
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular doping: accessing the first carborane-substituted 1,2,3-triphospholanide via insertion of P− into a P−P bond†
Peter
Coburger
 a,
Hansjörg
Grützmacher
bc and
Evamarie
Hey-Hawkins
*a
a,
Hansjörg
Grützmacher
bc and
Evamarie
Hey-Hawkins
*a
aLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bETH Zurich, Lab. für Anorganische Chemie, Vladimir-Prelog-Weg 1-5/10, 8093 Zürich, Switzerland
cLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-sen University, 135 West Xingang Road, Guangzhou 510275, China
First published on 4th February 2019
Abstract
Insertion of a P− anion into a P–P bond yielding the first carborane-substituted 1,2,3-triphospholanide 1 was achieved by treating a carborane-substitued 1,2-diphosphetane with sodium phosphaethynolate. The triphospholanide 1 can serve as a versatile nucleophilic building block for unprecedent functionalised triphospholanes and carborane-substituted polyphosphanes.
Polyphosphanes and polyphosphanides have received much interest due to their structural diversity, reactivity and coordination properties.1 Alkyl and aryl groups have been predominantly used as substituents at the phosphorus centre. ortho-Carborane has been very rarely employed as substituent despite its interesting properties such as a flexible Ccluster–Ccluster bond, its electron-withdrawing effect, the possibility of the hydridic B–H hydrogen atoms to participate in metal bonding, and the possibility of converting the closo-cluster structure into an anionic nido-carborane.2 In fact, the only examples of carborane derivatives containing more than one P–P bond are the 1,2,3-triphospholanes Ia–d reported by us (Fig. 1). The respective triphospholanides are absent in the literature,3 but structurally related compounds with more common backbones have been published. Pietschnig et al. reported a series of triphospha-[3]ferrocenophanes IIa–f;4 here, the respective triphosphanide can be accessed by deprotonation of IId5 which opens up further possibilities for functionalisation to polyphosphane derivatives. Schmidpeter et al. obtained a phenylene-substituted triphospholanide IIIvia degradation of P4 with 1,2-phenylenebis(phenylphosphanide). Reactivity studies showed that III is a versatile nucleophilic building block; in particular, a tetraphosphane could be obtained via reaction with chlorodiphenylphosphane.6 Here, we present the triphospholanide 1, which is formed upon treatment of a strained carborane-substituted 1,2-diphosphetane7 with sodium phosphaethynolate,8 and its conversion to new carborane-containing triphospholane derivatives.
 | ||
| Fig. 1 Triphospholane and triphospholanide derivatives. Counterions not shown. | ||
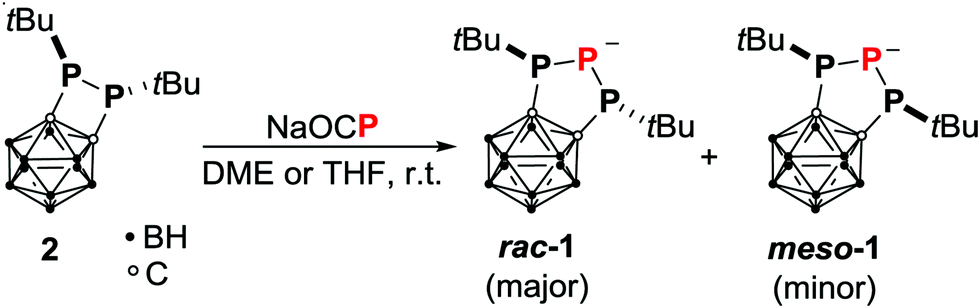
