel: +98 21 66165332
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 4th December 2018
Abstract
A series of ten rhenium tricarbonyl complexes (C1–C10), bearing halogen-substituted bidentate N,N-donor ligands with halogens or trifluoromethyl groups (X = –F, –Cl, –Br, and –CF3) in different positions on the aromatic rings were synthesized and characterized by FT-IR and 1H-NMR spectroscopy and their solid-state structures were determined by single crystal X-ray diffraction. The resultant complexes Re(CO)3(N,N)X display an octahedral coordination geometry around the central Re atom, and in all the complexes the Re(CO)3 unit adopts a fac geometry. The metal-bound halogen atoms along with the halogen-substituted ligands were used to fine-tune the electron density of the halogen σ-hole on the coordinated halide that is involved in halogen–halogen and other intermolecular interactions. This series of compounds was used to explore the range of possible intermolecular interactions involving rhenium coordinated halides. These supramolecular interactions include: (i) halogen bonding through the metal-bound halogen with the carbon-bound halogen (Re–X⋯X–C), (ii) halogen bonding through the carbon-bound halogen with another carbon-bound halogen (C–X⋯X–C), (iii) dipolar interactions through carbonyl–carbonyl (CO⋯CO), (iv) C–H⋯X hydrogen bonding interactions, (v) C–H⋯O hydrogen bonding interactions, (vi) halogen (X)⋯heteroatom (N, O) interactions and (vii) metal-bond carbonyl lone pair with aromatic π-ring interaction, Re–CO(lone pair)⋯π(aromatic ring). In each case the molecular electrostatic potential and non-covalent interaction index were calculated. Crystal packing analyses using Hirshfeld surface calculation confirmed that metal-bound halogen is more effective than carbon-bound halogen in the formation of intermolecular interactions. Complexes C8 and C10 featured interesting intramolecular Re–CO(lone pair)⋯π interactions, the presence of which were confirmed by molecular orbital and non-covalent interaction index (NCI) calculations.
Introduction
The assembly of molecular complexes in the solid-state to form supramolecular arrays is governed by a range of competing non-covalent interactions, and the structures of the resultant assemblies are based on the directionality, strength and specificity of the interactions involved.1,2 As such, the structural importance of non-covalent interactions has attracted extensive interest in the areas of crystal engineering,3 molecular recognition,4 protein folding,5 enzymatic reactions,5 supramolecular chemistry6 and crystal structure prediction.7 The competition between different types of non-covalent interactions in the solid-state and knowledge of the strengths of such interactions is at the heart of crystal engineering.8 The competition or cooperative behavior between hydrogen bonds and halogen bonds in the solid-state has been studied in various systems.9 Among the different non-H-bond mediated non-covalent interactions which have received extensive attention is halogen bonding. It is clearly defined as a non-covalent interaction between an electron deficient halogen atom and an electron donor halogen.10–12Politzer et al. showed the existence of an electropositive crown so-called “σ-hole” – a zone of low electron density displaying a positive electrostatic potential of the halogen atom directed to the electron donor.13,14 On the other hand, it has been shown that the halogen bonding is not merely electrostatic in nature but the contributing factors are a convolution of electrostatic,14 polarization,15 charge transfer16 and dispersion17,18 forces and the contribution of each of these factors is directly affected by the molecular components involved in the interactions. The extensive studies by Metrangolo, Resnati, and Brammer and coworkers have revolutionized the conception of halogen bonding and made it applicable in different areas of chemistry in which molecular recognition plays a central role.19–23
Most of the studies were based on the halogen interactions in organic systems but the seminal work by Brammer and co-workers systematically examined the potential of metal-bound halogens such as the halometallate anion (M–X) with halocarbon (C–X) groups for halogen bonding.24–26 From large numbers of crystallographic observations, it has been substantiated that the metal-bound halogens and halide anions are strongly nucleophilic compared to carbon-bound halogens which have only a very weakly nucleophilic character in hydrogen bonding interactions, establishing a hierarchy of hydrogen bond strengths as D–H⋯X− > D–H⋯X–M ≫ D–H⋯X–C (D = N, O, C).20,24 This can also be extended to halogen bonding interactions between M–X′ and C–X (M–X′⋯X–C in which X′ = X or X′ ≠ X) since a study was performed using metal–organic tectons which bring coordination bonds into competition with directional intermolecular non-covalent interactions.27
Additionally, non-covalent interactions involving π-systems such as π(aromatic)⋯lp(lone pair), have been extensively studied in recent decades. Originally, the existence of such interaction was studied in biological systems and it has been shown that such interactions are of great importance in the structures of biological macromolecules.28 To this effect, the lp⋯π interaction involving the carbonyl group is of considerable interest in metal carbonyl complexes recently and it has been found that some metal-carbonyl complexes feature interesting intra- and/or intermolecular M–CO(lone pair)⋯π(arene) interactions.29,30 The π-systems involved are usually π-acidic aromatic rings with electron-withdrawing halo-, nitro- and cyano-functional groups or electron-deficient aromatic heterocycles.31
Another interesting intra- and intermolecular interaction is the n → π* interaction which has been extensively studied in biomolecules. Such an interaction was reported for the first time very recently by Echeverria32 and Mooibroek et al.,33 in transition metal complexes containing carbonyl ligands which surprisingly stabilize precise molecular conformations due to the overlap between donor and acceptor complexes. The ground work of such interaction in organic carbonyl compounds was investigated by Burgi and Dunitz in 1975.34
Herein, we report the design, synthesis, characterization, structural chemistry and theoretical calculations of a series of new chloro- and bromo-rhenium(I) tricarbonyl complexes bearing halogen and/or trifluoromethyl substituted N,N-donor ligands. We chose the complexes containing the ubiquitous “Re(CO)3X” fragment because of the rich redox and luminescent properties that these complexes possess,35 and because of the diverse structural chemistry that they exhibit.36 The strategy behind this design was to facilitate the possibility of different intermolecular interactions between the ranges of functional groups in these complexes.
Experimental
General
All chemicals used were analytical reagent grade. All solvents purchased from Merck were reagent grade and purified by standard techniques where required. Commercially available Re(CO)5Cl and Re(CO)5Br were obtained from Aldrich and were used as received. The 1H-NMR spectra (Fig. S1–S8, ESI†), were recorded using BRUKER AVANCE 500 MHz spectrometer in CDCl3. FT-IR spectra (Fig. S9–S16†) in the region of 4000–400 cm−1 were recorded in KBr pellets with a Shimadzu IR Prestige-21 FTIR spectrophotometer. The elemental analyses were performed using a LECO CHN instrument. The syntheses and crystal structures of the halogen-substituted bidentate N,N-ligands (Scheme 1) and the 32ClFenReBr complex (abbreviated as C1), have been reported previously.37–42 The preparation of all of the Re(I)-tricarbonyl complexes has been achieved using literature methods.43 | ||
| Scheme 1 Synthesis pathway of C1–C10. | ||
Crystallisations
Crystals were obtained by vapour diffusion of n-hexane into the dichloromethane solution of the complexes under ambient conditions.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), was added an equimolar amount of Re(CO)5Cl (0.55 mmol, 0.20 g). The mixture was heated at reflux for 3 h. The solution was concentrated to 5 mL and n-hexane was added to precipitate the complex quantitatively. Calc.: C, 35.28; H, 1.87; N, 4.33. Found: C, 35.31; H, 1.86; N, 4.35. FTIR (KBr, cm−1): 2021, 1917 and 1882 (CO); 1624 (C
:2), was added an equimolar amount of Re(CO)5Cl (0.55 mmol, 0.20 g). The mixture was heated at reflux for 3 h. The solution was concentrated to 5 mL and n-hexane was added to precipitate the complex quantitatively. Calc.: C, 35.28; H, 1.87; N, 4.33. Found: C, 35.31; H, 1.86; N, 4.35. FTIR (KBr, cm−1): 2021, 1917 and 1882 (CO); 1624 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 1H-NMR (CDCl3, δppm): 4.15–4.35 (m, CH2); 7.30–8.20 (m, aromatic H); 8.90 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.10 (m, CH2); 4.30–4.40 (m, CH2); 7.20–7.30 (m, aromatic H); 7.70–7.85 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.10 (m, CH2); 4.30–4.40 (m, CH2); 7.20–7.30 (m, aromatic H); 7.70–7.85 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.15 (m, CH2); 4.35–4.45 (m, CH2); 7.40–7.50 (m, aromatic H); 7.70–7.80 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 3.97–4.13 (m, CH2); 4.19–4.34 (m, CH2); 7.20–7.27 (m, aromatic H); 7.80–7.87 (m, aromatic H); 7.93–7.96 (m, aromatic H); 8.77 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.25 (m, CH2); 4.25–4.40 (m, CH2); 7.35–7.55 (m, aromatic H); 8.20–8.30 (m, aromatic H); 8.90 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.20 (m, CH2); 4.30–4.40 (m, CH2); 7.50–7.80 (m, aromatic H); 8.85 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.20 (m, CH2); 4.55–4.60 (m, CH2); 7.50–7.70 (m, aromatic H); 8.25 (s, aromatic H); 8.85 (s, iminic H).
N).
