
Asymmetric construction of dihydrobenzofuran-2,5-dione derivatives via desymmetrization of p-quinols with azlactones†
Lihua
Xie
 ,
Shunxi
Dong
,
Qian
Zhang
,
Xiaoming
Feng
and
Xiaohua
Liu
*
,
Shunxi
Dong
,
Qian
Zhang
,
Xiaoming
Feng
and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
First published on 4th December 2018
Abstract
The desymmetrization of p-quinols through a chiral bisguanidinium hemisalt catalyzed enantioselective Michael addition/lactonization cascade reaction with azlactones was reported. 3-Amino-benzofuran-2,5-diones containing a chiral amino acid residue were achieved with up to 99% ee and >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. An exploration of the structure of the catalyst bisguanidinium was undertaken, revealing a bifunctional catalytic model.
:1 dr. An exploration of the structure of the catalyst bisguanidinium was undertaken, revealing a bifunctional catalytic model.
The dihydrobenzofuran-2,5-dione skeleton and its analogues frequently occur in bioactive natural products, such as paeonilactone B, rezishanone A, fumimycin, and sorbicillactone A and B (Scheme 1a).1 Alkaloid sorbicillactone A, isolated from sponge-derived microorganisms, showed anti-leukemic activity without notable cytotoxicity.2 It is the first sorbicillinoid containing an amino acid residue and could be obtained by biotechnological production.3 For the asymmetric chemical construction of these polyfunctionalized scaffolds, the intramolecular 1,4-addition of 2,5-cyclohexadienone provides a straightforward process (Scheme 1b).3 For instance, Harned's group utilized stereoselective cyclization of cyclohexadienone tethered to the activated methylene group to get the bicyclic lactone.4 A similar strategy was employed for concise construction of sorbicillactone A and 9-epi-diastereomer after several transformations.5 Lu's group discovered a Rauhut–Currier reaction of allenonate to afford bicyclic α-allenic γ-butyrolactone in excellent enantioselectivity.6 Nevertheless, these processes could not directly introduce an amide substituent at the C3 position as shown in the structures in Scheme 1a.
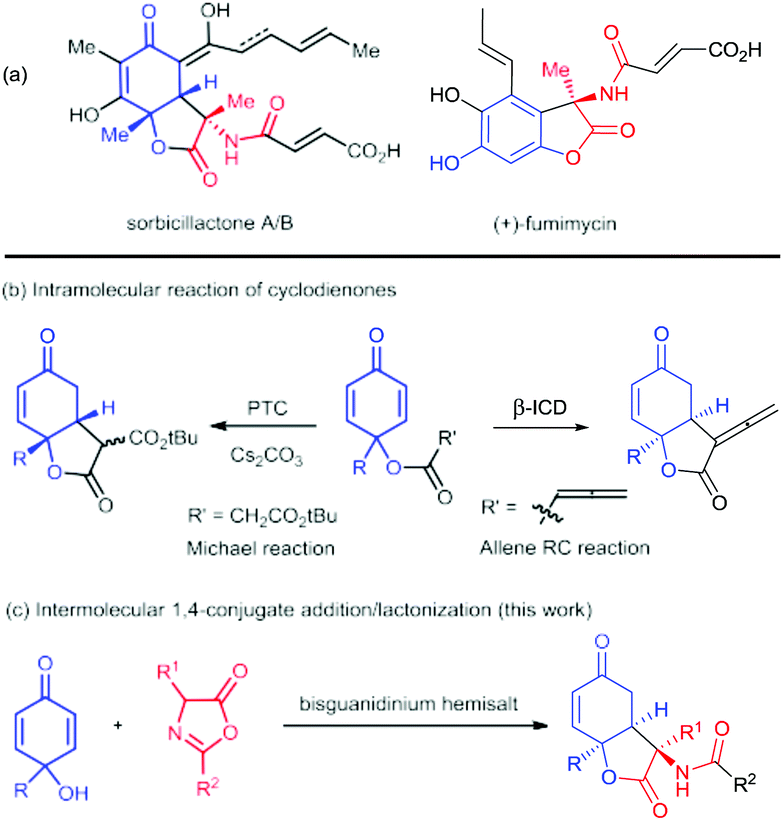 | ||
| Scheme 1 Representative natural products bearing a benzofuran-2,5-dione backbone and the synthesis strategies for dihydrobenzofuran-2,5-dione skeletons. | ||