The carborane-substituted diphosphetane 2 (Scheme 1) has proven to be a versatile synthon.3,7,9 Due to the electron-withdrawing effect of the ortho-carborane backbone, reactivities include the reductive cleavage of the P–P bond with elemental lithium and [K(thf)0.2][Co(η4-cod)2] (cod = 1,5-cyclooctadiene).9a,c,d Diphosphetane 2 also exhibits a considerable ring strain of 12.3 kcal mol−1.9e We therefore concluded that 2 should react with a suitable anionic phosphorus source to yield unprecedented carborane-substituted phosphanido species. Indeed, treatment of 2 with sodium phosphaethynolate in ethereal solvents afforded the triphospholanide 1 by formal insertion of P− into the P–P bond (Scheme 1). Reactions in which the phosphaethynolate anion (formally) inserts into an element–element bond are extremely scarce and limited to a few examples reported by Goicoechea et al. (insertion into a Si–Si bond),10 Weigand et al. (insertion into a Ga–P bond)11 and Driess et al. (insertion into a Si–C bond).12 To a certain extent, these reactions can be viewed as molecular doping reactions with P− as dopant.13
 | ||
| Scheme 1 Reaction of 2 with sodium phosphaethynolate. Counterion (Na+) not shown. | ||
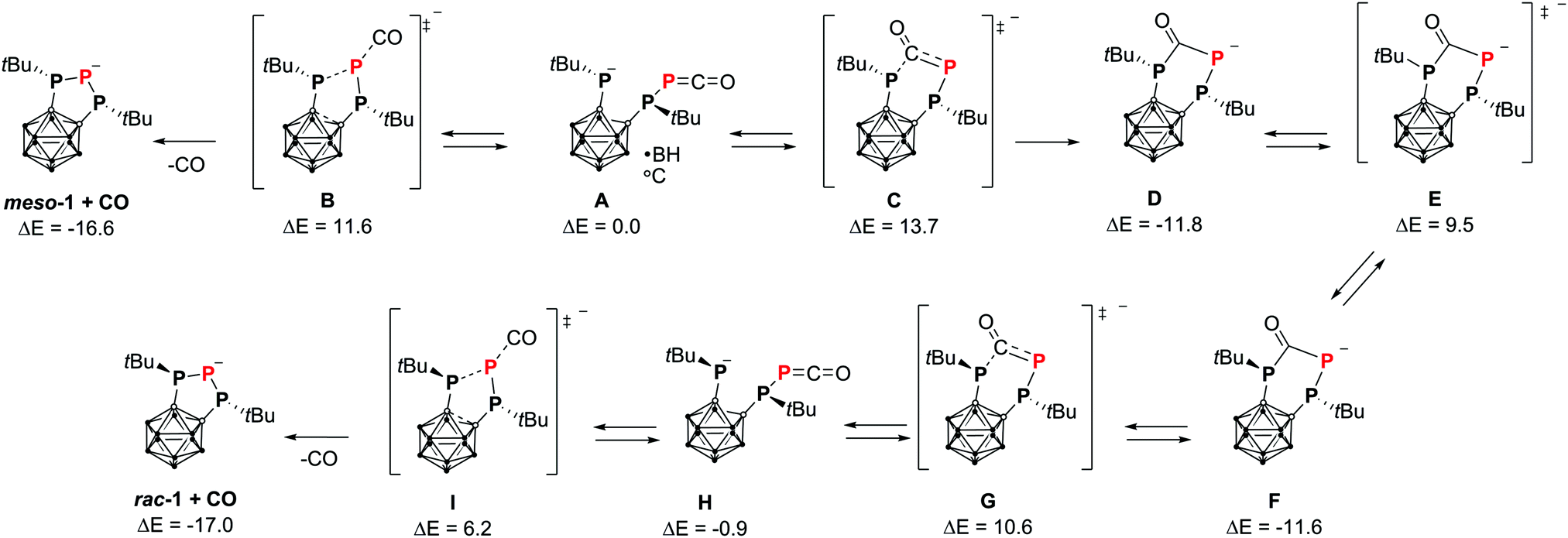
The orange carborane-substituted 1,2,3-triphospholanide 1 is formed as a diastereomeric mixture of the anions rac-1 (δ31P = 91.9 (d), −86.3 (t) ppm, 1JPP = 380 Hz) and meso-1 (δ31P = 81.3 (d), −105.6 (t) ppm, 1JPP = 280 Hz) in THF in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. In contrast, only traces of meso-1 are detectable when the reaction is carried out in DME. Additionally, an epimerisation barrier of 29.5 kcal mol−1 between rac-1 and meso-1 was calculated, whilst the energy difference between the two isomers was calculated to be only −0.1 kcal mol−1 (TPSS-D3BJ/def2-TZVP,14–16 see Computational details in the ESI†). These findings indicate that kinetic reasons are responsible for the formation of the observed diastereomeric mixture. A possible reaction mechanism was investigated employing DFT and ab initio methods (Scheme 2). Structures of isolated molecules were optimised with the TPSS-D3BJ14,15 functional and the def2-TZVP basis,15 and energies were calculated with the DLPNO-CCSD(T)17 method by Neese et al. and the def2-QZVPP basis16 (see Computational details in the ESI†). In the first step, the P–P bond in diphosphetane 2 is broken upon attack of the phosphaethynolate anion at one phosphorus atom forming the phosphanido-phosphaketene A. This step proceeds via a very low energy barrier (ΔE# = 6.8 kcal mol−1) and is slightly exothermic (ΔE = −3.3 kcal mol−1, see the ESI†). Notably, the reaction energy is significantly smaller than the ring strain of 2 (12.3 kcal mol−1)9e indicating that release of steric strain might strongly contribute to the driving force of the initial step. The phosphanido moiety in A can react intramolecularly with the phosphaketene moiety in two ways. One possibility is the elimination of carbon monoxide via transition state B forming the triphospholanide meso-1 irreversibly. The other possibility is the nucleophilic attack at the carbon atom of the phosphaketene moiety via transition state C forming the heterocyclic compound D. Due to the rather high activation energy of the backwards reaction (ΔE# = 25.5 kcal mol−1, corresponding to a t1/2 of 6.5 days at r.t.) this step is irreversible as well.