N). 1H-NMR (CDCl3, δppm): 4.15–4.35 (m, CH2); 7.30–8.20 (m, aromatic H); 8.90 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.10 (m, CH2); 4.30–4.40 (m, CH2); 7.20–7.30 (m, aromatic H); 7.70–7.85 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.10 (m, CH2); 4.30–4.40 (m, CH2); 7.20–7.30 (m, aromatic H); 7.70–7.85 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.00–4.15 (m, CH2); 4.35–4.45 (m, CH2); 7.40–7.50 (m, aromatic H); 7.70–7.80 (m, aromatic H); 8.75 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 3.97–4.13 (m, CH2); 4.19–4.34 (m, CH2); 7.20–7.27 (m, aromatic H); 7.80–7.87 (m, aromatic H); 7.93–7.96 (m, aromatic H); 8.77 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.25 (m, CH2); 4.25–4.40 (m, CH2); 7.35–7.55 (m, aromatic H); 8.20–8.30 (m, aromatic H); 8.90 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.20 (m, CH2); 4.30–4.40 (m, CH2); 7.50–7.80 (m, aromatic H); 8.85 (s, iminic H).
N). 1H-NMR (CDCl3, δppm): 4.10–4.20 (m, CH2); 4.55–4.60 (m, CH2); 7.50–7.70 (m, aromatic H); 8.25 (s, aromatic H); 8.85 (s, iminic H).
N).
X-ray crystallography
Single crystals of C2 to C10 suitable for X-ray diffraction analysis, were grown by slow vapor diffusion of n-hexane into dichloromethane solution of the complex. The ORTEP view of the complexes is shown in Fig. 1. Details of the crystal data collection and refinement parameters are summarized in Tables S1–S9.† X-ray intensity data were collected using the full sphere routine by φ and ω scans strategy on the Agilent SuperNova dual wavelength EoS S2 diffractometer with mirror monochromated Mo Kα radiation (λ = 0.71073 Å). For all data collections the crystals were cooled to 150(2) K using an Oxford Diffraction Cryojet low-temperature attachment. The data reduction, including an empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm,44 was performed using the CrysAlisPro software package.45,46 The crystal structures were solved by direct methods using the online version of AutoChem 2.0 in conjunction with OLEX2 suite of programs implemented in the CrysAlis software,47 and then refined by full-matrix least-squares (SHELXL2014) on F2.48 The non-hydrogen atoms were refined anisotropically. All of the hydrogen atoms were positioned geometrically in idealized positions and refined with the riding model approximation, with Uiso(H) = 1.2 Ueq(C). For the molecular graphics the program SHELXTL was used.49 All geometric calculations were carried out using the PLATON software.50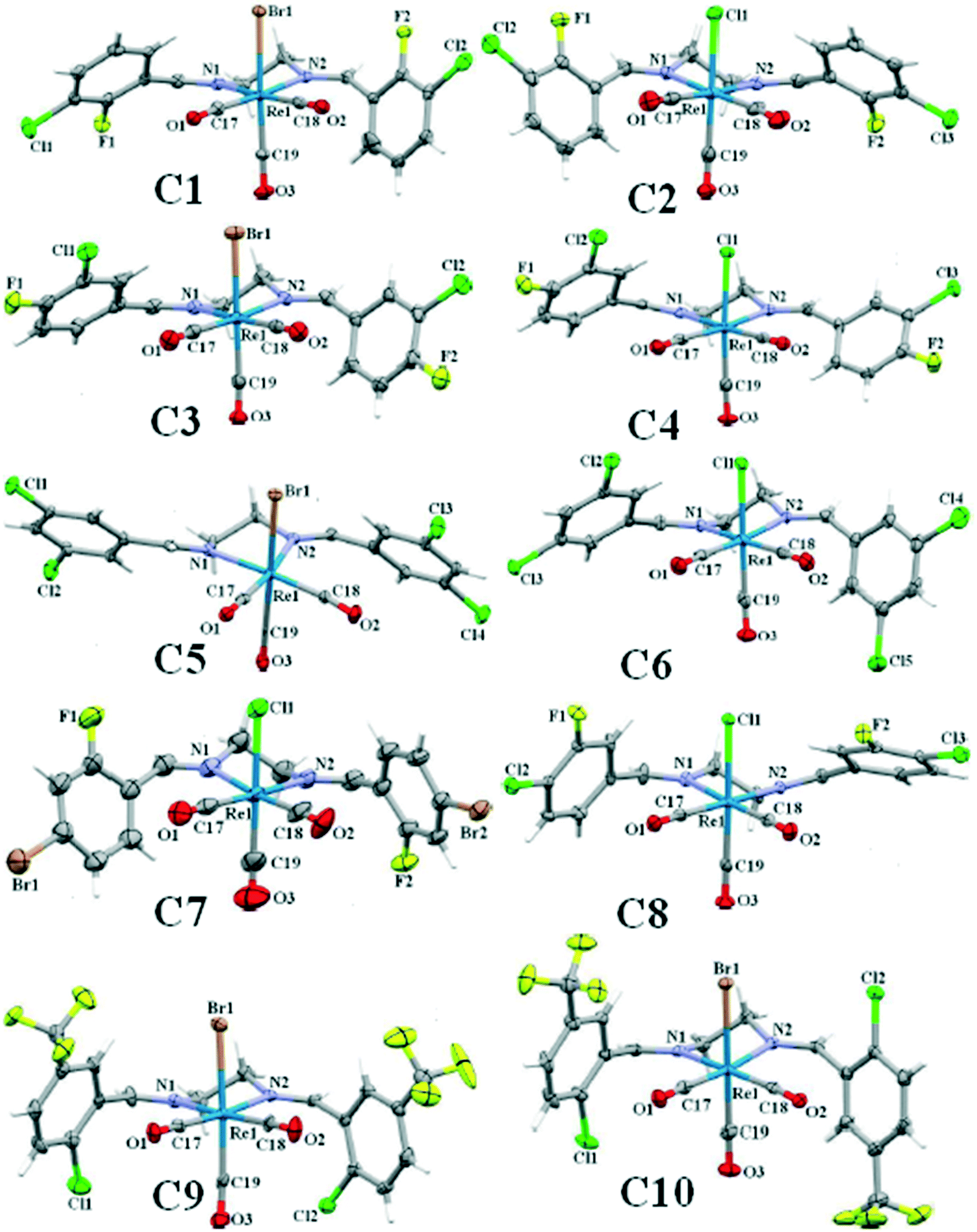 | ||
| Fig. 1 The ORTEP view and selected atom numbering scheme of C1–C10 with 40% ellipsoids probability. | ||
In complex C7, the rhenium atom shows positional disorder over two sites with refined site occupancy 0.927(6)/0.073(6). In complex C10, there was a solvent accessible volume of 385 Å is equal to almost 167 electrons which were not easy to model. The structure was squeezed and the contribution of these electrons in the unit cell was subtracted by back Fourier transform which was equal to almost two n-hexane molecules as solvent of recrystallization. CCDC 1871495–1871503 [C2 (1871495), C3 (1871497), C4 (1871498), C5 (1871496), C6 (1871497), C7 (18714501), C8 (18714500), C9 (18714502), C10 (18714503)] contain the supplementary crystallographic data for this paper.
Computational chemistry
All calculations were carried out using the Gaussian09 program51 using the BP86-D3/def2-SV(P) level of theory. For the calculations we have used the BP86 functional with the latest available Grimme correction functions for dispersion (D3). We have used the crystallographic coordinates as initial approximation for the theoretical analysis of the non-covalent interactions observed in the solid-state. This method has previously been successfully used to evaluate similar interactions.52–55 The interaction energies were computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were corrected for the basis set superposition error (BSSE) using the counterpoise method.56 The molecular electrostatic potential (MEP) for each complex was also done by the Gaussian09 program with BP86-D3 function using def2-TZVP basis set (Fig. S17–S26†).57 The non-covalent interaction index (NCI)58 of the intramolecular interactions was obtained by direct Gaussian cube file to NCI routine via total electron density function calculated with BP86-D3 level of theory using def2-TZVP basis set.In order to better understand the nature of some of the intermolecular interactions such as hydrogen bonding based on the electron density and its Laplacian at bond critical points, the ground-state electronic wavefunctions obtained from Gaussian09 using BP86 level of theory and def2-SVP basis set for all atoms were used for calculations on the topology of the theoretical electron density based on quantum theory of atoms in molecules (QTAIM), including both local and integral properties, with the aid of the program AIM2000.59
Hirshfeld surface analysis
The quantitative analysis of the intermolecular interaction based on Hirshfeld surfaces calculations was studied. The Hirshfeld surfaces60 such as dnorm and 2D fingerprint plots were generated using Crystal Explorer 3.1 and the crystallographically determined coordinates of the atoms in the structures.61 To keep internal consistency when comparing structures, the bond lengths of hydrogen atoms were normalized to standard neutron values (C–H = 1.083 Å, O–H = 0.983 Å, N–H = 1.009 Å). The normalized contact distance (dnorm) based on both de (the distance from a point on the surface to the nearest atom outside the surface) and di (the distance from a point on the surface to the nearest atom inside the surface) and van der Waals radii of the atom, given by eqn (1), enables the identification of the regions of particular importance to intermolecular interactions. The value of dnorm is negative (red colour) or positive (blue colour) when intermolecular contacts are shorter or longer than the van der Waals separations of the designated atoms, respectively.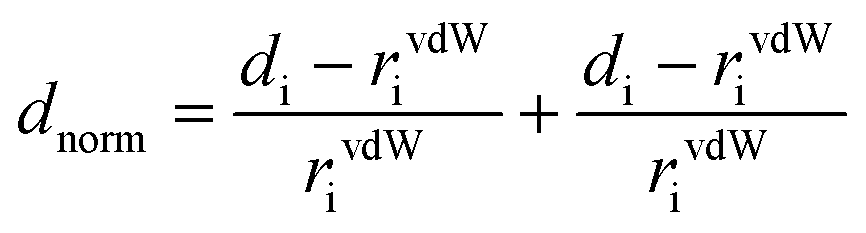 | (1) |
A fingerprint plot is a 2D map that describes the nature and the type of intermolecular interactions quantitatively experienced by a molecule in the crystalline moiety in a convenient graphical format, which is unique for a given crystal structure. In addition, the similarities and dissimilarities between related crystal structures can be identified.
