
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis and enzymatic extension of an N-GlcNAz asparagine building block†
Mikkel Haarslev Schröder
Marqvorsen
 *a,
Sivasinthujah
Paramasivam
b,
Ward
Doelman
a,
Antony John
Fairbanks
bc and
Sander Izaäk
van Kasteren
a
*a,
Sivasinthujah
Paramasivam
b,
Ward
Doelman
a,
Antony John
Fairbanks
bc and
Sander Izaäk
van Kasteren
a
aLeiden Institute of Chemistry (LIC), Division of Bio-Organic Chemistry, Einsteinweg 55, Leiden, The Netherlands. E-mail: m.h.s.marqvorsen@lic.leidenuniv.nl
bDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand
cBiomolecular Interaction Centre, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand
First published on 17th April 2019
Abstract
N-Azidoacetyl-D-glucosamine (GlcNAz) is a particularly useful tool in chemical biology as the azide is a metabolically stable yet accessible handle within biological systems. Herein, we report a practical synthesis of FmocAsn(N-Ac3GlcNAz)OH, a building block for solid phase peptide synthesis (SPPS). Protecting group manipulations are minimised by taking advantage of the inherent chemoselectivity of phosphine-mediated azide reduction, and the resulting glycosyl amine is employed directly in the opening of Fmoc protected aspartic anhydride. We show potential application of the building block by establishing it as a substrate for enzymatic glycan extension using sugar oxazolines of varying size and biological significance with several endo-β-N-acetylglucosaminidases (ENGases). The added steric bulk resulting from incorporation of the azide is shown to have no or a minor impact on the yield of enzymatic glycan extension.
N-Azidoacetyl-D-glucosamine (GlcNAz (1), Scheme 1A) is a carbohydrate motif that has proven to be a particularly useful tool in chemical biology; the azide is a metabolically stable1 yet accessible handle within biological systems, allowing bioorthogonal visualisation after metabolic labelling.2,3 In some recent examples, the GlcNAz motif has been demonstrated as a useful component of synthetic probes used in chemical biology studies of glycan biology. For example Vocadlo and co-workers synthesised a GlcNAz derivative (2-azidoacetamido-2-deoxy-5-fluoro-β-D-glucopyranosyl fluoride) and used it as an activity-based protein probe for successful isolation of a bacterial N-acetyl-β-D-glucosaminidase.4 In another study, Overkleeft and co-workers studied proteasomal recognition of fully synthetic O-GlcNAc and O-GlcNAz containing peptide inhibitors using activity-based protein profiling, showing minimal interference by the azide motif. Several other studies have utilized the related GalNAz motif for a variety of chemical biology studies.5–7
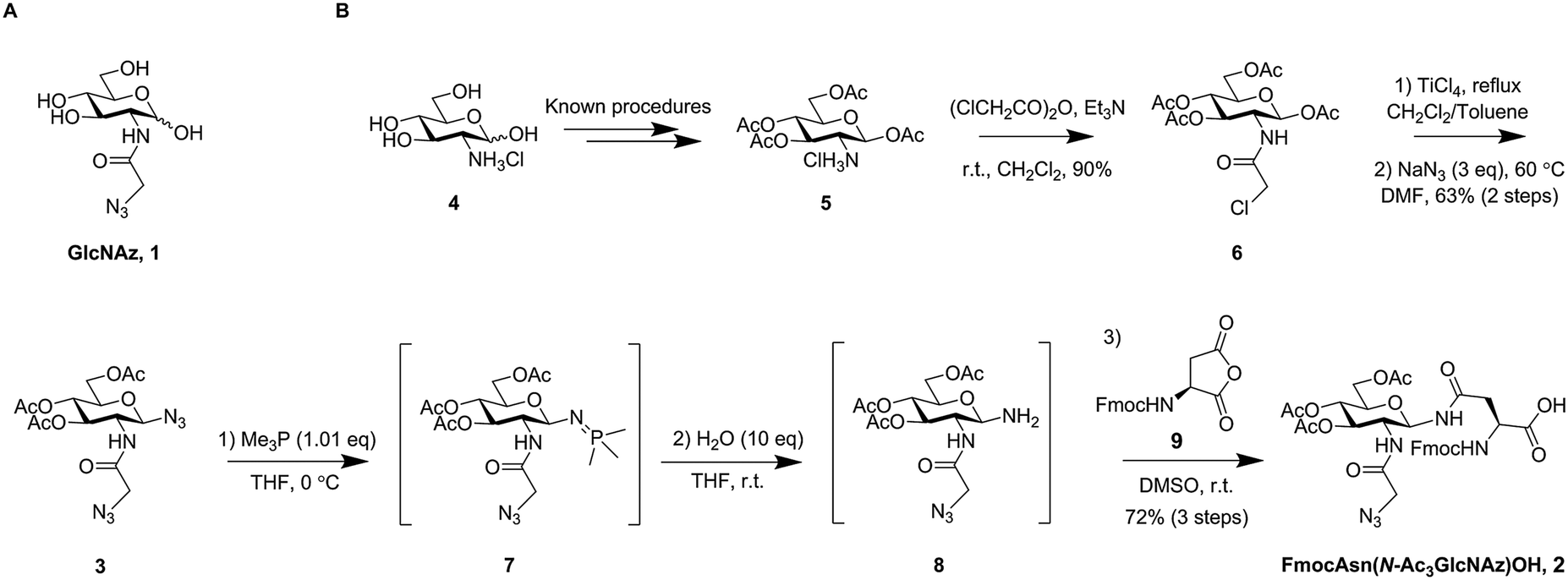 | ||
| Scheme 1 (A) The chemical structure of GlcNAz; (B) synthetic route and intermediates towards the target compound FmocAsn(N-Ac3GlcNAz)OH (2). | ||