:1 ratio. In contrast, only traces of meso-1 are detectable when the reaction is carried out in DME. Additionally, an epimerisation barrier of 29.5 kcal mol−1 between rac-1 and meso-1 was calculated, whilst the energy difference between the two isomers was calculated to be only −0.1 kcal mol−1 (TPSS-D3BJ/def2-TZVP,14–16 see Computational details in the ESI†). These findings indicate that kinetic reasons are responsible for the formation of the observed diastereomeric mixture. A possible reaction mechanism was investigated employing DFT and ab initio methods (Scheme 2). Structures of isolated molecules were optimised with the TPSS-D3BJ14,15 functional and the def2-TZVP basis,15 and energies were calculated with the DLPNO-CCSD(T)17 method by Neese et al. and the def2-QZVPP basis16 (see Computational details in the ESI†). In the first step, the P–P bond in diphosphetane 2 is broken upon attack of the phosphaethynolate anion at one phosphorus atom forming the phosphanido-phosphaketene A. This step proceeds via a very low energy barrier (ΔE# = 6.8 kcal mol−1) and is slightly exothermic (ΔE = −3.3 kcal mol−1, see the ESI†). Notably, the reaction energy is significantly smaller than the ring strain of 2 (12.3 kcal mol−1)9e indicating that release of steric strain might strongly contribute to the driving force of the initial step. The phosphanido moiety in A can react intramolecularly with the phosphaketene moiety in two ways. One possibility is the elimination of carbon monoxide via transition state B forming the triphospholanide meso-1 irreversibly. The other possibility is the nucleophilic attack at the carbon atom of the phosphaketene moiety via transition state C forming the heterocyclic compound D. Due to the rather high activation energy of the backwards reaction (ΔE# = 25.5 kcal mol−1, corresponding to a t1/2 of 6.5 days at r.t.) this step is irreversible as well.
 | ||
| Scheme 2 Calculated mechanism for the formation of meso-1 and rac-1. Relative energies are given in kcal mol−1. Counterions (Na+) not included. | ||
Subsequently, D can undergo epimerisation (E, F) followed by ring opening to form the phosphanido-phosphaketene H. Finally, elimination of CO via transition state I yields the triphospholanide rac-1. All transition states in the calculated mechanism are accessible at room temperature. In particular, the transition states B and C, which determine the ratio of meso-1 to rac-1, are close in energy in agreement with the formation of a diastereomeric mixture of 1. When explicit counterion and solvation effects are incorporated into the calculation, their energy is even closer (ΔE#(B) = 18.3 kcal mol−1, ΔE#(C) = 18.7 kcal mol−1, at the same level of theory, see the ESI† for further details).
