
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular switching of the self-assembly of cyclic peptide–polymer conjugates via host–guest chemistry†
Qiao
Song
 a,
Jie
Yang
a,
Julia Y.
Rho
a and
Sébastien
Perrier
*abc
a,
Jie
Yang
a,
Julia Y.
Rho
a and
Sébastien
Perrier
*abc
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: S.Perrier@warwick.ac.uk
bFaculty of Pharmacy and Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, VIC 3052, Australia
cWarwick Medical School, The University of Warwick, Coventry CV4 7AL, UK
First published on 10th April 2019
Abstract
A supramolecular strategy of switching the self-assembly of cyclic peptide–polymer conjugates using host–guest chemistry is proposed. The formation of tubular supramolecular polymers based on cyclic peptide–polymer conjugates can be controlled by reversibly attaching cucurbit[7]uril onto the cyclic peptide via host–guest interactions.
Supramolecular polymers are defined as polymeric arrays of monomeric units which are held together by highly directional and reversible non-covalent interactions,1–9 such as multiple hydrogen bonding,10–13 host–guest interactions,14–18 metal-coordination interactions,19,20 and donor–acceptor interactions.21,22 Among these, tubular supramolecular polymers assembled by cyclic peptide–polymer conjugates belong to a relatively new class of self-assembled supramolecular polymers.23–30 The alternating D- and L-amino acid configuration of the cyclic peptide leads to the formation of a flat ring-like structure. Between these cyclic peptide rings, there exist strong multiple hydrogen bonding interactions, leading to the formation of nanotubes.31,32 Conjugating hydrophilic polymers onto cyclic peptides will prevent their aggregation and improve their stability and solubility, forming tubular supramolecular polymers with a well-defined structure.33–37 However, in spite of the fact that the driving force is based on non-covalent multiple hydrogen bonding, studies on controlling the assembly and disassembly of the tubular supramolecular polymers are still limited.38–40
Cucurbit[n]urils (CB[n]s), an important class of macrocyclic hosts, can bind firmly with a variety of neutral or positively charged guests.41 Among them, CB[7], comprising 7 glycoluril units, has a height of ∼0.9 nm, an outer diameter of 1.6 nm, and an inner diameter of ∼0.7 nm. Considering that the distance between each cyclic peptide is reported to be 0.47 nm, it is anticipated that introducing CB[7] onto cyclic peptides could lead to the disassembly of the tubular supramolecular polymers due to the steric hindrance of the CB[7].42–44 In this regard, it is believed that we might be able to develop a strategy of tuning the self-assembly of cyclic peptide–polymer conjugates using CB based host–guest chemistry.
Herein, as a proof of concept, we designed and synthesized a cyclic peptide conjugated with a water-soluble polymer–poly(ethylene glycol) (PEG), which could form tubular supramolecular polymers in aqueous solution, Scheme 1. It has been reported that CB[7] binds strongly with a phenylalanine moiety, with the binding constant as high as 106 M−1.45 To this end, we introduced two phenylalanine moieties onto the cyclic peptide, enabling us to non-covalently incorporate two bulky CB[7] host molecules onto every cyclic peptide–polymer conjugate. Moreover, a competitive guest molecule (1-adamantanamine, ADA) can be added to disassociate the binding between the cyclic peptide–polymer conjugate and CB[7]. In this way, we are able to reversibly tune the self-assembly of the cyclic peptide–polymer conjugate.