As part of ongoing investigations into the antigen presentation biology of dendritic cells using bioorthogonal antigens,8–10 we sought to access peptide probes containing N-linked GlcNAz for a variety of chemical biology experiments. However, a literature survey revealed that such compounds had not been reported previously. Accordingly, we sought to develop a reliable and scalable synthetic approach to the suitably protected building block FmocAsn(N-Ac3GlcNAz)OH (2, Scheme 1B), which would allow its use in the solid phase peptide synthesis (SPPS) of glycopeptides and other biological probes. Herein, we report a practical synthesis of glycosyl amino acid 2, and demonstrate its usefulness by establishing it as a substrate for enzymatic glycan extension using sugar oxazolines11 of varying size and biological significance with several endo-β-N-acetylglucosaminidase enzymes (ENGases).12
A key synthetic challenge in the production of FmocAsn(N-Ac3GlcNAz)OH (2) is installation of the glycosyl amine for subsequent coupling to a selectively protected aspartic acid derivative in the presence of the 2-azidoacetamido group. Whilst more convoluted and labour-intensive protecting group based strategies might meet this challenge, we envisioned a synthetic route in which a diazide 3 (Scheme 1B) was converted directly to the corresponding glycosyl amine via selective azide reduction. That such an approach may prove feasible was supported by the work of Wong and co-workers, who have previously published methodology for the chemoselective Staudinger-type reduction of azides using Me3P in THF solution.13 Accordingly, diazide 3 was prepared using a scalable approach from the hydrochloride salt of glucosamine (4, Scheme 1B). The β-anomeric glycosyl acetate 5 was prepared without chromatographic purification, as reported by Bergmann and Zervas,14 and chloroacetylated to give 6. TiCl4 mediated glycosyl chloride formation15 followed by double substitution with sodium azide in DMF afforded the desired diazide 3 in 63% yield (Scheme 1B), corresponding to 57% yield overall from compound 5.
The Staudinger reduction of a glycosyl azide to afford the corresponding glycosyl amine has only been reported sporadically, mostly employing triphenylphosphine (or a resin bound version of it)16 as the reductant.16–20 We have previously reported the use of Me2PhP for reduction of a glycosyl azide.21 Other phosphines have been used to obtain phosphinimides as targets,22 either for direct transformation into other desired functionalities, e.g. via the traceless Staudinger ligation,23,24 or for use in other reactions in which the phosphinimides act as nucleophiles.25–27 Wong and co-workers found that selective azide reduction to afford amines was possible in some cases using Me3P. The selectivity seemed not to follow expectations based on steric considerations, but was instead predictable from the 1H NMR chemical shifts of the ipso-protons next to each individual azide.13 We found that, indeed, upon addition of Me3P (1 eq., 1 M in THF), a crude 1H NMR spectrum revealed almost instant and complete conversion of the electron poor anomeric azide to give phosphinimide 7, whilst leaving the azidoacetyl group unaffected. Subsequent addition of 10 equivalents of water to the crude reaction mixture and agitation at room temperature led to fast hydrolysis whilst avoiding competitive anomerisation, thus affording clean conversion to the desired β-anomeric glycosyl amine 8.