Results
Crystal structure description
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), showing only slight changes of the unit cell dimensions (Tables S1 and S2†). Due to the importance of the intra- and intermolecular interactions in the crystal structure, the information of the crystal packing of each complex with all involved interactions and the packing index was discussed in detail. The packing index was calculated by PLATON.50 In each structure, the bite angle, the angle formed by the coordination of the bidentate N,N-donor ligand to metal centre (N1–Re1–N2), was reported.
), showing only slight changes of the unit cell dimensions (Tables S1 and S2†). Due to the importance of the intra- and intermolecular interactions in the crystal structure, the information of the crystal packing of each complex with all involved interactions and the packing index was discussed in detail. The packing index was calculated by PLATON.50 In each structure, the bite angle, the angle formed by the coordination of the bidentate N,N-donor ligand to metal centre (N1–Re1–N2), was reported.
| Complex | C 2 | C 3 | C 4 | C 5 | C 6 | C 7 | C 8 | C 9 | C 10 |
|---|---|---|---|---|---|---|---|---|---|
| Empirical formula | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re | C19H12Cl3F2N2O3Re |
| Colour | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow | Pale-yellow |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P |
P |
P21/c |
P |
P21/c |
P |
P |
P21/c | P21/c |
| θ max (°) | 27.5 | 27.5 | 27.5 | 29.3 | 27.5 | 27.5 | 29.2 | 26.0 | 30.8 |
| a (Å) | 7.1480(2) | 7.3600(1) | 15.4530(5) | 7.1653(3) | 7.1210(1) | 7.3756(4) | 7.3006(5) | 7.2547(4) | 9.6938(1) |
| b (Å) | 9.2830(2) | 9.2080(3) | 7.0730(2) | 10.1441(5) | 33.7880(7) | 8.8914(4) | 11.5980(7) | 26.9040(16) | 12.9012(2) |
| c (Å) | 17.0670(6) | 16.5900(5) | 23.5290(8) | 16.1968(10) | 9.4780(2) | 16.9636(10) | 12.6789(8) | 12.6293(7) | 22.2519(3) |
| α (°) | 105.7160(9) | 101.508(1) | 90 | 86.971(4) | 90 | 98.004(4) | 104.105(5) | 90 | 90 |
| β (°) | 98.767(1) | 94.970(2) | 126.1351(11) | 82.270(4) | 96.0240(8) | 90.442(5) | 97.855(5) | 93.127(6) | 91.119(1) |
| γ (°) | 95.9490(14) | 98.653(2) | 90 | 71.257(4) | 90 | 105.113(4) | 96.998(5) | 90 | 90 |
| V (Å3) | 1064.83(5) | 1081.25(5) | 2076.98(12) | 1104.65(10) | 2267.86(7) | 1062.4(1) | 1017.88(12) | 2461.3(2) | 2782.33(6) |
| Z | 2 | 2 | 4 | 2 | 4 | 2 | 1 | 4 | 4 |
| D calc (Mg m−3) | 2.017 | 2.123 | 2.096 | 2.177 | 1.991 | 2.300 | 2.111 | 2.136 | 1.889 |
| μ (mm−1) | 6.123 | 7.754 | 6.278 | 7.817 | 5.971 | 9.647 | 6.405 | 6.850 | 6.060 |
| F (000) | 616 | 652 | 1232 | 684 | 1296 | 688 | 616 | 1496 | 1496 |
| Index ranges | −15 ≤ h ≤ 15 | −9 ≤ h ≤ 9 | −20 ≤ h ≤ 17 | −8 ≤ h ≤ 9 | −8 ≤ h ≤ 9 | −8 ≤ h ≤ 9 | −9 ≤ h ≤ 10 | −8 ≤ h ≤ 8 | −13 ≤ h ≤ 13 |
| −17 ≤ k ≤ 19 | −11 ≤ k ≤ 11 | −9 ≤ k ≤ 8 | −12 ≤ k ≤ 13 | −43 ≤ k ≤ 27 | −43 ≤ k ≤ 27 | −15 ≤ k ≤ 15 | −33 ≤ k ≤ 33 | −18 ≤ k ≤ 18 | |
| −17 ≤ l ≤ 19 | −21 ≤ l ≤ 21 | −21 ≤ l ≤ 21 | −21 ≤ l ≤ 22 | −12 ≤ l ≤ 12 | −12 ≤ l ≤ 12 | −17 ≤ l ≤ 17 | −15 ≤ l ≤ 15 | −32 ≤ l ≤ 29 | |
| No. of measured reflns. | 15268 |
15821 |
9932 | 9299 | 13899 |
15534 |
8612 | 21636 |
36476 |
| No. of independent reflns./Rint | 4817 | 4940 | 4688 | 5040 | 5136 | 4862 | 4683 | 4820 | 8709 |
| No. of parameters | 271 | 271 | 271 | 271 | 271 | 281 | 271 | 325 | 325 |
| GOF | 1.064 | 1.279 | 1.030 | 1.031 | 1.035 | 1.023 | 1.019 | 1.098 | 1.061 |
| R 1 (observed data) | 0.0382 | 0.0272 | 0.0435 | 0.0353 | 0.0339 | 0.0527 | 0.0321 | 0.0331 | 0.0381 |
| wR2 (all data) | 0.0456 | 0.0339 | 0.0671 | 0.0410 | 0.0642 | 0.0727 | 0.0361 | 0.0442 | 0.0627 |
| Complex | C 2 | C 3 | C 4 | C 5 | C 6 | C 7 | C 8 | C 9 | C 10 |
|---|---|---|---|---|---|---|---|---|---|
| Re1–N1 | 2.190(4) | 2.209(4) | 2.200(5) | 2.185(44) | 2.214(3) | 2.194(8) | 2.205(3) | 2.195(4) | 2.197(4) |
| Re1–N2 | 2.204(4) | 2.216(4) | 2.227(6) | 2.203(4) | 2.209(4) | 2.193(8) | 2.214(3) | 2.208(4) | 2.196(4) |
| C17–O1 | 1.152(7) | 1.156(6) | 1.159(9) | 1.150(6) | 1.163(5) | 1.149(12) | 1.145(5) | 1.149(5) | 1.156(6) |
| C18–O2 | 1.136(7) | 1.160(6) | 1.154(7) | 1.146(6) | 1.155(5) | 1.156(14) | 1.150(5) | 1.151(6) | 1.155(6) |
| C19–O3 | 1.133(7) | 1.142(7) | 1.144(8) | 1.107(6) | 1.151(6) | 1.048(17) | 1.138(6) | 1.126(6) | 1.139(6) |
| Re1–Br1 | — | 2.6251(5) | — | 2.6196(6) | — | — | — | 2.628(1) | 2.628 (1) |
| Re1–Cl1 | 2.483(1) | — | 2.480(2) | — | 2.489(1) | 2.485(3) | 2.476(1) | — | — |
| N1–Re1–N2 | 77.76(16) | 77.62(15) | 78.4(2) | 77.64(14) | 77.04(13) | 77.7(3) | 77.32(12) | 77.10(14) | 77.19(14) |
| C17–Re1–C18 | 84.4(2) | 83.6(2) | 85.7(3) | 85.2(2) | 85.61(19) | 84.5(4) | 84.30(17) | 85.35(19) | 85.5(2) |
| N1–Re1–C18 | 176.1(2) | 98.15(18) | 176.6(2) | 176.69(18) | 174.90(17) | 174.1(4) | 176.53(14) | 175.04(17) | 99.06(17) |
| N2–Re1–C17 | 176.3(2) | 100.58(18) | 174.3(2) | 172.32(19) | 174.67(18) | 176.6(4) | 176.10(14) | 174.95(17) | 98.52(18) |
| Complex | D–H⋯A | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| Symmetry codes: (i) −1 + x, y, z (ii) x, 1 + y, z (iii) 1 − x, 1 − y, 1 − z (iv) −x, 1 − y, 1 − z (v) 1 − x, −y, 1 − z (vi) 1 + x, y, z (vii) x, y, −1 + z (viii) −1 + x, 1/2 − y, −1/2 + z (ix) 1 − x, 1 − y, 2 − z (x) −x, 1 − y, 2 − z (xi) 2 − x, 1 − y, −z (xii) x, 3/2 − y, 1/2 + z (xiii) −x, 1/2 + y, 1/2 − z. | ||||
| C1 | C2–H2⋯Br1 | 2.78 | 3.691(7) | 161 |
| C7–H7⋯Br1i | 2.91 | 3.846(7) | 170 | |
| C8–H8B⋯O2i | 2.47 | 3.263(10) | 137 | |
| C2 | C12–H12A⋯Cl1 | 2.62 | 3.520(6) | 163 |
| C10–H10A⋯Cl1i | 2.71 | 3.627(6) | 169 | |
| C7–H7A⋯O2ii | 2.33 | 3.245(7) | 168 | |
| C3 | C2–H2⋯Br1 | 2.86 | 3.764(5) | 164 |
| C7–H7⋯Br1i | 2.82 | 3.748(5) | 177 | |
| C10–H10⋯O1ii | 2.34 | 3.269(7) | 173 | |
| C15–H15⋯O3iii | 2.56 | 3.192(8) | 125 | |
| C4 | C2–H2A⋯Cl1 | 2.62 | 3.491(9) | 156 |
| C7–H7A⋯Cl1ii | 2.61 | 3.519(7) | 165 | |
| C5 | C6–H6⋯O3iii | 2.50 | 3.353(6) | 152 |
| C7–H7⋯O3iv | 2.60 | 3.240(6) | 127 | |
| C8–H8A⋯Br1v | 2.93 | 3.780(5) | 147 | |
| C8–H8A⋯O1vi | 2.53 | 3.318(6) | 138 | |
| C14–H14⋯Cl2 | 2.83 | 3.744(5) | 169 | |
| C6 | C2–H2A⋯Cl1 | 2.64 | 3.531(5) | 161 |
| C7–H7A⋯Cl1vi | 2.71 | 3.639(4) | 175 | |
| C8–H8B⋯O2vi | 2.52 | 3.369(6) | 168 | |
| C10–H10A⋯O1vii | 2.45 | 3.369(6) | 168 | |
| C14–H14A⋯O3viii | 2.42 | 3.259(7) | 150 | |
| C7 | C5–H5⋯O3iv | 2.55 | 3.255(16) | 132 |
| C8–H8B⋯O2ii | 2.41 | 3.122(12) | 130 | |
| C9–H9A⋯O1i | 2.54 | 3.222(12) | 128 | |
| C8 | C12–H12⋯Cl1 | 2.57 | 3.461(5) | 159 |
| C5–H5⋯O2ix | 2.57 | 3.290(6) | 135 | |
| C6–H6⋯O3x | 2.60 | 3.376(6) | 142 | |
| C7–H7⋯F1i | 2.37 | 3.291(5) | 173 | |
| C10–H10⋯F2i | 2.54 | 3.402(5) | 154 | |
| C16–H16⋯F2i | 2.55 | 3.384(5) | 149 | |
| C9 | C7–H7⋯F1vi | 2.46 | 3.171(6) | 134 |
| C9–H9B⋯F2xi | 2.42 | 3.195(6) | 136 | |
| C15–H15⋯Br1xii | 2.88 | 3.696(5) | 148 | |
| C10 | C4–H4⋯O3xiii | 2.54 | 3.224(10) | 129 |
32ClFenReCl (C2). In the molecular structure of C2 the bite angle of the N,N-donor ligand is 77.77(17)°. One of the substituted arene rings (C11–C16), is significantly rotated comparing to the other arene ring due to the intramolecular C12–H12A⋯Cl1 hydrogen bonding, making a S(7) ring. The crystal packing shows intermolecular π⋯π interactions with centroid to centroid distances of 3.644(3) to 3.966(4) Å. The crystal packing shows intermolecular C7–H7A⋯O2 and C10–H10A⋯Cl1 hydrogen bonding in which the Cl1 atom is a bifurcated acceptor.