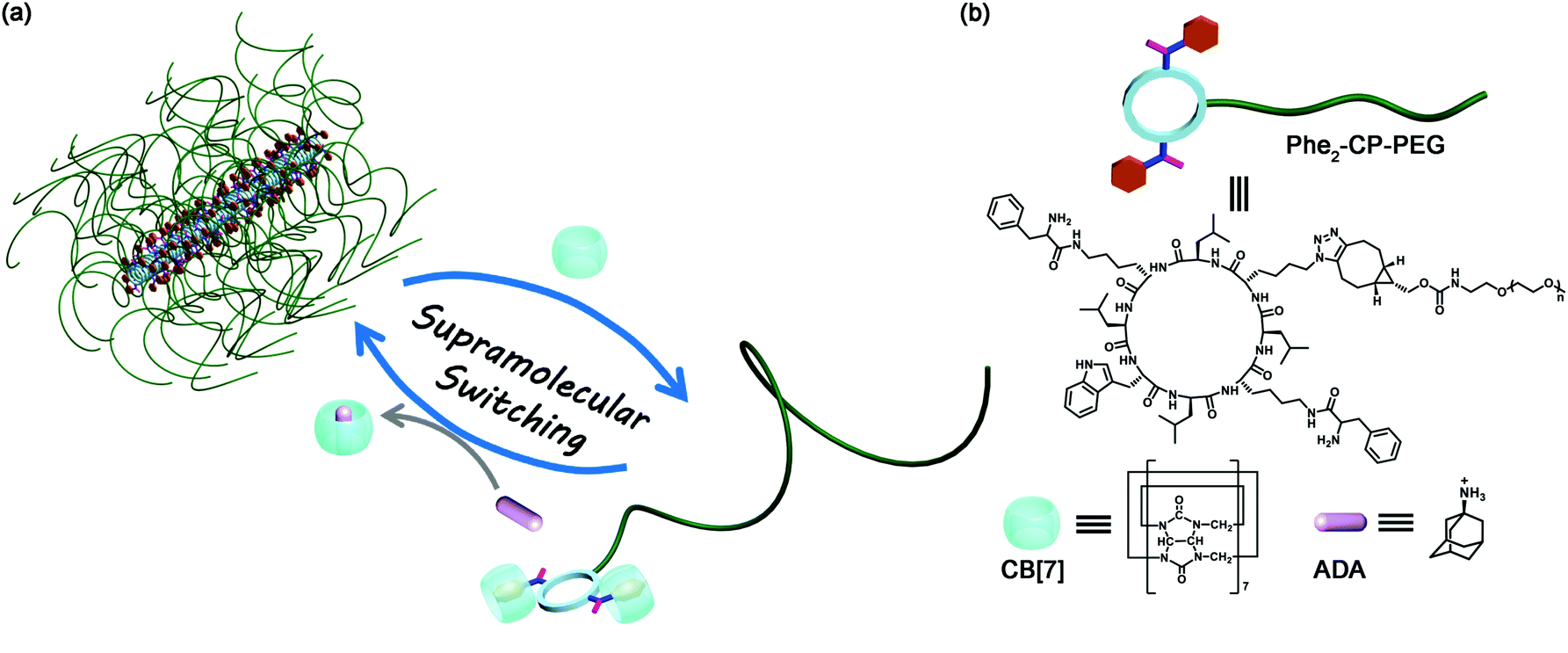 | ||
| Scheme 1 (a) Schematic representation of the reversible self-assembly of a cyclic peptide–polymer conjugate via host–guest chemistry; (b) chemical structures of cyclic peptide–polymer conjugate Phe2-CP-PEG, CB[7], and ADA. | ||
A linear peptide with the sequence of H2N-L-Lys(Boc)-D-Leu-L-Lys(N3)-D-Leu-L-Lys(Boc)-D-Leu-L-Trp(Boc)-D-Leu-COOH was first synthesized by solid-phase peptide synthesis using Fmoc-deprotection chemistry. The cyclic peptide (H2N)2-CP-N3 was then synthesized by a cyclization reaction of the linear peptide under dilute conditions, followed by deprotection of the protecting groups (see the ESI,† S2). The two ‘guest’ phenylalanine moieties were introduced onto the cyclic peptide via a two-step synthesis (Phe2-CP-N3), which was confirmed by 1H NMR spectroscopy and ESI-MS (see the ESI,† S2 and Fig. S4).