We sought to maximise the practicality of the approach by coupling glycosyl amine 8 with aspartic anhydride28 (9, Scheme 1B) to afford glycosyl amino acid building block 2 for SPPS. Crude glycosyl amine 8 was therefore dissolved in DMSO and Fmoc aspartic anhydride (9) was added. Indeed, after only 1 hour at room temperature all of the glycosyl amine had disappeared to give the desired product. Purification by precipitation was most practical on large scale, and a 5 mmol reaction afforded the desired FmocAsn(N-Ac3GlcNAz)OH (2) in a 72% yield over three steps without any column chromatography.
GlcNAc-asparagine conjugates, either as discrete glycosyl amino acids or incorporated into larger peptides or proteins, act as efficient handles for the enzymatic attachment of a variety of N-glycan structures.29 Numerous examples30 of their application for the production of a variety of biologically active glycopeptides31 and glycoproteins32 exist. The utility of the FmocAsn(N-Ac3GlcNAz)OH amino acid 2 would therefore be significantly increased if the carbohydrate portion of this building block could be elaborated into more extended N-glycan structures for later applications as biological probes.
Previously we,22 and others,33 have reported on the tolerance of ENGase enzymes13 to structural modifications of the acceptor for ENGase-catalysed glycosylation reactions using N-glycan oxazolines as donors.12,34 However, these studies did not investigate modification of the GlcNAc 2-acetamido group.
Although structural information is available on several of the family GH85 ENGases,35 a particular concern was that the incorporation of an azide might significantly impair the ability of ENGases to attach N-glycan structures. Before application of the GlcNAz building block for the construction of more extended peptide structures, it was therefore deemed prudent to examine the ability of the monomeric glycosyl amino acid to act as an acceptor in a series of ENGase-mediated reactions.