The intermolecular C7–H7A⋯O2 hydrogen bond connects the neighboring molecules into one-dimensional infinite chain along the b-axis. Alternatively, the chains are further consolidated by the intermolecular C10–H10A⋯Cl1 hydrogen bond, Cl1⋯Cl3 halogen bonding, O2⋯Cl2 and Cl2⋯C7 short contacts, forming a two-dimensional sheet running parallel to ab-plane (Fig. 2). The Cl1⋯Cl3 halogen bonding interaction is attractive. The packing index of C2 is 68.2%.
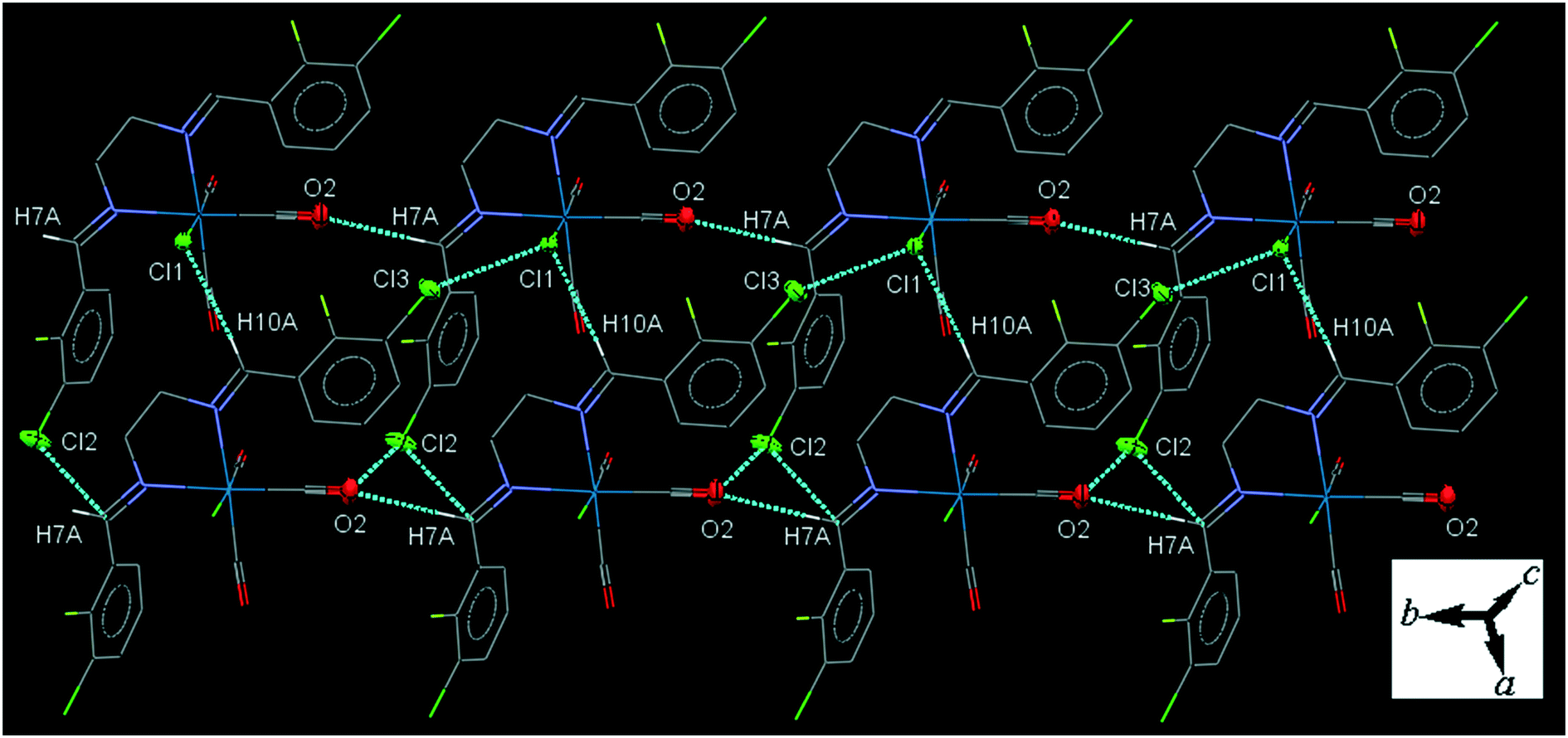 | ||
| Fig. 2 The crystal packing of C2 viewed almost down the c-axis showing one-dimensional chains of molecules running along the b-axis and further consolidated by the intermolecular interactions into a sheet parallel to ab-plane. The H-atoms not involved in the interactions omitted for clarity. | ||
34ClFReBr (C3). One of the substituted arene rings (C1–C6) in C3, is significantly rotated comparing to the other due to the intramolecular C2–H2⋯Br1 hydrogen bonding, making a S(7) ring. The intermolecular C7–H7⋯Br1 and C15–H15⋯O3 supported by antiparallel dipolar CO⋯CO interactions linked molecules into a column parallel to ac-plane which are further connected by C10–H10⋯O1 interactions along the b-axis, forming a three-dimensional network (Fig. 3). The interesting feature of the crystal structure is the individual dimer formation by the intermolecular Cl2⋯F2 interactions which are more connected by the intermolecular Cl1⋯C7 and C2⋯C2 short contacts (Fig. S27†). The packing index of C3 is 67.9%.
 | ||
| Fig. 3 Part of the crystal packing of C3 viewed down the b-axis, showing the connection of neighboring molecules through C–H⋯O, C–H⋯Cl and antiparallel CO⋯CO interactions. | ||
34ClFReCl (C4). The bite angle of the ligand is 78.4(2)°. One of the substituted arene rings (C11–C16), is significantly rotated comparing to the other arene ring due to the intramolecular C2–H2A⋯Cl1 hydrogen bonding, making a S(7) ring. The intermolecular C7–H7A⋯Cl1 hydrogen bonding linked neighboring molecules into one-dimensional extended chain along the b-axis which was supported by the weak intermolecular O2⋯C7 short contacts (Fig. 4).
 | ||
| Fig. 4 Part of the crystal packing of C4 viewed down the c-axis, showing one-dimensional extended chain along the b-axis through intermolecular C–H⋯Cl and C⋯O interactions. | ||
Pairs of weak intermolecular C15–H15A⋯O1 forming a head-to-tail individual dimers which are further connected to each other through the intermolecular C10–H10A⋯F1 interactions (Fig. S28†). The crystal packing also shows Cl2⋯Cl2 interactions which are shorter than the sum of the van der Waals radius of Cl [1.75 Å] atoms. The packing index of C4 is 69.9%.
35ClenReBr (C5). The bite angle of the ligand is 77.63(16)°. The orientation of the halogen-substituted arene ring is different to that in the other complexes with a torsion angle of almost 10°. The intermolecular C4–H4⋯Cl3 and C14–H14⋯Cl2 interactions link neighboring head-to-tail arranged molecules into a one-dimensional extended chain along the c-axis which are interconnected further by the weak intermolecular Cl1⋯Cl3 halogen bond and C2–H2⋯Br1 to form a sheet (Fig. 5). Pair of intermolecular C7–H7⋯O3 and C6–H6⋯O1 hydrogen bonds form a centrosymmetric dimer, linking molecules into an infinite chain along the a-axis (Fig. S29†). The packing index of C5 is 69.9%.