Conjugating a water-soluble polymer to the cyclic peptide helps improve the solubility of the cyclic peptide and prevent the further lateral aggregation of the formed tubular supramolecular polymers. Here, PEG (5000 g mol−1) was chosen to conjugate to Phe2-CP-N3 through the highly efficient strained alkyne/azide group ligation. The cyclic peptide–polymer conjugate Phe2-CP-PEG was easily synthesized by reacting Phe2-CP-N3 with mPEG-BCN and purified by precipitation in a mixed solvent.
The conjugate Phe2-CP-PEG was thoroughly characterized by HPLC, GPC and 1H NMR spectroscopy. As shown in Fig. S6 (ESI†), the retention time of Phe2-CP-PEG was 16.6 min, and no trace of unreacted Phe2-CP-N3 was observed at 17.2 min, indicating that all the Phe2-CP-N3 was conjugated with mPEG-BCN. GPC analysis gave an increased Mn,GPC of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1 with a narrow dispersity (Đ) of 1.06 for the Phe2-CP-PEG conjugate, while the Mn,GPC of mPEG-BCN is 10800 g mol−1, as shown in Fig. S7 (ESI†). Finally, the 1H NMR spectrum of Phe2-CP-PEG was measured in TFA-d and proton signals ascribed to the cyclic peptide and PEG could be clearly assigned (Fig. S8, ESI†).
800 g mol−1 with a narrow dispersity (Đ) of 1.06 for the Phe2-CP-PEG conjugate, while the Mn,GPC of mPEG-BCN is 10800 g mol−1, as shown in Fig. S7 (ESI†). Finally, the 1H NMR spectrum of Phe2-CP-PEG was measured in TFA-d and proton signals ascribed to the cyclic peptide and PEG could be clearly assigned (Fig. S8, ESI†).
Hydrogen bonding interactions between the amide bonds of cyclic peptides are the driving force to form tubular supramolecular polymers. The self-assembly of Phe2-CP-PEG in aqueous solution was first studied by diffusion-ordered NMR spectroscopy (DOSY). DOSY is a widely-used technique in the field of supramolecular chemistry to directly confirm the formation of assemblies in solution.46 As shown in Fig. 1(a), the average diffusion coefficient of Phe2-CP-PEG in D2O was measured to be 2.2 × 10−11 m2 s−1, while the diffusion coefficient of mPEG-BCN was 1.0 × 10−10 m2 s−1 (Fig. S10, ESI†). Such a decrease in the diffusion coefficient indicates the formation of large assemblies by Phe2-CP-PEG conjugate. Simplistically assuming that all the assemblies are hydrodynamically spherical, the hydrodynamic radius Rh is estimated to be 11.2 nm based on the Stokes–Einstein equation (Fig. S13 and Table S1, ESI†).