The acetate protecting groups were removed from triacetate 2, to give the acceptor FmocAsn(N-GlcNAz)OH (10) as a substrate for ENGase-catalysed reactions (Scheme 2). Triol 10 was then subjected to a series of glycosylation reactions using several ENGase enzymes (WT Endo A,36 WT Endo M,37 and Endo M N175Q38), and a variety of N-glycan oxazolines as donors (disaccharide oxazoline 11,39 tetrasaccharide oxazoline 12,40 and decasaccharide oxazoline 13;41Table 1 and Scheme 2). The yields of the corresponding glycosylation reactions using FmocAsn(N-GlcNAc)OH (i.e. the analogous acceptor without the azide) are given for comparison where data is available.
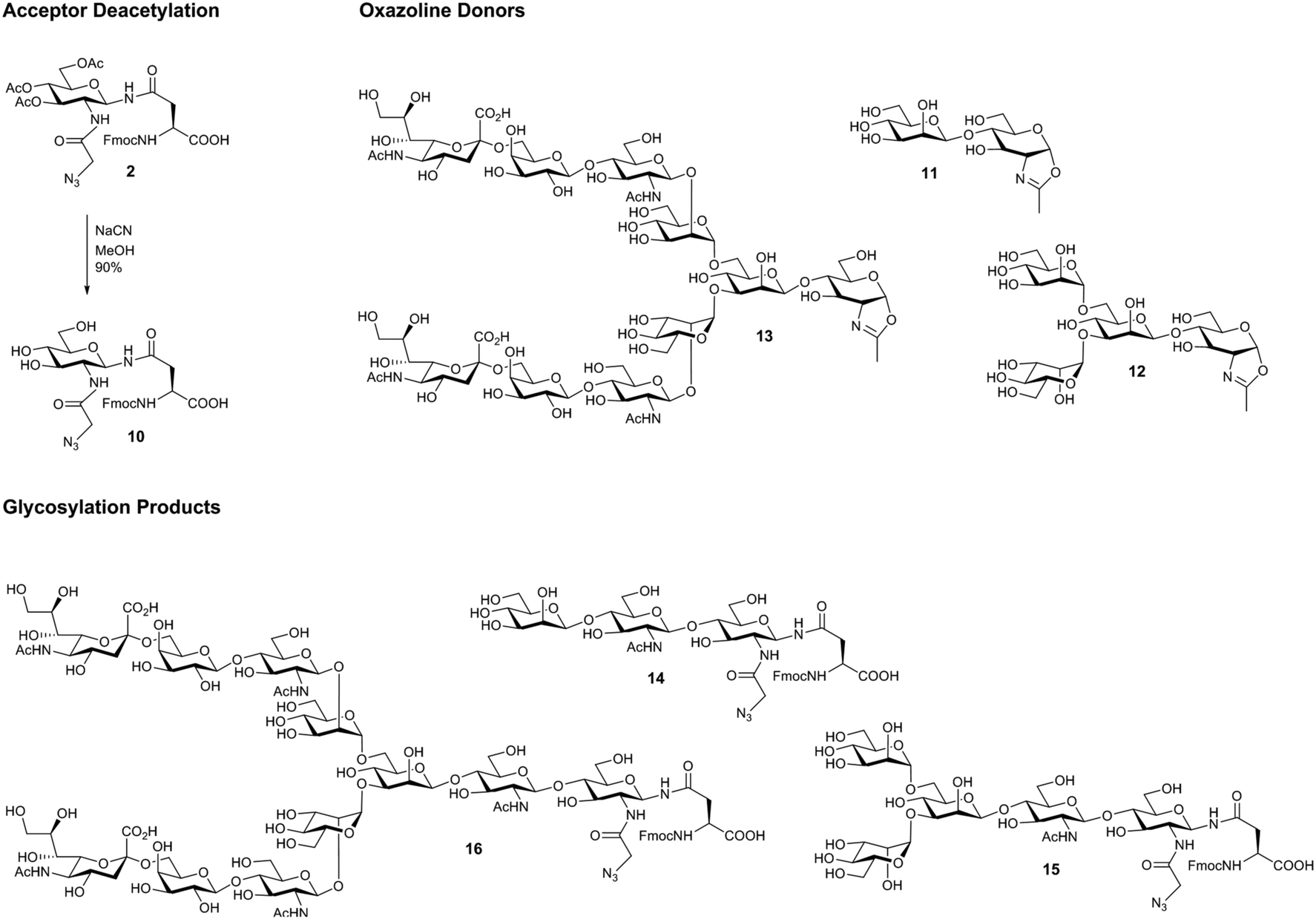 | ||
| Scheme 2 Deacetylation of FmocAsn(N-Ac3GlcNAz)OH 2, and oxazoline substrates and products of ENGase-catalysed glycosylation reactions. | ||
| Entry | Oxazoline donor | Product | ENGase | Yielda (%) | % Yield with corresponding GlcNAc acceptora |
|---|---|---|---|---|---|
| a Determined by integration of product and acceptor HPLC peaks. b See ref. 41. Note the acceptor was the methyl ester and not the free acid. c See ref. 22. | |||||
| 1 | 11 | 14 | WT Endo M | 90 | 95b |
| 2 | 11 | 14 | N175Q Endo M | 6 | — |
| 3 | 11 | 14 | WT Endo A | 94 | >95b |
| 4 | 12 | 15 | WT Endo M | 16 | 36b/17c |
| 5 | 12 | 15 | N175Q Endo M | 60 | 69c |
| 6 | 12 | 15 | WT Endo A | 63 | 67b |
| 7 | 13 | 16 | WT Endo M | 20 | 30c |
| 8 | 13 | 16 | N175Q Endo M | 78 | 78c |
Time course studies were also performed (see ESI†) revealing that neither the trisaccharide (14) nor the pentasaccharide (15) products were hydrolytic substrates for WT Endo A. Additionally, unsurprisingly the Endo M N175Q glycosynthase mutant did not hydrolyse any of the reaction products.
In all cases it can be seen that the azide-containing glycosyl amino acid was an effective acceptor substrate; all of the glycosylation yields were comparable to the corresponding GlcNAc acceptor, though slightly lower in two cases (entries 5 and 7). This series of experiments confirms that the ENGase enzymes tested (WT Endo A, WT Endo M, and the glycosynthase Endo M N175Q) are all able to tolerate the increased steric bulk of the azido-substituted acceptor within their active site. The excellent yield produced for glycosylation using the complex bi-antennary decasaccharide oxazoline 13 using the N175Q Endo M mutant to give 16 is particularly noteworthy.