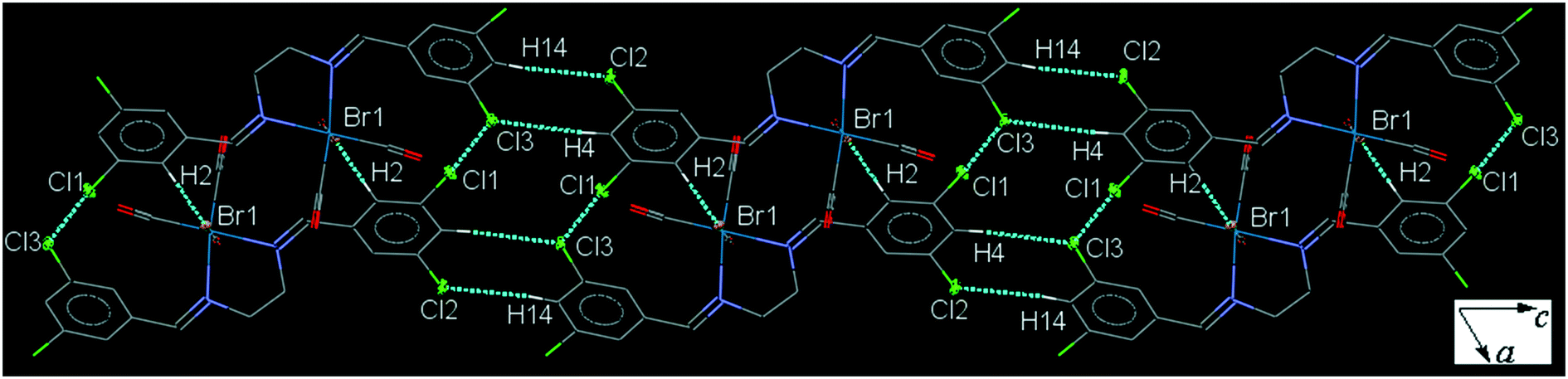 | ||
| Fig. 5 Part of the crystal packing of C5, viewed down the b-axis, showing parallel chain formation through the intermolecular C–H⋯Cl interactions which are by Cl⋯Cl short contacts. | ||
35ClenReCl (C6). The bite angle of the ligand is 77.04(13)°. The intramolecular C2–H2A⋯Cl1 interaction makes a S(7) ring. The intermolecular C7–H7A⋯Cl1 and C8–H8B⋯O2 interactions link neighboring molecules into one-dimensional extended chains along the a-axis. The chains are further connected by the intermolecular C14–H14A⋯O3 and C10–H10A⋯O1 interactions along b and c-axis, respectively, forming a three-dimensional network. The interesting feature of the crystal packing is the intermolecular Cl1⋯Cl3 [3.425(2) Å] halogen bonding supported by the intermolecular Cl2⋯C7 interactions (Fig. 6). The packing index of C6 is 67.3%.
 | ||
| Fig. 6 Part of the crystal packing of C6, showing connection of molecules into a parallel chain through the intermolecular C⋯Cl and Cl⋯Cl interactions. | ||
42BrFReCl (C7). The bite angle of the ligand is 77.7(3)°. Pair of intermolecular C5–H5⋯O3 interactions makes a head-to-tail centrosymmetric dimer. The intermolecular C8–H8B⋯O2 and C9–H9A⋯O1 connect the neighboring molecules into one-dimensional extended chains along the b- and a-axis, respectively, which are supported further by the intermolecular CO(π)⋯Br and C⋯Br interactions. The interesting feature of the crystal packing is the weak intermolecular C15–H15⋯Cl1 interaction and F1⋯F2 halogen bonding which link neighboring molecules into an extended chain in the [110] direction, furthermore the chains are interconnected head-to-tail through the intermolecular centrosymmetric Cl1⋯Br2 halogen bonding (Fig. 7). The packing index of C7 is 66.5%.
 | ||
| Fig. 7 Part of the crystal packing of C7, showing interconnected chains of neighboring molecules through the intermolecular Cl⋯Br halogen bond along [110] direction. | ||
43ClFReCl (C8). The bite angle of the ligand is 77.32(13)°. The intramolecular C12–H2⋯Cl1 interaction makes a S(7) ring. The intermolecular C7–H7⋯F1 and C16–H16⋯F1 interactions, link neighboring molecules into a one-dimensional extended chain along the a-axis. The running head-to-tail chains are further interconnected through the intermolecular centrosymmetric C5–H5⋯O2 and C6–H6⋯O3 hydrogen bonds further supported by the intermolecular Cl2⋯O2 and Cl3⋯C10 short contacts, forming a three-dimensional network (Fig. S30†). The interesting feature of the structure is the intramolecular Re–CO⋯π(arene) interactions which has been explained in the theoretical section based on the molecular orbital calculations. Another interesting feature of the crystal structure is the intermolecular Cl3⋯O2 contact and Cl2⋯Cl2 halogen bond which connects neighboring molecule into a one-dimensional herringbone pattern (Fig. 8). The packing index of C8 is 71.2%.
 | ||
| Fig. 8 Part of the crystal packing of C8, showing one-dimensional herringbone extended chain of molecules through the intermolecular Cl⋯O and Cl⋯Cl contacts. | ||
ClFReBr (C9). The bite angle of the ligand is 77.09(14)°. The intermolecular C15–H15⋯Br1 interaction connects neighboring molecules into a one-dimensional head-to-tail zig-zag extended chain along the c-axis (Fig. 9). The intermolecular C7–H7⋯F1 interaction link neighboring molecules into a one-dimensional extended chain along the a-axis and further the parallel chains are connected together through the intermolecular C9–H9B⋯F2 interaction (Fig. S31†).
 | ||
| Fig. 9 Part of the packing of C9, showing zig-zag connection of molecules into one-dimensional extended chain along the c-axis through C–H⋯Br interaction. | ||
The intermolecular C4–H4⋯F6 contact, links neighboring molecule into a one-dimensional herringbone-type extended chain along the b-axis (Fig. 10). The interesting feature of the crystal packing is the intermolecular Cl⋯Cl interaction which is significantly shorter, [3.196(2) Å], than the sum of the van der Waals radius of Cl atoms [3.50 Å]. The packing index of C9 is 68.0%.
 | ||
| Fig. 10 Part of the crystal packing of C9, showing one-dimensional herringbone-type extended chain along the b-axis through the intermolecular C4–H4⋯F6 contact. | ||
ClFReBr′ (C10). The bite angle of the ligand is 77.1(2)°. The intermolecular C4–H4⋯O3 interaction connects neighboring molecules into a one-dimensional head-to-tail extended chain along the b-axis which is supported by the very short Cl1⋯O2 contact (Fig. 11). An interesting feature of the crystal packing is the intermolecular C–F⋯π interaction which was further supported by the weak intermolecular C⋯H and O⋯H contacts (Fig. S32†). Additionally, the structure is uniquely supported by the intramolecular Re–CO⋯π(arene) interactions which are discussed in the theoretical section based on the molecular orbital calculations. The packing index of C10 is 68.0%.