Azlactones are important amino acid precursors which have been used to synthesize different natural and unnatural amino acid derivatives as well as others.7 Nucleophilic addition/lactonization cascade or cycloaddition of azlactone has been designed for the construction of various heterocyclic compounds.8 Our group has utilized chiral guanidine catalysts to promote several cascade reactions of azlactone for the synthesis of hydrocoumarin and oxazoline bearing an amino substituent.8d–h We proposed an intermolecular 1,4-conjugate addition/lactonization cascade between para-quinols9 and azlactones, which allows the concomitant formation of contiguous stereocenters including an amino acid residue (Scheme 1c). Herein, we utilized chiral bisguanidinium hemisalt for the desymmetrization of cyclohexadienone10via enantioselective nucleophilic addition of azlactone, following the lactonization with the 6-hydroxyl group, giving 3-amino-benzofuran-2,5-diones in good yield with high diastereo- and enantioselectivity. It provides a straightforward access to the key chiral structure of the natural product sorbicillactone.
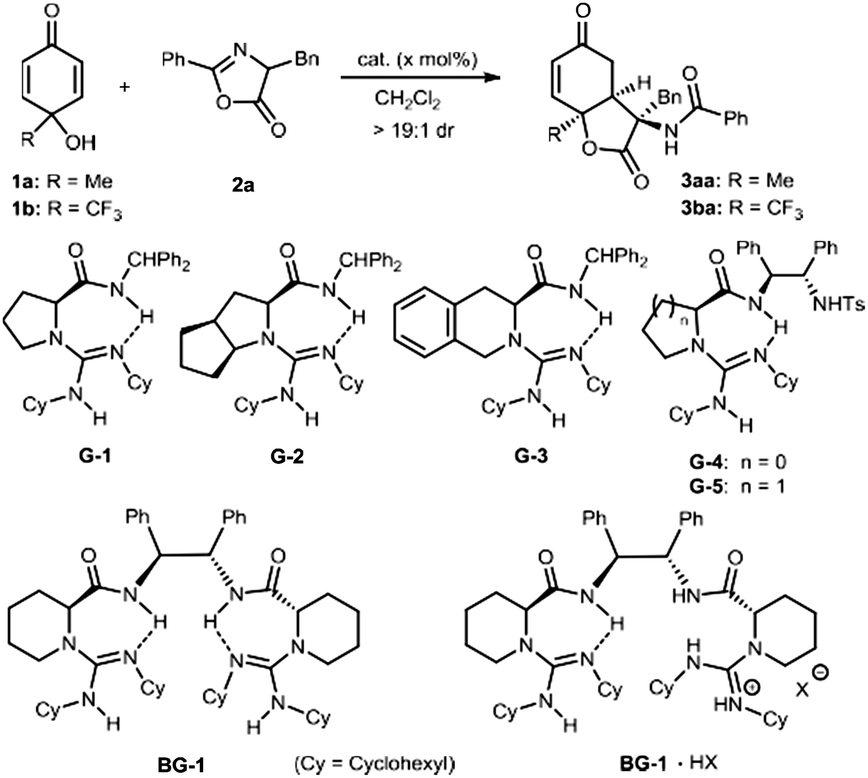
We started our investigation by studying the desymmetrization of p-quinol 1a with azlactone 2a, using bifunctional chiral guanidine catalysts developed by our group due to their applicability in a variety of azlactone-involved transformations.8d–h Despite the base sensitivity that cyclohexadienones generally exhibit,11 the cascade reaction occurred in the presence of different chiral guanidine-amides,12 which afforded the desired 3-amino-dihydrobenzofuran-2,5-dione 3aa in high diastereoselectivity (Table 1, entries 1–7). Single guanidine-amides G1–G3 bearing distinct amino acid backbones accelerated the reaction in low yield and enantioselectivity (entries 1–3). When guanidines G4 and G5 with an additional sulfonamide substituent were used, the enantioselection reversed and the ee value increased a lot (entries 4–5). Switching to bisguanidine BG-1 improved the yield and enantioselectivity slightly (entry 6). To our delight, the bisguanidinium hemisalt BG-1·HBArF4 enabled the reaction in 64% yield and 86% ee (entry 7). This indicates that the formation of a guanidinium salt can tune the conformation of the bisguanidine-amide, creating a suitable chiral environment for activation and discrimination of the substrate. The enantioselectivity of the reaction increased to 99% ee after the reaction temperature dropped from 30 °C to −10 °C, although the reactivity