These positive preliminary results indicate that incorporation of glycosyl amino acid 2 into larger peptide structures, and subsequent ENGase-mediated extension of glycan structure should provide a reliable route to glycopeptide probes which incorporate azide functionality as a biorthogonal handle suitable for a variety of purposes (vide supra). The syntheses of such glycopeptides as tools for investigation of antigen presentation processes are currently in progress, and the results will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Bakkum, T. van Leeuwen, A. J. C. Sarris, D. M. van Elsland, D. Poulcharidis, H. S. Overkleeft and S. I. van Kasteren, ACS Chem. Biol., 2018, 13, 1173–1179 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902 CrossRef CAS.
- K. A. Stubbs, A. Scaffidi, A. W. Debowski, B. L. Mark, R. V. Stick and D. J. Vocadlo, J. Am. Chem. Soc., 2008, 130, 327–335 CrossRef CAS PubMed.
- Q. Li, Z. Li, X. Duan and W. Yi, J. Am. Chem. Soc., 2014, 136, 12536–12539 CrossRef CAS PubMed.
- L. Wen, Y. Zheng, K. Jiang, M. Zhang, S. M. Kondengaden, S. Li, K. Huang, J. Li, J. Song and P. G. Wang, J. Am. Chem. Soc., 2016, 138, 11473–11476 CrossRef CAS PubMed.
- C. M. Gampe, H. Tsukamoto, E. H. Doud, S. Walker and D. Kahne, J. Am. Chem. Soc., 2013, 135, 3776–3779 CrossRef CAS PubMed.
- J. D. McIntosh, M. A. Brimble, A. E. S. Brooks, P. R. Dunbar, R. Kowalczyk, Y. Tomabechi and A. J. Fairbanks, Chem. Sci., 2015, 6, 4636–4642 RSC.
- D. M. van Elsland, E. Bos, W. de Boer, H. S. Overkleeft, A. J. Koster and S. I. van Kasteren, Chem. Sci., 2016, 7, 752–758 RSC.
- J. B. Pawlak, B. J. Hos, M. J. van de Graaff, O. A. Megantari, N. Meeuwenoord, H. S. Overkleeft, D. V. Filippov, F. Ossendorp and S. I. van Kasteren, ACS Chem. Biol., 2016, 11, 3172–3178 CrossRef CAS PubMed.
- A. J. Fairbanks, Beilstein J. Org. Chem., 2018, 14, 416–429 CrossRef CAS PubMed.
- A. J. Fairbanks, Chem. Soc. Rev., 2017, 46, 5128–5146 RSC.
- P. T. Nyffeler, C.-H. Liang, K. M. Koeller and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 10773–10778 CrossRef CAS PubMed.
- M. Bergmann and L. Zervas, Ber. Dtsch. Chem. Ges., 1931, 64, 975–980 CrossRef.
- O. Boutureira, G. J. L. Bernardes, M. Fernández-González, D. C. Anthony and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1432–1436 CrossRef CAS PubMed.
- R. S. Clark, S. Banerjee and J. K. Coward, J. Org. Chem., 1990, 55, 6275–6285 CrossRef CAS.
- E. Nuti, D. Cuffaro, F. D’Andrea, L. Rosalia, L. Tepshi, M. Fabbi, G. Carbotti, S. Ferrini, S. Santamaria, C. Camodeca, L. Ciccone, E. Orlandini, S. Nencetti, E. A. Stura, V. Dive and A. Rossello, ChemMedChem, 2016, 11, 1626–1637 CrossRef CAS PubMed.
- B. G. Reddy, K. P. Madhusudanan and Y. D. Vankar, J. Org. Chem., 2004, 69, 2630–2633 CrossRef CAS PubMed.
- F. Sajtos, L. Lázár, A. Borbás, I. Bajza and A. Lipták, Tetrahedron Lett., 2005, 46, 5191–5194 CrossRef CAS.