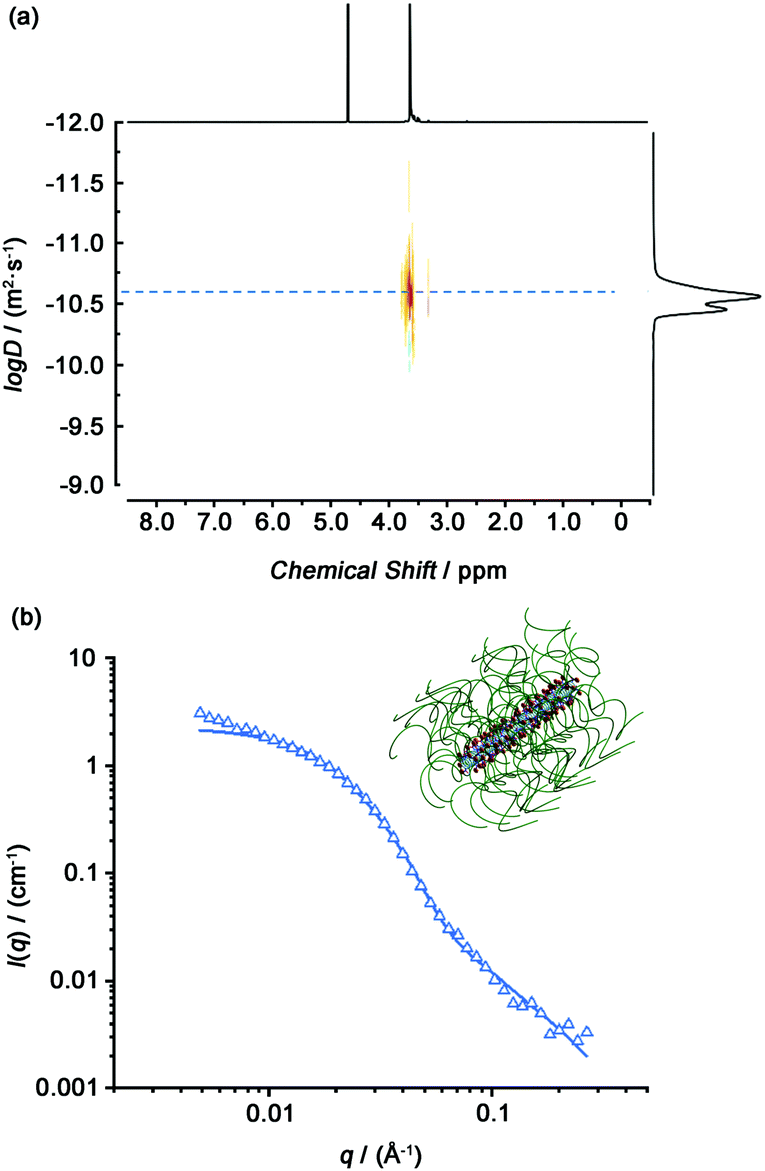 | ||
| Fig. 1 (a) DOSY spectrum of the Phe2-CP-PEG conjugate. (b) Reduced SANS scattering data for the Phe2-CP-PEG conjugate. The line corresponds to a fit to the hairy cylinder model. | ||
Small angle neutron scattering (SANS) is a powerful tool to characterize supramolecular systems to obtain information about structural parameters in solution. Here, we used SANS to confirm the formation of tubular supramolecular polymers in aqueous solution. Fig. 1(b) shows the reduced, corrected scattering data for Phe2-CP-PEG conjugate in D2O. Using SASfit software, a cylindrical micelle with attached polymer chains (‘‘CYL + CHAINS(RW)’’) (hairy cylinder) model was used, as fitting to a core–shell cylinder. From the fit, the average length of 16.8 ± 0.5 nm for each nanotube could be obtained, with a width of 6.4 ± 0.1 nm. Given that the distance between two cyclic peptides has been previously determined to be 0.47 nm, the average number of aggregation (Nagg) is calculated to be 36.
The host–guest interaction between Phe2-CP-PEG and CB[7] was established by 1H NMR spectroscopy. Initially mPEG-Phe was synthesized as a model compound. A clear upfield shift was observed for the phenyl groups by 1H NMR spectroscopy after being complexed with CB[7] (Fig. S11, ESI†). Phe2-CP-PEG conjugate showed similar changes. As indicated in Fig. 2(a), after adding CB[7], characteristic peaks at 7.73, 6.58 and 6.43 ppm were observed, which could be ascribed to the phenyl groups on the cyclic peptide encapsulated into the CB[7] cavity. Moreover, isothermal titration calorimetry (ITC) was used to thoroughly characterize the host–guest interactions between the conjugate and CB[7] (Fig. 2(b)). A two sequential-binding sites model was used to fit the data, yielding thermodynamic parameters for the interaction between the Phe2-CP-PEG conjugate and CB[7], K1 = (1.1 ± 0.2) × 106 M−1 and K2 = (7.1 ± 0.4) × 103 M−1. It is worth pointing out that the binding constant of the second step K2 is significantly lower than that of the first step K1, which is believed to be related to the disassembly of the tubular supramolecular polymers upon the addition of CB[7]. In other words, the second step K2 is in fact a combination of the binding between the phenylalanine moiety and CB[7] and the disassociation of the cyclic peptides.