decreased (entry 8). Furthermore, the identified catalyst BG-1·HBPh4 with variation of the counter anion was more effective in terms of reactivity, and the desired product 3aa was obtained in 71% yield with 99% ee (entry 9). In view of the importance of fluoro-containing compounds in the field of pharmaceuticals, agrochemicals and, other areas,13 we incorporated the trifluoromethyl group into p-quinol 1b. The reaction of 1b with azlactone 2a under identical reaction conditions revealed that it performed the reaction well, affording the corresponding product 3ba in 75% yield and 99% ee (entry 10). Interestingly, high yield with excellent enantioselectivity could be obtained even when the catalyst loading was reduced to 2.5 mol% and the amount of azlactone lowered to 1.5 equivalent (entries 12 and 13).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (x mol%) | 1 | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reactions were carried out with guanidine (2.5–10 mol%), 1 (0.10 mmol), and 2a (2.0 equiv.) in (1.0 mL) at 30 °C for 24 h. b Isolated yield. c Determined by HPLC analysis. d At −10 °C for 72 h. e 2a (1.5 equiv.). (HBArF4 = HB[3,5-(F3C)2C6H3]4). | ||||
| 1 | G-1 (10) | 1a | 17 | −5 |
| 2 | G-2 (10) | 1a | 24 | −10 |
| 3 | G-3 (10) | 1a | 11 | −11 |
| 4 | G-4 (10) | 1a | 22 | 40 |
| 5 | G-5 (10) | 1a | 23 | 41 |
| 6 | BG-1 (10) | 1a | 33 | 46 |
| 7 | BG-1·HBArF4 (10) | 1a | 64 | 86 |
| 8d | BG-1·HBArF4 (10) | 1a | 50 | 99 |
| 9d | BG-1·HBPh4 (10) | 1a | 71 | 99 |
| 10d | BG-1·HBPh4 (10) | 1b | 75 | 99 |
| 11d | BG-1·HBPh4 (5) | 1b | 80 | 99 |
| 12d | BG-1·HBPh4 (2.5) | 1b | 98 | 98 |
| 13d,e | BG-1·HBPh4 (2.5) | 1b | 94 | 98 |
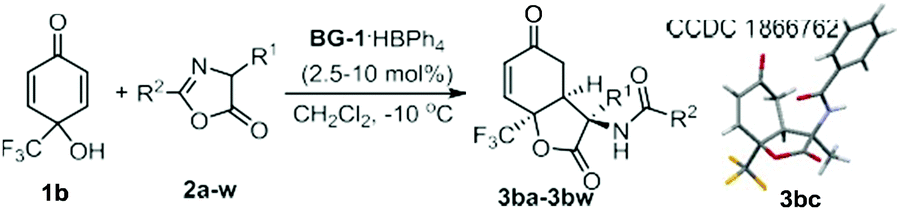
To further showcase the utility of this organocatalytic desymmetrization strategy, we next made efforts focused on azlactones, which bring different amino acid scaffolds into the dihydrobenzofuran-2,5-dione skeletons (Table 2). It was found that desymmetrization reaction triggered by 1,4-conjugate addition/lactonization of azlactones with 4-trifluoromethyl substituted p-quinol 1b performed well, delivering enantioenriched 3-amino substituted dihydrobenzofurandione with three contiguous stereogenic centres in good results. Initial investigation was conducted with azlactones 2b–2k derived from different amino acids. The azlactones synthesized from 2-amino-4-phenylbutanoic acid (2b), alanine (2c), leucine (2d), methionine (2e), and tryptophan (2f) tolerated the reaction well in the presence of 10 mol% of the catalyst BG-1·HBPh4. The desired lactone derivatives (3bb–3bf) were produced in 67–91% yield and 93–99% ee (entries 2–6). The electronic properties and position of the substituents on the aromatic rings of phenylalanine-derived azlactones 2g–2k had no influence on the enantioselectivity (98–99% ee), but a significant effect on the reactivity (64–99% yield) (entries 7–11). Furthermore, it showed that azlactones 2l–2w bearing aryl, heteroaryl, or alkyl substituents at the C2 position were suitable substrates to provide the desired dihydrobenzo-furandiones 3bl–3bw in excellent enantioselectivity (95–99% ee) and moderate to good yield (57–90% yield) (entries 12–23). The tolerance of a number of amino acid residues at the C3-position of the lactones 3 might present an impressive array of promising biological properties. Furthermore, the catalyst was applied to the reaction of para-aryl substituted quinols. The reaction was sluggish for p-quinols (1c–1e) at the previously optimized reaction condition, but several byproducts were detected after elevation of the reaction temperature to 30 °C. Products 3ca–3ea with phenyl, 4-cyanophenyl, and 4-bromophenyl could be isolated in 29–47% yield, but the enantioselectivity remained satisfactory. To demonstrate the practicality of the transformations, we carried out a gram-scale synthesis of the product 3ba. In the presence of 2.5 mol% of the chiral catalyst BG-1·HBPh4, 2.5 mmol of 1b and 3.75 mmol of azlactone 2a reacted well to afford the desired adduct 3ba in 92% yield (0.9876 g) with 98% ee.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (mol%) | R1, R2 | Yieldb (%) | eec (%) |
 a Unless otherwise noted, the reactions were carried out with BG-1·HBPh4 (2.5–10 mol%), 1b (0.10 mmol), and 2 (1.5 equiv.) in CH2Cl2 (1.0 mL) at −10 °C for 72 h.
b Isolated yield.
c Determined by HPLC and SFC analysis.
d The reactions were carried out with BG-1·HBPh4 (10 mol%), 1 (0.10 mmol), and 2a (1.5 equiv.) in CH2Cl2 (1.0 mL) at 30 °C for 72 h.
a Unless otherwise noted, the reactions were carried out with BG-1·HBPh4 (2.5–10 mol%), 1b (0.10 mmol), and 2 (1.5 equiv.) in CH2Cl2 (1.0 mL) at −10 °C for 72 h.
b Isolated yield.
c Determined by HPLC and SFC analysis.
d The reactions were carried out with BG-1·HBPh4 (10 mol%), 1 (0.10 mmol), and 2a (1.5 equiv.) in CH2Cl2 (1.0 mL) at 30 °C for 72 h.