- T. Nishiyama, Y. Kusumoto, K. Okumura, K. Hara, S. Kusaba, K. Hirata, Y. Kamiya, M. Isobe, K. Nakano, H. Kotsuki and Y. Ichikawa, Chem. – Eur. J., 2010, 16, 600–610 CrossRef CAS PubMed.
- Y. Tomabechi, M. A. Squire and A. J. Fairbanks, Org. Biomol. Chem., 2014, 12, 942–955 RSC.
- D. M. M. Jaradat, H. Hamouda and C. P. R. Hackenberger, Eur. J. Org. Chem., 2010, 5004–5009 CrossRef CAS.
- A. Bianchi and A. Bernardi, J. Org. Chem., 2006, 71, 4565–4577 CrossRef CAS PubMed.
- Y. He, R. J. Hinklin, J. Chang and L. L. Kiessling, Org. Lett., 2004, 6, 4479–4482 CrossRef CAS PubMed.
- L. Kovács, E. Ősz, V. Domokos, W. Holzer and Z. Györgydeák, Tetrahedron, 2001, 57, 4609–4621 CrossRef.
- L. Kovács, E. Ősz and Z. Györgydeák, Carbohydr. Res., 2002, 337, 1171–1178 CrossRef.
- K. J. Doores, Y. Mimura, R. A. Dwek, P. M. Rudd, T. Elliott and B. G. Davis, Chem. Commun., 2006, 1401 RSC.
- F. M. Ibatullin and S. I. Selivanov, Tetrahedron Lett., 2009, 50, 6351–6354 CrossRef CAS.
- T. B. Parsons, J. W. B. Moir and A. J. Fairbanks, Org. Biomol. Chem., 2009, 7, 3128–3140 RSC.
- For some reviews see: (a) C. Li and L.-X. Wang, Chem. Rev., 2018, 118, 8359–8413 CrossRef CAS PubMed; (b) L.-X. Wang and M. N. Amin, Chem. Biol., 2014, 21, 51–66 CrossRef CAS PubMed; (c) L.-X. Wang and J. V. Lomino, ACS Chem. Biol., 2012, 7, 110–122 CrossRef CAS PubMed; (d) A. J. Fairbanks, Pure Appl. Chem., 2013, 85, 1847–1863 CAS; (e) A. J. Fairbanks, C. R. Chim., 2011, 14, 44–58 CrossRef CAS.
- (a) Y. Tomabechi, G. Krippner, P. M. Rendle, M. A. Squire and A. J. Fairbanks, Chem. – Eur. J., 2013, 19, 15084–15088 CrossRef CAS PubMed; (b) J. D. McIntosh, M. A. Brimble, A. E. S. Brooks, P. R. Dunbar, R. Kowalczyk, Y. Tomabechi and A. J. Fairbanks, Chem. Sci., 2015, 6, 4636–4642 RSC; (c) H. Li, S. Singh, Y. Zeng, H. Song and L. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 895–898 CrossRef CAS PubMed; (d) L.-X. Wang, H. Song, S. Liu, H. Lu, S. Jiang, J. Ni and H. Li, ChemBioChem, 2005, 6, 1068–1074 CrossRef CAS PubMed; (e) W. Huang, X. Zhang, T. Ju, R. D. Cummings and L.-X. Wang, Org. Biomol. Chem., 2010, 8, 5224–5233 RSC; (f) J. V. Lomino, A. Naegeli, J. Orwenyo, M. N. Amin, M. Aebi and L.-X. Wang, Bioorg. Med. Chem., 2013, 21, 2262–2270 CrossRef CAS PubMed; (g) H. Hojo, H. Tanaka, M. Hagiwara, Y. Asahina, A. Ueki, H. Katayama, Y. Nakahara, A. Yoneshige, J. Matsuda, Y. Ito and Y. Nakahara, J. Org. Chem., 2012, 77, 9437–9446 CrossRef CAS PubMed; (h) H. Cai, J. Orwenyo, J. Guenaga, J. Giddens, C. Toonstra, R. T. Wyatt and L.-X. Wang, Chem. Commun., 2017, 53, 5453–5456 RSC.