 | ||
| Fig. 11 One-dimensional extended chain of C10 along the b-axis through intermolecular C–H⋯O and Cl⋯O interactions. | ||
Complexation energy, AIM and NCIPLOT data
To obtain a better understanding of the contribution of intermolecular interactions toward the crystal packing, it is important to obtain quantitative insights into the nature and energy of these interactions. We have focused on the theoretical calculation of the interesting supramolecular assemblies observed in the solid-state of all the structures described above on the basis of the dimers formed by the desired intermolecular interactions. The complexation energies of the important crystallographic fragments based on their related intermolecular interactions are summarized in Table 4; as a caveat, the energies quoted should not be treated as absolute but do put the interactions on a relative scale and provide “ball park” values for the energies. Some of the important crystallographic dimers formed by their related intermolecular interactions are shown in Fig. 12. As it is clear from Table 4, the dimers formed by the cooperative interactions and metal-bound halogen (e.g. dimer1 in C1; dimer4 in C2; dimer4 in C3; dimer2 in C4; dimer6 in C5; dimer3 in C6; dimer4 in C8 and dimer3 in C10) the interaction energies are significant and negative, confirming their strong binding motifs in the solid-state structures. In case of C9 and C10, the contribution of the trifluoromethyl group involved in the H⋯F interactions is also significant.| Complex | Dimers (intermolecular interactions) | BP86-D3/def2-SV(P) energy (kcal mol−1) |
|---|---|---|
| C 1 | Dimer1: H4⋯Br1, H6⋯O3, O2⋯F1 | −14.66 |
| Dimer2: C12⋯F2 | −19.58 | |
| C 2 | Dimer1: Cl1⋯Cl3, Cl2⋯O2 | −3.30 |
| Dimer2: CO⋯CO | −5.03 | |
| Dimer3: H7A⋯O2 | −6.55 | |
| Dimer4: H10A⋯Cl1 | −15.61 | |
| C 3 | Dimer1: H15⋯O3, H16⋯O3, CO⋯CO | −6.17 |
| Dimer2: F2⋯Cl2 | 0.05 | |
| Dimer3: H10⋯O1 | −7.28 | |
| Dimer4: H7⋯Br1, H8B⋯O2, C10⋯Cl2 | −16.53 | |
| C 4 | Dimer1: Cl2⋯Cl2 | −0.10 |
| Dimer2: H7A⋯Cl1, O2⋯C7, H8B⋯π | −18.98 | |
| Dimer3: F2⋯Cg | −2.66 | |
| Dimer4: H15A⋯O1 | −1.78 | |
| C 5 | Dimer1: Cl1⋯Cl3 | −8.27 |
| Dimer2: H14⋯Cl2 | −3.05 | |
| Dimer3: H7⋯O3 | −20.78 | |
| Dimer4: H5⋯O3 | −12.31 | |
| Dimer5: H8B⋯O1 | −13.91 | |
| Dimer6: H8A⋯Br1 | −29.66 | |
| C 6 | Dimer1: Cl1⋯Cl3 | −4.60 |
| Dimer2: C7⋯Cl2 | −16.61 | |
| Dimer3: H7A⋯Cl1, H8B⋯O2, C10⋯Cl4 | −16.45 | |
| Dimer4: H14⋯O3 | −1.82 | |
| C 7 | Dimer1: Br1⋯O3 | −7.21 |
| Dimer2: H5⋯O3 | −4.68 | |
| Dimer3: H8B⋯O2 | −6.61 | |
| Dimer4: H9A⋯O1 | −13.77 | |
| Dimer5: Cl1⋯Br2 | −10.98 | |
| Dimer6: F1⋯F2 | −4.61 | |
| Dimer7: Br2⋯Cg | −10.24 | |
| C 8 | Dimer1: Cg2⋯Cl2 | −9.82 |
| Dimer2: Cl2⋯Cl2 | 0.05 | |
| Dimer3: Cl2⋯O2 | −1.47 | |
| Dimer4: Cl1⋯C10 | −26.11 | |
| Dimer5: H5⋯O2 | −7.71 | |
| Dimer6: H6⋯O3 | −8.00 | |
| Dimer7: H10⋯F2, H16⋯F2 & H7⋯F1 | −15.56 | |
| Dimer8: H15⋯O2 | −16.32 | |
| C 9 | Dimer1: Cl1⋯Cl1 | −0.17 |
| Dimer2: H4⋯F6 | −2.86 | |
| Dimer3: H7⋯F1 | −15.35 | |
| Dimer4: H9B⋯F2 | −13.15 | |
| Dimer5: H15⋯Br1 | −9.72 | |
| C 10 | Dimer1: Cl2⋯O2 | −7.41 |
| Dimer2: F5⋯C7 | −4.28 | |
| Dimer3: H8A⋯O1, H8B⋯Br1, F3⋯C11 & C18⋯H9A | −18.13 | |
| Dimer4: Cl1⋯O2 & H4⋯O3 | −3.10 |
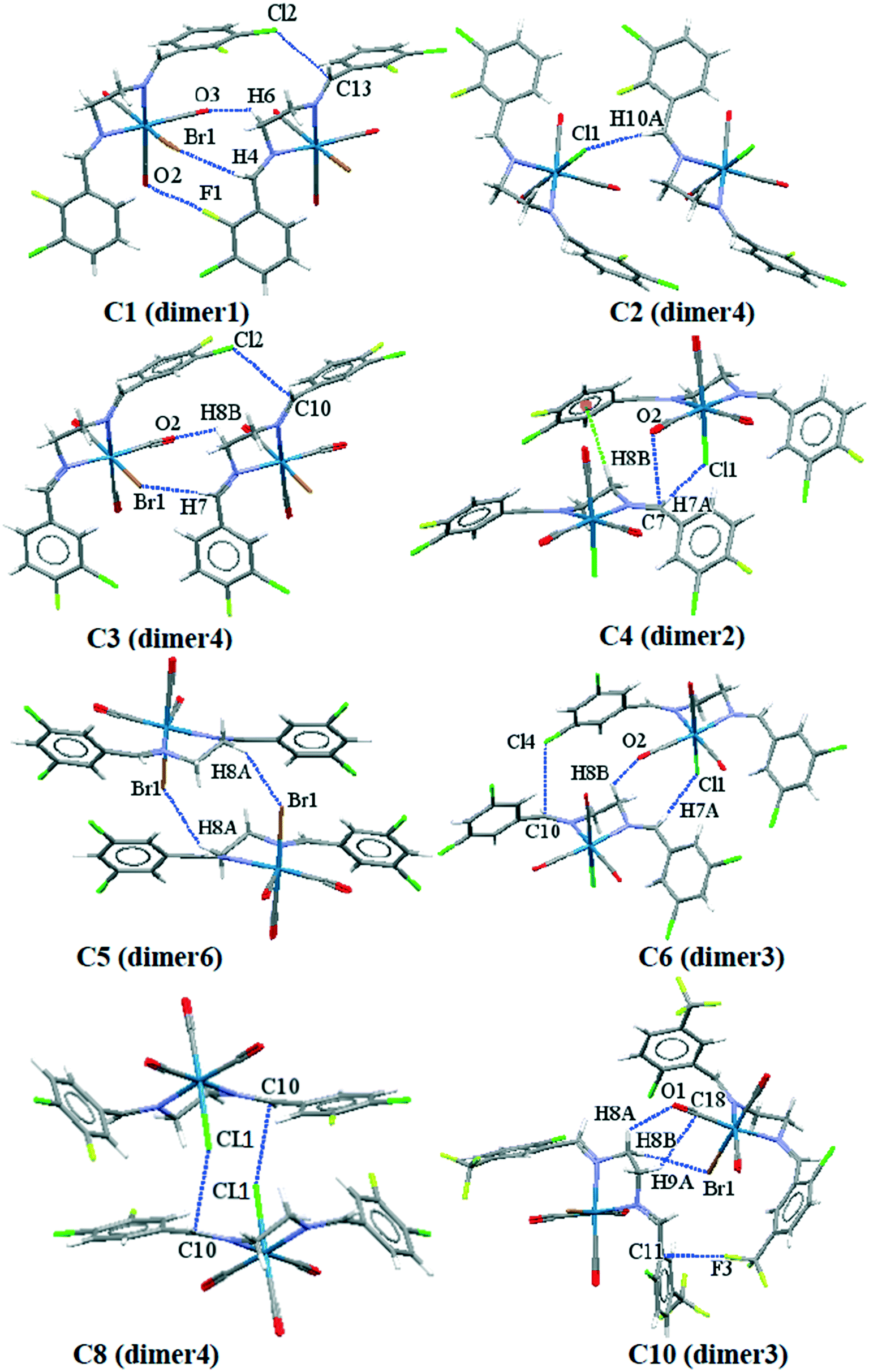 | ||
| Fig. 12 Some of the crystallographic dimers used for intermolecular interactions energy calculation. | ||
In all halogen–halogen interactions with the more linear type I contacts (dimer1 in C2, dimer2 in C3, dimer1 in C4, dimer1 in C6, dimer6 in C7, dimer2 in C8 and dimer2 in C9), in which regions with a partial positive charge on both halogens are in close proximity, the complexation energy is negligible and the interaction is not electrostatically favored. In type II contacts involving an approach of the region with a partial negative charge on one halogen atom to the region of partial positive charge on another, the interaction energy are significant (Table 4). The theoretical models of the important binding motifs in each complex are shown in Fig. S33a–j.† On the other hand, there are some non-classic hydrogen bonds in C2, C5 and C9 and also a halogen bonding in C8 which contribute significantly to the interaction energies. To pursue the nature of such interaction we used AIM calculations. The values of electron density, ρb, Laplacian of electron density, ∇2ρb, at the bond critical point (BCP) are the parameters that are usually used to characterize the nature of a chemical bond. These values for the BCP of the aforementioned hydrogen bonding and halogen bonding of the related dimers have been listed in Table 5. We have also calculated these dimers using other level of theory such as B97D3/def2SVP and MP2/def2-TZVP and the interaction energy is in the same range, giving reliability to the level of theory used. To better show the role of the metal-bound halogen in the intermolecular interactions in some of these complexes, the molecular electrostatic potential of C4, C6, C8 and C10 are shown in Fig. 13. It is clear that there is significant negative electrostatic charge at metal-bound halogen position comparing to the carbon-bound halogen with more electrophilic character.
| Interaction | ρ b | ∇ 2 ρ b |
|---|---|---|
| Cl1⋯H10A (dimer4 in C2) | 0.011 | 0.030 |
| Br1⋯H8A (dimer6 in C5) | 0.010 | 0.024 |
| Cl1⋯C10 (dimer4 in C8) | 0.008 | 0.032 |
| F1⋯H7 (dimer3 in C9) | 0.008 | 0.036 |
 | ||
| Fig. 13 The maps of electrostatic potential of C4, C6, C8 and C10 [isovalue at −0.045 (red) and 0.045 (blue)]. | ||
Another interesting feature of the crystal structure of these complexes is the presence of the intramolecular Re–CO(lone pair)⋯π(arene) interactions. In spite of their inherent weak nature, it has been shown that M–CO(lone pair)⋯π(arene) interactions are relevant in some transition metal carbonyl complexes which provide a measure of stability in their crystal structure.28–30 Besides the possibility of the different type of the intermolecular interactions in these complexes, we have tuned the electronic nature of the arene rings in the starting aldehydes by substituting different halogens at different positions. The shortest intramolecular Re–CO(lone pair)⋯π(arene) interactions were found in C8 and C10. In case of other complexes, the orientation of the substituted arene rings was affected by the other intermolecular interactions in the crystal packing so that there is not suitable short Re–CO⋯π contacts. In fact, the X-ray crystal structures of complexes C1–C10 show the Re–CO⋯π contacts from 3.059 to 3.586 Å. As in the previously studied systems,30 the shortest intramolecular Re–CO⋯π distance was found in C8 with fluoro and chloro substituents and in C10 with trifluoromethyl substituents which enhance the π-acidity of the aromatic rings. The crystallographic results were also rationalized by examining NCIPLOT graphical data and also from DFT calculations using molecular orbitals analyses (Fig. 14).