|
||||
| 1 | 2.5 | Bn, C6H5 | 3ba, 94 | 98 |
| 2 | 10 | Phenethyl, C6H5 | 3bb, 75 | 99 |
| 3 | 10 | Me, C6H5 | 3bc, 76 | 93 |
| 4 | 10 | iBu, C6H5 | 3bd, 77 | 99 |
| 5 | 10 | 2-(Methylthio)ethyl, C6H5 | 3be, 67 | 94 |
| 6 | 10 | 1H-Indol-3-yl, C6H5 | 3bf, 91 | 94 |
| 7 | 5 | 4-Chlorobenzyl, C6H5 | 3bg, 99 | 99 |
| 8 | 5 | 4-Bromobenzyl, C6H5 | 3bh, 83 | 99 |
| 9 | 5 | 4-Methylbenzyl, C6H5 | 3bi, 66 | 99 |
| 10 | 5 | 3-Methylbenzyl, C6H5 | 3bj, 73 | 99 |
| 11 | 5 | 3-Methoxybenzyl, C6H5 | 3bk, 64 | 98 |
| 12 | 5 | Bn, 4-EtC6H4 | 3bl, 82 | 99 |
| 13 | 2.5 | Bn, 4-MeC6H4 | 3bm, 87 | 99 |
| 14 | 2.5 | Bn, 4-BrC6H4 | 3bn, 90 | 95 |
| 15 | 2.5 | Bn, 4-ClC6H4 | 3bo, 86 | 97 |
| 16 | 5 | Bn, 4-MeOC6H4 | 3bp, 74 | 99 |
| 17 | 5 | Bn, 3,5-(Me)2C6H3 | 3bq, 77 | 97 |
| 18 | 10 | Bn, 2-naphthyl | 3br, 73 | 99 |
| 19 | 10 | Bn, 1-adamantyl | 3bs, 72 | 99 |
| 20 | 10 | Bn, 2-furyl | 3bt, 72 | 99 |
| 21 | 10 | Bn, 2-thienyl | 3bu, 67 | 99 |
| 22 | 10 | Bn, cyclopentyl | 3bv, 57 | 99 |
| 23 | 10 | Bn, cyclohexyl | 3bw, 60 | 99 |
It is noteworthy that only one diastereomer was detected in the catalytic asymmetric version in the investigated cases. Performing an X-ray crystallography analysis of the major enantiomer of the product 3aa allowed us to confirm both the cis-fused ring system and trans-position between the amide substituent and C6-proton (Fig. 1). X-ray analysis of the product 3bc confirmed its stereo-arrangement.15 The relative configuration of the three chiral centers is consistent with that in the natural product sorbicillactone. Previously, Harned and coworkers utilized the intramolecular addition of a tethered malonate to construct the lactone ring (Scheme 1b, left process) following installation of the amide nitrogen, but the 9-epi-sorbicillactone A was gotten as the major diastereomer.14 Our intermolecular strategy provides a direct route for the diastereo- and enantioselective introduction of the amide unit into the lactone structure. The products have a variety of synthetic handles for subsequent derivatization. Bromination of 3aa followed by elimination formed vinyl bromide 4aa, allowing the incorporation of a variety of functional groups though cross-coupling. Indeed, epoxide 5ba was yielded as a single diastereomer bearing five contiguous stereocenters by using tert-BuNH2 as the catalyst of epoxidation (Scheme 2). The absolute configuration of the epoxide was determined by X-ray crystallography analysis.15
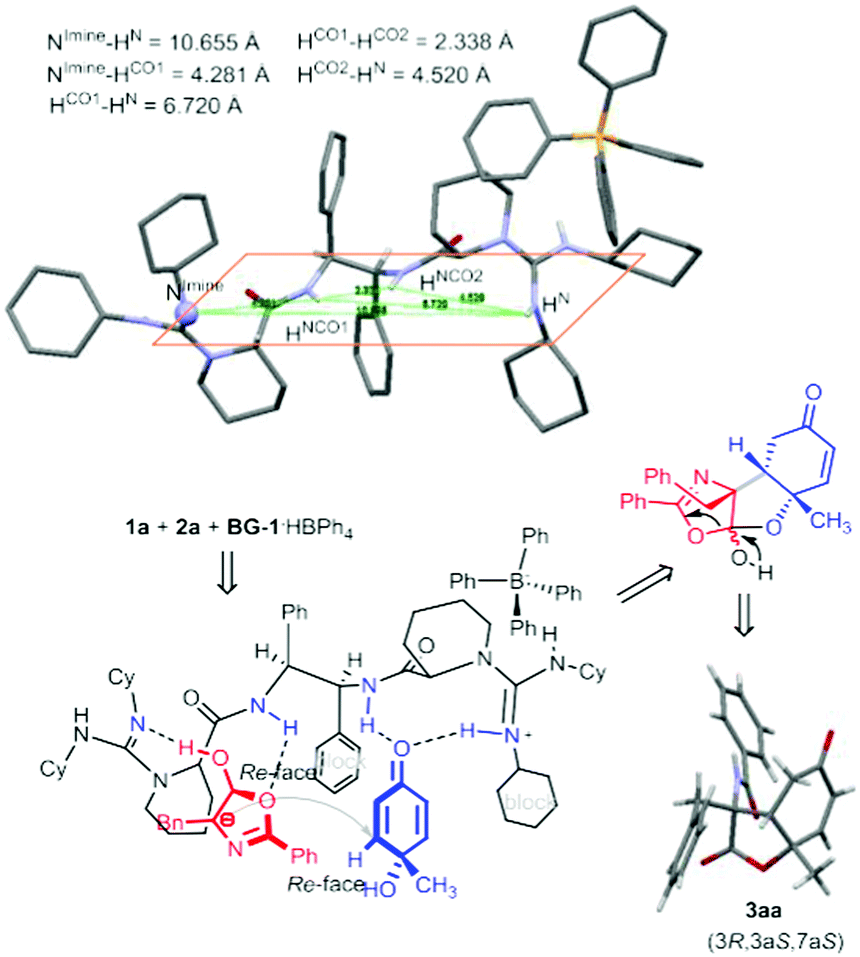 | ||
| Fig. 1 ORTEP representation of the X-ray crystal structure of the catalyst BG-1·HBPh4 and proposed model for the cascade reaction. | ||
 | ||
| Scheme 2 Transformation and X-ray crystal structure of the products. | ||