- (a) P. Priyanka, T. B. Parsons, A. Miller, F. M. Platt and A. J. Fairbanks, Angew. Chem., Int. Ed., 2016, 55, 5058–5061 CrossRef CAS PubMed; (b) T. Yamaguchi, M. N. Amin, C. Toonstra and L.-X. Wang, J. Am. Chem. Soc., 2016, 138, 12472–12485 CrossRef PubMed; (c) B. Li, H. Song, S. Hauser and L.-X. Wang, Org. Lett., 2006, 8, 3081–3084 CrossRef CAS PubMed; (d) H. Ochiai, W. Huang and L.-X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef CAS PubMed; (e) M. N. Amin, W. Huang, R. M. Mizanur and L.-X. Wang, J. Am. Chem. Soc., 2011, 133, 14404–14417 CrossRef CAS PubMed; (f) Q. Yang, Y. An, S. Zhu, R. Zhang, C. M. Loke, J. F. Cipollo and L.-X. Wang, ACS Chem. Biol., 2017, 12, 1665–1673 CrossRef CAS PubMed.
- (a) Y. Tomabechi, Y. Odate, R. Izumi, K. Haneda and T. Inazu, Carbohydr. Res., 2010, 345, 2458–2463 CrossRef CAS PubMed; (b) J. Q. Fan, L. H. Huynh, B. B. Reinhold, V. N. Reinhold, K. Takegawa, S. Iwahara, A. Kondo, I. Kato and Y. C. Lee, Glycoconjugate J., 1996, 13, 643–652 CrossRef CAS.
- M. Fujita, S.-i. Shoda, K. Haneda, T. Inazu, K. Takegawa and K. Yamamoto, Biochim. Biophys. Acta, 2001, 1528, 9–14 CrossRef CAS.
- (a) Z. Ling, M. D. L. Suits, R. J. Bingham, N. C. Bruce, G. J. Davies, A. J. Fairbanks, J. W. B. Moir and E. J. Taylor, J. Mol. Biol., 2009, 389, 1–9 CrossRef CAS PubMed; (b) J. Yin, L. Li, N. Shaw, Y. Li, J. K. Song, W. Zhang, C. Xia, R. Zhang, A. Joachimiak, H.-C. Zhang, L.-X. Wang, Z.-J. Liu and P. Wang, PLoS One, 2009, 4, e4658 CrossRef PubMed; (c) D. W. Abbott, M. S. Macauley, D. J. Vocadlo and A. B. Boraston, J. Biol. Chem., 2009, 284, 11676–11689 CrossRef CAS PubMed.
- K. Takegawa, K. Yamabe, K. Fujita, M. Tabuchi, M. Mita, H. Izu, A. Watanabe, Y. Asada, M. Sano, A. Kondo, I. Kato and S. Iwahara, Arch. Biochem. Biophys., 1997, 338, 22–28 CrossRef CAS PubMed.
- K. Fujita, K. Kobayashi, A. Iwamatsu, M. Takeuchi, H. Kumagai and K. Yamamoto, Arch. Biochem. Biophys., 2004, 432, 41–49 CrossRef CAS PubMed.
- (a) M. Umekawa, C. Li, T. Higashiyama, W. Huang, H. Ashida, K. Yamamoto and L.-X. Wang, J. Biol. Chem., 2010, 285, 511–521 CrossRef CAS PubMed; (b) M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L.-X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469–4479 CrossRef CAS PubMed.
- T. W. D. F. Rising, T. D. W. Claridge, N. Davies, D. P. Gamblin, J. W. B. Moir and A. J. Fairbanks, Carbohydr. Res., 2006, 341, 1574–1596 CrossRef CAS PubMed.
- T. W. D. F. Rising, C. D. Heidecke, J. W. B. Moir, Z. Ling and A. J. Fairbanks, Chem. – Eur. J., 2008, 14, 6444–6464 CrossRef CAS PubMed.
- (a) M. Umekawa, T. Higashiyama, Y. Koga, T. Tanaka, M. Noguchi, A. Kobayashi, S.-I. Shoda, W. Huang, L.-X. Wang, H. Ashida and K. Yamamoto, Biochim. Biophys. Acta, 2010, 1800, 1203–1209 CrossRef CAS PubMed; (b) A. Seko, M. Koketsu, M. Nishizono, Y. Enoki, H. R. Ibrahim, L. R. Juneja, M. Kim and T. Yamamoto, Biochim. Biophys. Acta, 1997, 1335, 23–32 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc02051a |
| This journal is © The Royal Society of Chemistry 2019 |