 | ||
Fig. 14 The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O(lone-pair)⋯π interactions (top, a and b), NCIPLOT (middle, c and d) and the molecular orbitals (bottom, e–g) of C8 and C10, respectively. The arrows in e–g show the contribution of the lone pair of the oxygen atom. O(lone-pair)⋯π interactions (top, a and b), NCIPLOT (middle, c and d) and the molecular orbitals (bottom, e–g) of C8 and C10, respectively. The arrows in e–g show the contribution of the lone pair of the oxygen atom. | ||
Fig. 14c and d show a detailed non-covalent interactions map using reduced density gradient iso-surface (NCI-RDG) with the color scale chosen to be, −0.03 < ρ × sign(λ2) < 0.03 a.u., for the short intramolecular CO⋯π interactions in C8 and C10. The iso-surface plot clearly shows the presence of the weak intramolecular interactions. In order to confirm this behavior we have carried out a theoretical calculation using Gaussian09 program with BP86-D3 function and def2-TZVP basis set. In particular, we have calculated all molecular orbitals of complexes C8 and C10 in order to clarify the differences based on the molecular orbital interactions to explain their short CO⋯π interactions. Interestingly, in some of the calculated molecular orbitals of the substituted arene ring we have found small but noteworthy contributions from the atomic orbitals of the interacting CO. This confirms that the π-antibonding orbitals (H–3 in C8 and H–4 in C10) and π-bonding orbital (H–3 in C10) have atomic contributions from the CO co-ligand (Fig. 14e–g). Therefore, it can be concluded that there are lone-pair⋯π interactions in complexes C8 and C10. We have also investigated these intramolecular interactions by AIM through bond path between the carbonyl groups of complexes C8 and C10 with the related arene rings. In both cases, there are bond paths with the related BCP between the carbon atom of the carbonyl groups and carbon atom of the arene rings with ρb = 0.01 a.u. and ∇2ρb = 0.032 a.u. in C8 and C10 (Fig. S34†), confirming the closed shell interactions but because of the long distance of the oxygen atom of the carbonyl group from the carbon atom of the arene ring, AIM does not show bond path between them although the presence of these weak interactions were confirmed by NCI index and molecular orbital calculations. In case of molecular orbital calculations we also found the contribution of the atomic orbitals of the fragments in these complexes by dividing each complex to six fragments, namely, the metal centre (Re), the axial X group (–Cl in C8 and –Br in C10), the axial CO group, the equatorial CO groups, and the diimine ligand (Table 6). In C8, it can be seen from Table 6 that the fragment orbital of COeq2 (p orbital of oxygen) and the ligand (π* orbital) have 1.3 and 94.2 percentage in H–3 and they can interact to each other (Fig. 14e). In C10, the fragment orbital of COeq1 (p orbital of oxygen) contributed 2.9% in H–3 and interact with the right hand side arene ring. Of the 40.3% of the ligand in H–3, only 26% is located on right hand side arene ring, 7% on the left one and the rest on ethylenediimine segment. On the other hand, in H–4, the fragment orbital of COeq2 (p orbital of oxygen) contributed 0.7% to the interaction with left hand side arene ring (Fig. 14g) with 88.4% contribution of the diimine ligand of which almost 86% is donated by the arene ring and the rest is only located on the ethylenediamine segment.
| Complex | Re1 | X | COax | COeq1 | COeq2 | Ligand |
|---|---|---|---|---|---|---|
| ax = axial, eq1 = right hand side equatorial to X, eq2 = left hand side equatorial to X, Ligand = NN-ligand. | ||||||
| C8 | X = Cl | |||||
| H–3 | 2 | 1.9 | 0.1 | 0.4 | 1.3 | 94.2 |
| C10 | X = Br | |||||
| H–3 | 14.4 | 33.8 | 2.4 | 2.9 | 1.2 | 40.3 |
| H–4 | 3.3 | 6.4 | 0.9 | 0.3 | 0.7 | 88.4 |
Furthermore, we also studied the nature and relative strength of the halogen-bonding interactions in all complexes using a reduced density gradient iso-surface (NCI-RDG) with respect to ρ × sign(λ2) by color scale. The intermolecular interaction iso-surfaces generated by NCIPLOT for s = 0.3 and −0.03 < ρ × sign(λ2) < +0.03 [colour scale: attractive (blue) → repulsive (red)] and the corresponding plots of ρ × sign(λ2) vs. reduced density gradient highlighting the halogen bonding were shown in the ESI† (Fig. S35a–o).
Hirshfeld surface studies
In order to obtain a deeper understanding of the intermolecular interactions in the molecular packing of the complexes, an analysis of their Hirshfeld surfaces was conducted. The Hirshfeld contact surfaces as dnorm for complexes C1–C10 and their 2D fingerprint plots are shown in Fig. S36 and S37,† respectively. The information presented in Table 3 is shown pictorially in the Hirshfeld surface plots (Fig. S36†) to aid the analysis of the intermolecular interactions. According to the Hirshfeld surface analysis, the intermolecular H⋯H contacts, i.e. dispersive interactions, contribute 13% of the total number of contacts in the crystal packing of C1. The shortest H⋯H contact appears as broad spikes at de + di ≈ 2.2 Å in the fingerprint plot. The Cl⋯H contact makes a 21% contribution in the fingerprint plot but the shortest contact between Cl and H is longer than their sum of the van der Waals radii, therefore, it is not the main intermolecular short contact in the crystal packing of C1. The structure of C1 is dominated by Br⋯H (9.3%) and O⋯H (8%) contacts. The Br⋯H contact in the fingerprint plot is shown as wings having sharp spikes with the shortest de + di ≈ 2.8 Å. The shortest O⋯H contacts are appeared in the fingerprint plot as characteristic sharp spikes at de + di ≈ 2.4 Å. In a similar manner to C1, the main contribution to the intermolecular interactions of C2 in the fingerprint plot arises from O⋯H contact and metal-bound halogen (Cl) with 24.3 and 21.3% in the crystal packing. Both contacts feature characteristic sharp spikes at de + di ≈ 2.2 Å. The intermolecular H⋯H contacts contribute almost 12% of the total number of contacts in the crystal packing. Further inspection of other intermolecular contacts in the structure of C2 revealed a portion of C⋯C, C⋯O, Cl⋯O and C⋯Cl contacts with less than 5% contribution. By introducing Cl and F in 3 and 4 positions of the arene rings of C4, again H⋯O and H⋯Br are dominant interactions in the crystal packing as it is shown in the fingerprint plot with 21.1 and 7.1% of the total number of contacts, respectively. They are appeared as sharp spikes at de + di ≈ 2.2 Å for H⋯O and 2.75 Å for H⋯Br. The fingerprint plot of C3 features a weak Cl⋯F interaction as sharp spikes at de + di ≈ 3.0 Å but with negligible contribution (4.3%) of the total number of contacts. In the fingerprint plot of C4 characteristic sharp spikes at de + di ≈ 2.5 Å, arise from the interaction of the metal coordinated chloride with hydrogen (H⋯Cl), having 24.5% of the total number of contacts in the crystal packing. The main contribution in the fingerprint plot of C5 arises from H⋯O, H⋯Br and H⋯Cl interactions with overall 18.4, 9.6 and 19.5% contribution to the surface in the crystal packing. They appear as sharp spikes at de + di ≈ 2.4 Å for H⋯O, ≈2.80 Å for H⋯Br and at ≈2.70 Å for H⋯Cl. In the crystal packing of C5 the metal-coordinated halogen (Br) and substituted Cl2 atoms contribute to H⋯Br and H⋯Cl interactions. The fingerprint plot of C6 corresponds to H⋯Cl and H⋯O contacts with a pair of superimposed sharp spikes with an overall 28% and 19% contribution to the surface, respectively. The spikes related to H⋯Cl and H⋯O is located at de + di ≈ 2.6 Å and 2.31 Å, respectively. The weak Cl⋯Cl contact less than the sum of the van der Waals radii of Cl was contributed to the Hirshfeld surface (7%) of C6 at de + di ≈ 3.4 Å. In these three pairs (C1–C6) it can be seen that by changing the metal-coordinated halogen the fingerprint plot change significantly. The most significant intermolecular interaction in the crystal packing of C7 is non-classic C–H⋯O interactions which are appeared by superimposed sharp spikes with an overall 17.6% contribution to the surface at de + di ≈ 2.4 Å. The fingerprint plot of C8 is favored by the intermolecular H⋯Cl, H⋯O and H⋯F contacts with an overall 21%, 18% and 14% contribution to the surface. The H⋯Cl and H⋯O interactions in the fingerprint plot appeared as wing at de + di ≈ 2.9 Å and 2.6 Å, respectively. The H⋯F contacts are shown as superimposed sharp and long spikes at de + di ≈ 2.2 Å in the fingerprint plot. The H⋯F contacts contribution in the structure of C8 is the most significant comparing to the rest of the structures. The most significant intermolecular contact in the fingerprint plot of C9 is H⋯F and H⋯Br with an overall 27.4 and 7.4% contribution to the surface which appear at de + di ≈ 2.36 Å and 2.8 Å, respectively. The fingerprint plot of C10 features the presence of short O⋯H and Cl⋯O contacts with overall 10.7% and 7% contribution to the Hirshfeld surface appearing at de + di ≈ 2.5 Å and 3.0 Å, respectively. The sharp superimposed spikes at 3.0 Å indicate the significant short Cl⋯O contact. It is noteworthy to mention that in C1–C6 and C9, the metal-coordinated halogen with nucleophilic character, as shown in the electrostatic maps (MEP, Fig. S17–S26†), is more effective in the intermolecular contacts, directing the crystal packing, comparing to the carbon-substituted halogen atoms.Conclusions
In conclusion, nine new rhenium(I) tricarbonyl complexes bearing halogen-substituted NN-donor ligands were synthesized and structurally characterized by X-ray diffraction, FT-IR, 1H-NMR and elemental analyses. The detailed analysis of the crystal packing along with their interaction energies of the crystallographically generated dimers connected by the related intermolecular interactions confirmed that the metal-bound halogen (Re–X), is more effective than organic halogen (C–X) in directing the crystal packing. This evaluation was also confirmed using molecular electrostatic potential (MEP) surface calculations at metal-bond halogen which was also confirmed by the Hirshfeld surface analyses. A rare intramolecular M–CO(lone-pair)⋯π interactions were found in two complexes (C8 and C10) with short intramolecular metal-bound carbonyl and arene ring which were confirmed by non-covalent interaction index (NCI) and molecular orbital calculations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
RK is thankful to Sharif University of Technology research Council for the research facility and to Bath University for a visiting lectureship. PRR is grateful to the Engineering and Physical Sciences Research Council (EPSRC) for continued funding (EP/K004956/1).References
- (a) H. A. Sparkes, P. R. Raithby, E. Clot, G. P. Shields, J. A. Chisholm and F. H. Allen, CrystEngComm, 2006, 8, 563–570 RSC; (b) F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- J. M. Lehn, Science, 1985, 227, 849 CrossRef CAS PubMed.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- C. B. Aakeröy, A. Rajbanshi, Z. J. Li and J. Desper, CrystEngComm, 2010, 12, 4231–4239 RSC.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- G. R. Desiraju, Perspectives in Supramolecular Chemistry, John Wiley & Sons, Chichester, UK, 1996, vol. 2 Search PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8(8), 5392–5398 RSC.