To gain some insights into this cascade process, we obtained the X-ray crystal structure of the chiral catalyst BG-1·HBPh4. Fig. 1 shows the actual three-dimensional architecture of the bisguanidinium hemisalt. In comparison with the information obtained from X-ray crystal structures of bisguanidine BG-1 and the corresponding double salt,16 special arrangement of BG-1·HBPh4 reveals some differences. The two intramolecular hydrogen-bonds disappear after one equivalent of HBPh4 is incorporated. Accordingly, the relative position of the substituents tethered to the amide units changes. The two phenyl groups rotate to the anti-position, releasing the steric hindrance in the gauche conformation in BG-1. The rotation leads to the existence of three equidirectional hydrogen bonds, including two NH of amides and one NH of guanidinium. The bulky Ph4B− anion locates nearby the guanidinium unit through electrostatic attraction, creating steric hindrance at the top side. The imine-nitrogen of the basic guanidine unit turns inside towards the hydrogen-bond-network.
Due to the instability of the two reactants in the presence of guanidine catalyst, a detailed confirmation of a catalyst–substrate adduct was not possible. We probed 1H NMR (see the ESI† for details) of a commixture of catalyst and substrates 1b and 2a. A significant upfield shift of the CH resonance on quinol 1b, and the benzyl CH2 resonance and C4-H resonance on azlactone 2a by approximately 0.05–0.15 ppm is observed. This indicates the multiple intermolecular H-bonding between the bisguanidinium salt and substrates in the transition state. When 4-MeO substituted cyclohexadienone was used instead of p-quinol 1b, the corresponding Michael reaction did not occur. This indicates that the synergic lactonization step might accelerate the initial conjugation addition step. With these experimental observations in hand, we proposed a bifunctional catalyst model that is consistent with the significant levels of diastereo- and enantioselectivity. Fig. 1 shows catalyst–substrate interaction with the guanidinium hemisalt maintaining its conformation as the crystal. The basic guanidine unit accelerates the enolization of azlactone, contacting the formed intermediate as a hydrogen-acceptor; meanwhile the vicinal amide bonds to the other oxygen atom via H-bonding. The guanidinium salt and neighboring amide provide double hydrogen-bonds to contact the carbonyl group of p-quinol 1a. An intermolecular conjugate addition between azlactone and p-quinol might take place via a Re–Re face contact. The phenyl group and cyclohexyl group underneath raise steric hindrance to discriminate the para-substituents of p-quinol, thus desymmetrization of p-quinol occurs. Next, the OH group attacks the carbonyl group of the azlactone, performing lactonization to give 3-amino-benzofuran-2,5-dione (3R,3aS,7aS)-3aa.