In the next step, the reactivity of triphospholanide 1 towards several electrophiles (Scheme 3) was probed. Protonation of 1 with water in n-hexane afforded the secondary triphospholane 1-H in good yields (72%). Triphospholane 1-H is obtained as a colourless solid and can be stored and handled under ambient conditions and can be deprotonated again quantitatively with nBuLi at 0 °C to yield 1. Therefore, we used 1-H as a convenient source of 1 in further reactivity studies. 1 can be converted to the respective 2-chlorotriphospholane 1-Cl by treatment with hexachloroethane as chloronium ion source in n-hexane. 1-Cl was isolated as an air-stable colourless solid in moderate yield (39%). Triphospholanide 1 can also serve as a building block for more complex polyphosphanes. Thus, the 2:1 reaction with dichlorophenylphosphane affords the heptaphosphane 3 as a colourless solid in 63% yield. 3 is the first carborane-substituted polyphosphane with more than three phosphorus atoms. Of the possible stereoisomers (two rac and two meso isomers),4 only one meso isomer is formed in the case of 1-Cl and 3 as confirmed by single-crystal X-ray diffraction studies (vide infra). In contrast, triphospholane 1-H is always obtained as a mixture of a rac and two meso isomers, even when triphospholanide 1 was prepared in DME and thus contained only traces of meso-1.
 | ||
| Scheme 3 Reactions of 1 with electrophiles. Counterion (Na+) in rac/meso-1 not shown. | ||
Additional NMR spectroscopic investigations proved that the isomers slowly form an equilibrium in solution with the meso isomers being the major compounds (see the ESI†). Overall, the observed stereochemistry of the triphospholane derivatives is in good agreement with a detailed theoretical study (ESI†). Single crystals of the triphospholane derivatives rac-1-H, 1-Cl and 3 suitable for X-ray structure analysis were obtained.‡ The molecular structures of rac-1-H, 1-Cl and 3 are depicted in Fig. 2, and selected bond lengths and angles are summarised in Table 1. The C2P3 ring in rac-1-H adopts a distorted, non-symmetrical envelope conformation with two different P–P bond lengths [P1–P3: 2.238(2) Å, P2–P3: 2.192(3) Å] within the standard range of P–P single bonds.18rac-1-H also displays a non-symmetric conformation in solution as indicated by the presence of two distinct resonances accounting for the different tert-butylphosphanyl groups in the 31P{1H} NMR spectrum (see the ESI†). meso-1-Cl crystallises in the monoclinic space group C2/c; the C2P3 ring adopts an envelope conformation. Overall, the structure of the C2P3 rings in rac-1-H and 1-Cl is rather relaxed with bond lengths and angles similar to the ones observed in triphospholanes (Fig. 1).3,18 This is also the case for the triphospholane rings in 3. However, they markedly differ from each other [e.g. P1–P3–P2 is 102.86(2)°, whilst P6–P5–P7 is 92.25(2)°, Table 1]. This unsymmetrical conformation might be adopted to minimise steric repulsion between the substituents on the phosphorus atoms. Additionally, weak attractive interactions between P4 and P1, P4 and P2, P4 and P6, P4 and P7, and P3 and P5 are indicated by their rather short distances [3.2070(7)–3.5687(7) Å, Fig. S4, ESI†] which are well within the sum of their van der Waals radii (3.6 Å).19 The presence of attractive interactions is further supported by an NBO analysis where Wiberg bond indices (WBI) of 0.02 to 0.04 are found between the respective phosphorus atoms (see Computational details in the ESI†).