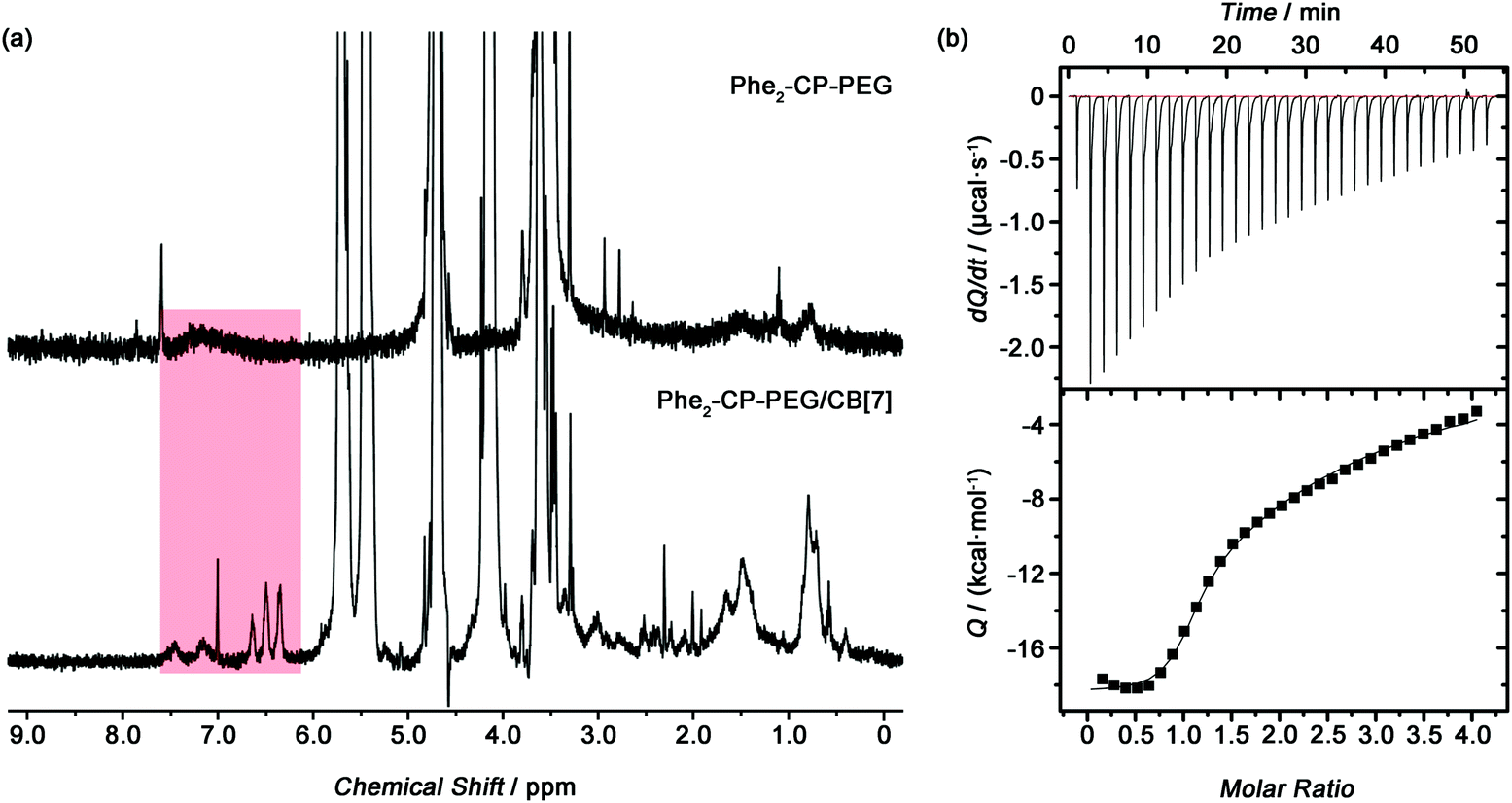 | ||
| Fig. 2 (a) 1H NMR spectra of Phe2-CP-PEG and Phe2-CP-PEG/CB[7] (400 MHz, D2O). (b) ITC data for titration of Phe2-CP-PEG (0.1 mM) with CB[7] (2.0 mM) in DI water at 25 °C. Data were fitted using a two sequential-binding sites model. | ||
To investigate whether introducing bulky CB[7] onto the cyclic peptide could lead to the disassembly of the tubular supramolecular polymers formed by the Phe2-CP-PEG conjugate, DOSY was conducted with Phe2-CP-PEG in the presence of excess CB[7] in D2O (Fig. 3(a)). The diffusion coefficient was measured to be 6.5 × 10−11 m2 s−1, significantly higher than that of the Phe2-CP-PEG conjugate. The Rh is estimated to be 3.8 nm based on the Stokes–Einstein equation. The disassembly of the tubular supramolecular polymers was further proved by SANS. As shown in Fig. 3(b), whilst a hairy cylinder model fit is not valid anymore, a polymer chain model (DozierStar) gives a better fitting, suggesting that the tubular supramolecular polymers underwent disassembly due to the steric hindrance caused by CB[7] between the cyclic peptides.
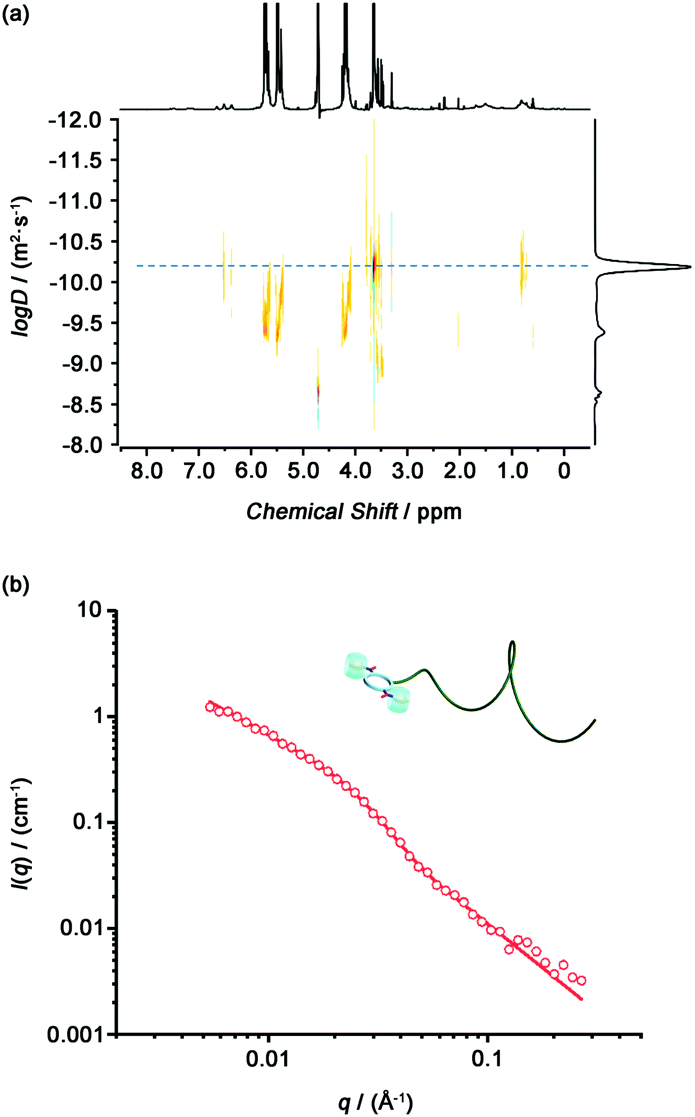 | ||
| Fig. 3 (a) DOSY spectrum of Phe2-CP-PEG/CB[7]. (b) Reduced SANS scattering data for Phe2-CP-PEG/CB[7]. The line corresponds to a fit to the polymer chain model. | ||