In summary, we have developed a highly diastereo- and enantioselective addition/lactonization of azlactones to p-quinols. The generation of 3-amino-benzofuran-2,5-diones bearing three stereogenic centers serves to synthesize potential biologically active compounds for building non-natural sorbicillactone A homologue libraries. Our results include the study of azlactones derived from different amino acids and aldehydes, and prochiral p-quinols containing different para-substituents, revealing mechanistic consequences. From the examination of the crystal structure of the bisguanidinium hemisalt, we also disclosed that the catalyst works as a bifunctional catalyst and bonds the reactants through multiple hydrogen-bonds. The catalytic principle may be extended equally well to other classes of asymmetric organocatalysis. Further application of these kinds of catalysts to other reactions is underway.
We thank Prof. Zhihua Mao of SiChuan University for single crystal analysis. We appreciate the National Natural Science Foundation of China (No. 21625205, 21332003), and National Program for Support of Top-Notch Young Professionals for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Matsuzaki, H. Tahara, J. Inokoshi and H. Tanaka, J. Antibiot., 1998, 51, 1004 CrossRef CAS; (b) G. Bringmann, G. Lang and T. A. M. Gulder, Tetrahedron, 2005, 61, 7252 CrossRef CAS; (c) Y.-J. Kwon, M.-J. Sohn, C.-J. Zheng and W.-G. Kim, Org. Lett., 2007, 9, 2449 CrossRef CAS PubMed.
- (a) A. M. Harned and K. A. Volp, Nat. Prod. Rep., 2011, 28, 1790 RSC; (b) J. J. Meng, X. H. Wang, D. Xu, X. X. Fu, X. P. Zhang, D. W. Lai, L. G. Zhou and G. Z. Zhang, Molecules, 2016, 21, 715 CrossRef.
- (a) J. Z. Zhang, J. Wu, Z. W. Yin, H. S. Zeng, K. Khanna, C. Huc and S. P. Zheng, Org. Biomol. Chem., 2013, 11, 2939 RSC; (b) M. G. Edwards, M. N. Kenworthy, R. R. A. Kitson, A. Perry, M. S. Scott, A. C. Whitwood and R. J. K. Taylor, Eur. J. Org. Chem., 2008, 4769 CrossRef CAS; (c) S. Takizawa, T. M.-N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed., 2012, 51, 5423 CrossRef CAS.
- R. T. Aburto, K. A. Kalstabakken, K. A. Volp and A. M. Harned, Org. Biomol. Chem., 2011, 9, 7849 RSC.
- K. A. Volp and A. M. Harned, Org. Lett., 2011, 13, 4486 CrossRef CAS PubMed.
- W. J. Yao, X. W. Dou, S. Wen, J. E. Wu, J. J. Vittal and Y. X. Lu, Nat. Commun., 2016, 7, 13024 CrossRef.
- (a) B. M. Trost and K. Dogra, J. Am. Chem. Soc., 2002, 124, 7256 CrossRef CAS; (b) M. Terada, K. Moriya, K. Kanomata and K. Sorimachi, Angew. Chem., Int. Ed., 2011, 50, 12586 CrossRef CAS; (c) P. P. de Castro, A. G. Carpanez and G. W. Amarante, Chem. – Eur. J., 2016, 22, 10294 CrossRef CAS; (d) M. Zhang, C.-J. Yu, J.-Q. Xie, X.-D. Xun, W.-S. Sun, L.-A. Hong and R. Wang, Angew. Chem., Int. Ed., 2018, 57, 4921 CrossRef CAS.