 | ||
| Fig. 2 Molecular structures of rac-1-H, 1-Cl and 3 with ellipsoids at the 50% probability level. Hydrogen atoms (other than P3-H1 in rac-1-H), disorder within the C2P3 ring and the tert-butyl groups (in case of 1-H), co-crystallised n-hexane (1-Cl) and toluene (3) are omitted for clarity. | ||
| rac-1-H | 1-Cl | 3 | |
|---|---|---|---|
| C1–C2 (C3–C4) | 1.680(4) | 1.689(3) | 1.699(2) (1.675(3)) |
| P1–P3 (P5–P6) | 2.238(2) | 2.2292(8) | 2.2189(6) (2.2311(6)) |
| P2–P3 (P5–P7) | 2.192(3) | 2.2227(9) | 2.2108(6) (2.2276(6)) |
| P3–P4/Cl1 (P4–P5) | — | 2.0846(7) | 2.2428(6) (2.2444(6)) |
| P1–P3–P2 (P6–P5–P7) | 97.26(9) | 93.62(3) | 102.86(2) (92.25(2)) |
| P1–C1–C2 (P6–C3–C4) | 112.6(2) | 114.7(1) | 118.2(1) (114.6(1)) |
| P2–C2–C1 (P7–C4–C3) | 117.9(2) | 114.3(1) | 118.1(1) (113.5(1)) |
| P3–P1–C1 (P5–P6–C3) | 100.2(1) | 95.68(7) | 99.51(6) (94.79(5)) |
| P3–P2–C2 (P5–P7–C4) | 100.9(1) | 95.68(7) | 99.84(6) (95.63(5)) |
| P3–P4–P5 | — | — | 99.08(2) |
The 31P{1H} NMR spectra of the triphospholane derivatives are in accordance with their solid-state structures with 1JPP coupling constants of 1-H and 1-Cl in the range of those observed for the triphospholanes Ia–d (ESI†).3 In solution, meso-3 displays four multiplets in the 31P{1H} NMR spectrum at room temperature which cannot be assigned easily because the NMR spectra of meso-3 are strongly temperature dependent indicating hindered rotations at room temperature (see the ESI,† Fig. S6 and S7). The multiplets corresponding to the nuclei P3, P4 and P5 become remarkably simple at elevated temperatures. Subsequently, the signals could be simulated as the X and YY′ part of the expected AA′BB′XYY′ spin system at 60 °C, yielding the following chemical shifts and coupling constants: δA = δA′ = 62.9 ppm, δB = δB′ = 59.2 ppm, δX = –4.2 ppm, δY = δY′ = −16.9 ppm; JAA′ = JBB′ = JAB′ = JBA′ = 0 Hz, JAB = JA′B′ = 5 Hz, JBY′ = JB′Y = 20 Hz, JAY′ = JA′Y = 34 Hz, JYY′ = 86 Hz, JBX = JB′X = 180 Hz, JAX = JA′X = 188 Hz, JXY = JXY′ = −188 Hz, JBY = JB′Y′ = −225 Hz, JAY = JA′Y′ = −233 Hz (see Fig. S8, ESI,† for a comparison of the experimental and the simulated spectrum). The large 2JPP coupling constants between P4 and P1, P2, P6 and P7 (180 and 188 Hz) may indicate attractive interactions between these phosphorus atoms even in solution. Significant couplings could also be observed in a 31P–31P COSY experiment (Fig. S9, ESI†). Through-space interactions between phosphorus atoms are also commonly observed in carborane-substituted bisphosphanes and result in coupling constants of similar magnitudes.2c
In summary, the first anionic carborane-substituted triphospholanide 1 was obtained by reacting diphosphetane 2 with the phosphaethynolate anion. Possible mechanisms of this reaction in which OCP− acts as source for the molecular dopant P− were revealed by DFT calculations. Triphospholanide 1 is a valuable synthon and can be easily converted to 1-H, 1-Cl and 3 upon reaction with the respective electrophiles. Especially 1-H and 1-Cl are promising stable starting materials for further reactivity studies due to the presence of a reactive P–H or P–Cl bond which will eventually allow to incorporate these as building blocks in polymers and other materials. These studies are presently underway.
We gratefully acknowledge financial support from the Studienstiftung des deutschen Volkes (doctoral grant for P. C.). Further support came from the Lehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-sen university, the National Natural Science Foundation of China (NSFC) (project 21720102007), and the ETH Zürich, (project 0-20406-18). We thank Gabriele Hierlmeier (University of Regensburg) for help with the synthesis of sodium phosphaethynolate.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS; (b) M. Baudler and K. Glinka, Chem. Rev., 1994, 94, 1273–1297 CrossRef CAS; (c) S. Gómez-Ruiz and E. Hey-Hawkins, Coord. Chem. Rev., 2011, 255, 1360–1386 CrossRef; (d) K.-O. Feldmann and J. J. Weigand, J. Am. Chem. Soc., 2012, 134, 15443–15456 CrossRef CAS PubMed; (e) A. Wiśniewska, R. Grubba, Ł. Ponikiewski, M. Zauliczny and J. Pikies, Dalton Trans., 2018, 47, 10213–10222 RSC.