More importantly, due to the dynamic nature of the host–guest interactions causing the disassembly of the tubular supramolecular polymers, the tubular supramolecular polymers could be reversibly reformed. Herein, ADA, which has a much stronger binding strength than a phenylalanine moiety, was used as a competitive guest to dissociate the binding between Phe2-CP-PEG and CB[7]. As shown by 1H NMR spectroscopy (Fig. S12, ESI†), after adding ADA into the solution of Phe2-CP-PEG and CB[7], peaks at 7.73, 6.58 and 6.43 ppm vanished and a broad peak at ∼7.2 ppm was observed instead, suggesting that CB[7] was dissociated from Phe2-CP-PEG and therefore the tubular supramolecular polymers were reformed.
In conclusion, we have successfully fabricated tubular supramolecular polymers on the basis of cyclic peptide–polymer conjugates. By introducing binding sites of CB[7] onto the cyclic peptide, the self-assembly of a cyclic peptide–polymer conjugate could be easily tuned using CB based host–guest chemistry. Considering that various functional polymers could be conjugated onto cyclic peptides to construct tubular supramolecular polymers with various functionalities, it is anticipated that this type of supramolecular host–guest strategy will provide a new perspective towards fine control of the structure and function of such tubular supramolecular polymers.
The Royal Society Wolfson Merit Award (WM130055; SP), the European Research Council (TUSUPO 647106; QS, JYR and SP) and the Horizon 2020 Marie-Sklodowska-Curie action (TSPBNTM, JY) are acknowledged for funding. The authors also thank Dr Robert Dalgliesh (ISIS, Oxford, UK), as well as Dr Edward Mansfield and Mr Sean Ellacott for assistance with SANS. We also appreciate the help of Dr Ivan Prokes with DOSY.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254 CrossRef CAS.