- (a) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed; (b) Z. Han, Y. Zhao, J. Peng, A. Tian, Q. Liu, J. Ma, E. Wanga and N. Hu, CrystEngComm, 2005, 7(63), 380–387 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- L. Maugeri, J. Asencio-Hernandez, T. Lebl, D. B. Cordes, A. M. Z. Slawin, M.-A. Delsuc and D. Philp, Chem. Sci., 2016, 7, 6422–6428 RSC.
- L. Brammer, G. M. Espallargas and H. Adams, CrystEngComm, 2003, 5(60), 343–345 RSC.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44, 57–64 CrossRef.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- M. Hennemann, J. S. Murray, K. E. Riley, P. Politzer and T. Clark, J. Mol. Model., 2012, 18, 2461–2469 CrossRef CAS PubMed.
- M. Palusiak, J. Mol. Struct., 2010, 945, 89–92 CrossRef CAS.
- K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2008, 4, 232–242 CrossRef CAS.
- A. J. Stone, J. Am. Chem. Soc., 2013, 135, 7005–7009 CrossRef CAS.
- P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835–5838 CrossRef CAS.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1(4), 277–290 CrossRef CAS.
- G. M. Espallargas, F. Zordan, L. A. Marin, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem. – Eur. J., 2009, 15, 7554–7568 CrossRef PubMed.
- (a) Halogen Bonding: Fundamentals and Applications, ed. G. Metrangolo and G. Resnati, Springer, Berlin, 2008 Search PubMed; (b) L. Meazza, J. Martí-Rujas, G. Terraneo, C. Castiglioni, A. Milani, T. Pilati, P. Metrangolo and G. Resnati, CrystEngComm, 2011, 13, 4427–4435 RSC.
- J. Martí-Rujas, L. Meazza, G. Keat Lim, L. Terraneo, T. Pilati, K. D. M. Harris, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2013, 52, 13444–13448 CrossRef PubMed.
- L. Brammer, G. M. Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- L. Brammer, Faraday Discuss., 2017, 203, 485–507 RSC.
- F. Zordan, G. M. Espallargas and L. Brammer, CrystEngComm, 2006, 8, 425–431 RSC.
- S. Haddad, F. Awwadi and R. D. Willet, Cryst. Growth Des., 2003, 3, 501 CrossRef CAS.
- (a) J. C. Calabrese, D. B. Jordan, A. Boodhoo, S. Sariaslani and T. Vannelli, Biochemistry, 2004, 43, 11403–11416 CrossRef CAS; (b) E. J. Stollar, J. L. Gelpi, S. Velankar, A. Golovin, M. Orozco and B. F. Luisi, Proteins, 2004, 57, 1–8 CrossRef CAS PubMed; (c) T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC; (d) M. Eali and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef PubMed.
- (a) J. Zukerman-Schpector, I. Haiduc and E. R. T. Tiekink, Chem. Commun., 2011, 47, 12682–12684 RSC; (b) J. Zukerman-Schpector, I. Haidu and E. R. T. Tiekink, Adv. Organomet. Chem., 2012, 60, 49–92 CrossRef CAS; (c) C.-Q. Wan, X.-D. Chen and T. C. W. Mak, CrystEngComm, 2008, 10, 475–478 RSC.
- C. Murcia-García, A. Bauzá, G. Schnakenburg, A. Frontera and R. Streubel, CrystEngComm, 2015, 17, 1769–1772 RSC.
- Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650–4652 CrossRef CAS PubMed.
- J. Echeverria, Chem. Commun., 2018, 54, 3061–3064 RSC.
- M. T. Doppert, H. van Overeem and T. J. Mooibroek, Chem. Commun., 2018, 54, 12049–12052 RSC.
- P. Murray-Rust, H. B. Burgi and J. D. Dunitz, J. Am. Chem. Soc., 1975, 97, 921–928 CrossRef CAS.
- (a) J. R. Shaw and R. H. Schmehl, J. Am. Chem. Soc., 1991, 113, 389–394 CrossRef CAS; (b) S. Ranjan, S.-Y. Lin, K.-C. Hwang, Y. Chi, W.-L. Ching, C.-S. Liu, Y.-T. Tao, C.-H. Chien, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2003, 42, 1248–1255 CrossRef CAS PubMed; (c) Z. Si, J. Li, B. Li, F. Zhao, S. Liu and W. Li, Inorg. Chem., 2007, 46, 6155–6163 CrossRef CAS PubMed.
- (a) Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed; (b) J. Shakeri, H. Farrokhpour, H. Hadadzadeh and M. Joshaghani, RSC Adv., 2015, 5, 41125–41134 RSC; (c) L. E. Hatcher, CrystEngComm, 2018, 20, 5990–5997 RSC.
- (a) J. C. Calabrese, D. B. Jordan, A. Boodhoo, S. Sariaslani and T. Vannelli, Biochemistry, 2004, 43, 11403–11416 CrossRef CAS; (b) E. J. Stollar, J. L. Gelpi, S. Velankar, A. Golovin, M. Orozco and B. F. Luisi, Proteins, 2004, 57, 1–8 CrossRef CAS PubMed; (c) M. Eali and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef PubMed; (d) T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- H.-K. Fun and R. Kia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1916 CrossRef CAS PubMed.
- H.-K. Fun and R. Kia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o2131 CrossRef CAS PubMed.
- H.-K. Fun and R. Kia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1941 CrossRef CAS PubMed.
- H.-K. Fun and R. Kia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o2169 CrossRef PubMed.
- H.-K. Fun and R. Kia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1722 CrossRef CAS PubMed.
- R. Kia and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 65, m192 CrossRef PubMed.
- J. M. Villegas, S. R. Stoyanov, W. Huang and D. P. Rillema, Dalton Trans., 2005, 1042–1051 RSC.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 64, 887–897 CrossRef.
- SuperNova Eos S2 System: Empirical absorption correction, 2011, CrysAlis-Software package, Oxford Diffraction Ltd Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS and Agilent (2012). AutoChem 2.0, in conjunction with OLEX2. Agilent Technologies UK Ltd, Yarnton, Oxfordshire, England.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng and D. J. Sonnenb, Gaussian 09, Revision A.02 Search PubMed.
- A. Bauzá, A. Terrón, M. Barceló-Oliver, A. García-Raso and A. Frontera, Inorg. Chim. Acta, 2016, 452, 244–250 CrossRef.
- D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 11, 1958–1972 CrossRef.
- M. Mirzaei, H. Eshtiagh-Hosseini, Z. Bolouri, Z. Rahmati, A. Esmaeilzadeh, A. Hassanpoor, A. Bauza, P. Ballester, M. Barceló-Oliver, J. T. Mague, B. Notash and A. Frontera, Cryst. Growth Des., 2015, 15, 1351–1361 CrossRef CAS.
- P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) NCIplot, version 3.0., Department of Chemistry, Duke University, USA, 2011, http://www.chem.duke.edu/~yang/Software/softwareNCI.html Search PubMed; (b) E. R. Johnson, S. Keinan, S. P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (c) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- F. Biegler-König and J. J. Schönbohm, Comput. Chem., 2002, 23(15), 1489–1494 CrossRef PubMed.
- (a) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer, version 3.0, University of Western Australia, Crawley, Australia, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1871495–1871503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01860j |
| ‡ This paper is dedicated to the memory of Dr Frank H. Allen, a mentor, friend and one of the pioneers of the study of intermolecular interactions in the solid-state. |
| This journal is © The Royal Society of Chemistry 2019 |