- (a) G.-F. Li, W.-S. Sun, J.-Y. Li, F.-J. Jia, L. Hong and R. Wang, Chem. Commun., 2015, 51, 11280 RSC; (b) X.-Y. Yu, J.-R. Chen, Q. Wei, H.-G. Cheng, Z.-C. Liu and W.-J. Xiao, Chem. – Eur. J., 2016, 22, 6774 CrossRef CAS PubMed; (c) C. Ma, J.-Y. Zhou, Y.-Z. Zhang, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5398 CrossRef CAS ; for our previous study relating to guanidine-amide promoted reactions with azlactones, see: ; (d) S. X. Dong, X. H. Liu, X. H. Chen, F. Mei, Y. L. Zhang, B. Gao, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2010, 132, 10650 CrossRef CAS; (e) S. X. Dong, X. H. Liu, Y. L. Zhang, L. L. Lin and X. M. Feng, Org. Lett., 2011, 13, 5060 CrossRef CAS; (f) K. R. Yu, X. H. Liu, X. B. Lin, L. L. Lin and X. M. Feng, Chem. Commun., 2015, 51, 14897 RSC; (g) Q. Zhang, S. S. Guo, J. Yang, K. R. Yu, X. M. Feng, L. L. Lin and X. H. Liu, Org. Lett., 2017, 19, 5826 CrossRef CAS; (h) S. Ruan, X. B. Lin, L. H. Xie, L. L. Lin, X. M. Feng and X. H. Liu, Org. Chem. Front., 2018, 5, 32 RSC.
- For selected examples on catalytic asymmetric [3 + 2] or [3 + 3] cyclizations of p-quinol analogues: (a) L. H. Liao, C. Shu, M. M. Zhang, Y. J. Liao, X. Y. Hu, Y. H. Zhang, Z. J. Wu, W. C. Yuan and X. M. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10471 CrossRef CAS PubMed; (b) C.-S. Wang, R.-Y. Zhu, Y.-C. Zhang and F. Shi, Chem. Commun., 2015, 51, 11798 RSC; (c) X.-X. Sun, H.-H. Zhang, G.-H. Li, L. Meng and F. Shi, Chem. Commun., 2016, 52, 2968 RSC.
- For selected reviews on desymmetrization of prochiral dienone: (a) G. Maertens, M.-A. Ménard and S. Canesi, Synthesis, 2014, 1573 Search PubMed; (b) K. A. Volp and A. M. Harned, Tetrahedron, 2014, 70, 9571 CrossRef PubMed.
- For selected examples: (a) M. T. Corbett and J. S. Johnson, Chem. Sci., 2013, 4, 2828 RSC; (b) S. N. Reddy, V. R. Reddy, S. Dinda, J. B. Nanubolu and R. Chandra, Org. Lett., 2018, 20, 2572 CrossRef CAS.
- (a) W. D. Cao, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2018, 29, 1201 CrossRef CAS; (b) S. X. Dong, X. M. Feng and X. H. Liu, Chem. Soc. Rev., 2018, 47, 8525 RSC.
- Y. Zhou, J. Wang, Z. N. Gu, S. N. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS.
- K. A. Volp and A. M. Harned, in Strategies and Tactics in Organic Synthesis, ed. M. Harmata, Elsevier, London, 2015, vol. 11, pp. 253–308 Search PubMed.
- CCDC 1836787 (3aa), 1866762 (3bc), 1847525 (BG-1·HBPh4), and 1851705 (5bc)†.
- X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1836787, 1866762, 1847525 and 1851705. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc08985j |
| This journal is © The Royal Society of Chemistry 2019 |