- J.-M. Lehn, Polym. Int., 2002, 51, 825 CrossRef CAS.
- T. De Greef, M. Smulders, M. Wolffs, A. Schenning, R. Sijbesma and E. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196 CrossRef CAS PubMed.
- E. Krieg, M. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042 RSC.
- T. Aida, E. Meijer and S. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- J. Fox and S. Rowan, Macromolecules, 2009, 42, 6823 CrossRef CAS.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971 CrossRef CAS PubMed.
- R. Sijbesma, F. Beijer, L. Brunsveld, B. Folmer, J. Hirschberg, R. Lange, J. Lowe and E. Meijer, Science, 1997, 278, 1601 CrossRef CAS PubMed.
- B. Folmer, R. Sijbesma, R. Versteegen, J. van der Rijt and E. Meijer, Adv. Mater., 2000, 12, 874 CrossRef CAS.
- R. Sijbesma and E. Meijer, Chem. Commun., 2003, 5 RSC.
- T. Park, E. Todd, S. Nakashima and S. Zimmerman, J. Am. Chem. Soc., 2005, 127, 18133 CrossRef CAS PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982 CrossRef CAS PubMed.
- Y. Liu, H. Yang, Z. Wang and X. Zhang, Chem. – Asian J., 2013, 8, 1626 CrossRef CAS PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- G. Yu, X. Zhao, J. Zhou, Z. Mao, X. Huang, Z. Wang, B. Hua, Y. Liu, F. Zhang, Z. He, O. Jacobson, C. Gao, W. Wang, C. Yu, X. Zhu, F. Huang and X. Chen, J. Am. Chem. Soc., 2018, 140, 8005 CrossRef CAS PubMed.