- (a) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (b) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (c) A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS.
- A. Kreienbrink, S. Heinicke, T. T. Duong Pham, R. Frank, P. Lönnecke and E. Hey-Hawkins, Chem. – Eur. J., 2014, 20, 1434–1439 CrossRef CAS PubMed.
- S. Borucki, Z. Kelemen, M. Maurer, C. Bruhn, L. Nyulászi and R. Pietschnig, Chem. – Eur. J., 2017, 23, 10438–10450 CrossRef CAS PubMed.
- S. Isenberg, L.-M. Frenzel, C. Bruhn, R. Pietschnig, S. Isenberg, L.-M. Frenzel, C. Bruhn and R. Pietschnig, Inorganics, 2018, 6, 67 CrossRef.
- A. Schmidpeter, G. Burget and W. S. Sheldrick, Chem. Ber., 1985, 118, 3849–3855 CrossRef CAS.
- A. Kreienbrink, M. B. Sárosi, E. G. Rys, P. Lönnecke and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2011, 50, 4701–4703 ( Angew. Chem. , 2011 , 123 , 4798–4800 ) CrossRef CAS PubMed.
- H. Grützmacher and J. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 ( Angew. Chem. , 2018 , 130 , 17214–17240 ) CrossRef PubMed.
- (a) A. Kreienbrink, P. Lönnecke, M. Findeisen and E. Hey-Hawkins, Chem. Commun., 2012, 48, 9385–9387 RSC; (b) P. Coburger, J. Schulz, J. Klose, B. Schwarze, M. B. Sárosi and E. Hey-Hawkins, Inorg. Chem., 2017, 56, 292–304 CrossRef CAS PubMed; (c) P. Coburger, S. Demeshko, C. Rödl, E. Hey-Hawkins and R. Wolf, Angew. Chem., Int. Ed., 2017, 56, 15871–15875 ( Angew. Chem. , 2017 , 129 , 16087–16091 ) CrossRef CAS PubMed; (d) J. Schulz, A. Kreienbrink, P. Coburger, B. Schwarze, T. Grell, P. Lönnecke and E. Hey-Hawkins, Chem. – Eur. J., 2018, 24, 6208–6216 CrossRef CAS PubMed; (e) P. Coburger, R. Aures, P. Schulz and E. Hey-Hawkins, ChemPlusChem, 2018, 83, 1057–1064 CrossRef CAS.
- T. P. Robinson, M. J. Cowley, D. Scheschkewitz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 683–686 ( Angew. Chem. , 2015 , 127 , 693–696 ) CrossRef CAS PubMed.
- F. Hennersdorf, J. Frötschel and J. J. Weigand, J. Am. Chem. Soc., 2017, 139, 14592–14604 CrossRef CAS PubMed.
- M. Driess, A. Hermannsdorfer and D. W. Stephan, Chem. Commun., 2018, 54, 13523–13526 RSC.
- (a) J. Li, G. D’Avino, A. Pershin, D. Jacquemin, I. Duchemin, D. Beljonne and X. Blase, Phys. Rev. Mater., 2017, 1, 025602 CrossRef; (b) R. Fujimoto, Y. Yamashita, S. Kumagai, J. Tsurumi, A. Hinderhofer, K. Broch, F. Schreiber, S. Watanabe and J. Takeya, J. Mater. Chem. C, 2017, 5, 12023–12030 RSC; (c) I. Salzmann and G. Heimel, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 208–222 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed; (b) D. G. Liakos, M. Sparta, M. K. Kesharwani, J. M. L. Martin and F. Neese, J. Chem. Theory Comput., 2015, 11, 1525–1539 CrossRef CAS PubMed.
- A. H. Cowley, Chem. Rev., 1965, 65, 617–634 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR investigations, computational details. CCDC 1888315–1888317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00205g |
| ‡ rac-1-H co-crystallises with meso-1-H a as minor component and additionally displays disorder. Consequently, the precision of the bond lengths and angles is lower in comparison to 1-Cl and 3. However, they are in good agreement with the optimised geometry of rac2-1-H in the gas phase (see the ESI† for the Cartesian coordinates). |
| This journal is © The Royal Society of Chemistry 2019 |