- C. Li, Chem. Commun., 2014, 50, 12420 RSC.
- J. Beck and S. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS PubMed.
- A. Wild, A. Winter, F. Schlutter and U. Schubert, Chem. Soc. Rev., 2011, 40, 1459 RSC.
- S. Burattini, H. Colquhoun, J. Fox, D. Friedmann, B. Greenland, P. Harris, W. Hayes, M. Mackay and S. Rowan, Chem. Commun., 2009, 6717 RSC.
- Y. Tian, Y. Han, Z. Yang and F. Wang, Macromolecules, 2016, 49, 6455 CrossRef CAS.
- R. Chapman, M. Danial, M. Koh, K. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023 RSC.
- J. Couet, J. Jeyaprakash, S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297 CrossRef CAS PubMed.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2011, 2, 1956 RSC.
- R. Hourani, C. Zhang, R. van der Weegen, L. Ruiz, C. Li, S. Keten, B. A. Helms and T. Xu, J. Am. Chem. Soc., 2011, 133, 15296 CrossRef CAS PubMed.
- T. Xu, N. Zhao, F. Ren, R. Hourani, M. T. Lee, J. Y. Shu, S. Mao and B. A. Helms, ACS Nano, 2011, 5, 1376 CrossRef CAS PubMed.
- H. Borner, Prog. Polym. Sci., 2009, 34, 811 CrossRef.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2014, 14, 143 CrossRef PubMed.
- M. Ghadiri, J. Granja, R. Milligan, D. Mcree and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. Kim, E. Choi, M. Delgado, J. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. Weinberger, K. Wilcoxen and M. Ghadiri, Nature, 2001, 412, 452 CrossRef CAS PubMed.
- M. Danial, C. Tran, P. Young, S. Perrier and K. Jolliffe, Nat. Commun., 2013, 4, 2780 CrossRef PubMed.
- C. D. Spicer, C. Jumeaux, B. Gupt and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574 RSC.
- J. C. Brendel, J. Sanchis, S. Catrouillet, E. Czuba, M. Z. Chen, B. M. Long, C. Nowell, A. Johnston, K. A. Jolliffe and S. Perrier, Angew. Chem., Int. Ed., 2018, 57, 16678 CrossRef CAS PubMed.
- J. Y. Rho, J. C. Brendel, L. R. MacFarlane, E. D. H. Mansfield, R. Peltier, S. Rogers, M. Hartlieb and S. Perrier, Adv. Funct. Mater., 2018, 28, 1704569 CrossRef.
- Z. Wang, Y. Li, Y. Huang, M. P. Thompson, C. L. M. LeGuyader, S. Sahua and N. C. Gianneschi, Chem. Commun., 2015, 51, 17108 RSC.
- S. Catrouillet, J. C. Brendel, S. Larnaudie, T. Barlow, K. A. Jolliffe and S. Perrier, ACS Macro Lett., 2016, 5, 1119 CrossRef CAS.
- S. C. Larnaudie, J. C. Brendel, K. A. Jolliffe and S. Perrier, ACS Macro Lett., 2017, 6, 1347 CrossRef CAS.
- L. Sun, Z. Fan, Y. Wang, Y. Huang, M. Schmidta and M. Zhang, Soft Matter, 2015, 11, 3822 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef CAS PubMed.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285 CrossRef CAS PubMed.
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342 RSC.
- Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975 RSC.
- J. W. Lee, H. H. L. Lee, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2015, 119, 4628 CrossRef CAS PubMed.
- Y. Liu, Z. Wang and X. Zhang, Chem. Soc. Rev., 2012, 41, 5922 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc01914f |
| This journal is © The Royal Society of Chemistry 2019 |