
Molecular tectonics: from a rigid achiral organic tecton to 3D chiral Co and Fe coordination networks†
Patrick
Larpent
,
Abdelaziz
Jouaiti
*,
Nathalie
Kyritsakas
and
Mir Wais
Hosseini
 *
*
Molecular Tectonics Laboratory, UMR UDS-CNRS, 7140 & icFRC, Université de Strasbourg, F-67000, France. E-mail: hosseini@unistra.fr; Fax: +33 368851325; Tel: +33 368851323
First published on 4th December 2018
Abstract
An achiral organic tecton bearing four coordinating sites of the pyridyl type leads to the formation of iso-structural 3D helical coordination polymers when combined with Co(SCN)2 and Fe(SCN)2 achiral neutral complexes. Their formation occurs during the self-assembly process in the solid state, which leads to crystals composed of homochiral coordination polymers.
Chirality is present at all scales and plays fundamental roles both in biology1 and in chemistry. Over the past decade, coordination polymers (CPs)2 or metal–organic frameworks (MOFs)3 have attracted considerable attention due to their potential applications as functional materials.4 In recent years, significant efforts dealing with chiral helical architectures,5 especially of the MOF type offering helical channels,6 have been reported owing to their promising applications in chiral separation and chiral synthesis.7 The use of CPs as heterogeneous chiral supports for enantioselective separation and/or enantio-specific transformations8 is of prime importance. Using such crystalline materials, one may achieve stereospecific reactions in a confined chiral environment.9 Furthermore, the chiral space may be used to induce specific enantioselective interactions and thus specific chiral recognition between the coordination network and one enantiomer of a racemic mixture, leading to enantiomeric separation.10
The design and formation of such supports is a challenging task. For the formation of chiral CPs, different strategies have been explored. For homochiral CPs, the most reliable approach is to combine enantio-pure organic chiral tectons with metal centers or achiral metal complexes as connecting nodes. Homochiral CPs may also be obtained by combining both enantio-pure organic tectons and metal complexes. Another approach is based on post-synthetic modification. Indeed, achiral CPs can be chemically transformed into homochiral CPs by reaction between functional groups of the network and enantio-pure reagents.11 Finally, one may use achiral organic and inorganic tectons.12 In the latter case, homochiral CPs may be formed through spontaneous resolution during the crystallization process.13 However, even though isolated single crystals are enantio-pure, the bulk phase is typically encountered as a mixture of enantiomorphic crystals (racemic conglomerate). To overcome this issue, chiral templates such as solvents or counter-cations might be used to dictate the formation of one enantiomer over the other.14 Seeding with the desired enantiomer is also an efficient path for harvesting chiral-pure bulk phases. While spontaneous resolution for molecular crystals has been often observed and deracemization techniques for racemic conglomerates have been well developed, such behaviors for crystalline CPs are still rather rare and barely explored.15
Among chiral assemblies, a helix is a pertinent example of an inherently chiral motif. We have explored the design and formation of helical coordination networks using a variety of achiral as well as chiral organic tectons and metal cations or complexes.16
Here, we report on the synthesis of the achiral organic tetradentate coordinating tecton 1 and on the formation of 3D homochiral coordination networks in the presence of Co(SCN)2 and Fe(SCN)2 (Scheme 1).
 | ||
| Scheme 1 The achiral organic tecton 1 leads under self-assembly conditions to the formation of homochiral 3D coordination networks in the presence of M(SCN)2 (M = Co(II) or Fe(II)). Side view (a) and top view (b) of the enantiomerically pure double stranded helical architecture interconnected into a 3D coordination network (c). | ||
Tecton 1 is a tetrakis-monodentate ligand based on an aryl core bearing four neutral coordinating sites of the pyridyl type. Three out of the four 4-pyridyl moieties occupy the meta positions on the aryl unit, whereas the fourth one, a 4-ethynyl pyridyl unit, is connected to one of the three ortho positions (Scheme 1).
Tecton 1 was synthesised using two different reaction pathways. The first one was based on a [4+2] cycloaddition reaction between one equivalent of 1,4-di(4-pyridyl)-1,3-diacetylene and two equivalents of 4-ethynylpyridine in the presence of Pd2(dba)3 (Scheme 2). The diyne was prepared in 93% yield by a homo-coupling reaction between two 4-ethynlypyridine units using CuCl2 in oxygenated media.17
 | ||
| Scheme 2 Reaction scheme for the formation of tecton 1via a [4+2] cycloaddition reaction. | ||
Although this synthetic approach is original, the obtained yield of the desired compound 1 appeared to be rather low. Attempts to increase it by changing the reaction conditions were unsuccessful.
To overcome this issue, a new stepwise synthetic strategy was implemented. The latter is much more efficient and allows harvesting good amounts of tecton 1 in three steps. It is based on the use of 2,4,6-tribromoaniline as the starting material (Scheme 3). The first step leading to compound 2 consists of introducing the three 4-pyridyl units by Suzuki coupling reactions between 2,4,6-tribromoaniline and 4-pyridylboronic acid in the presence of Pd(II). This step proceeds with an overall yield of 71%. In the next step, the –NH2 group of compound 2 was transformed into an –I group. The reaction of 2 with NaNO2 in HBF4 at −5 °C produced the corresponding diazonium salt, which was further reacted with KI to afford compound 3 in 86% yield. Finally, a Sonogashira coupling reaction between compound 3 and 4-ethynylpyridine in the presence of catalytic amounts of Pd(II) and CuI yielded the targeted tecton 1 in 70% yield.
 | ||
| Scheme 3 Reaction scheme developed for the synthesis of tecton 1 starting with 2,4,6-tribromoaniline. | ||
The combination of tecton 1 with M(SCN)2 (M = Co or Fe) as an octahedral square 4-connected node leads to the formation of crystals in both cases. X-ray diffraction on single crystals revealed the formation of iso-structural 3D homochiral coordination networks.
For the Co(II) based network 1·Co(SCN)2, upon slow diffusion of a MeOH solution of Co(SCN)2 into a CHCl3 solution of tecton 1, light pink needle-shaped single crystals were obtained overnight.
1·Co(SCN)2 crystallizes in the trigonal system with the chiral space group P3121. The crystal is composed of the organic tecton 1, Co2+ cation, SCN− anion and solvent molecules. Owing to the disorder of the latter, the squeeze command was applied to solve the structure.
In the crystal, each Co2+ cation, adopting a distorted Oh coordination geometry, acts as a 4-connecting node, as the two apical positions are occupied by NCS− anions (dCo–N of 2.11 Å and N–Co–N angle of 179°). The square base of the octahedron is occupied by four pyridyl units belonging to four different organic tectons 1via N–Co bonds (dCo–N in the 2.16–2.20 Å range) (Fig. 1).
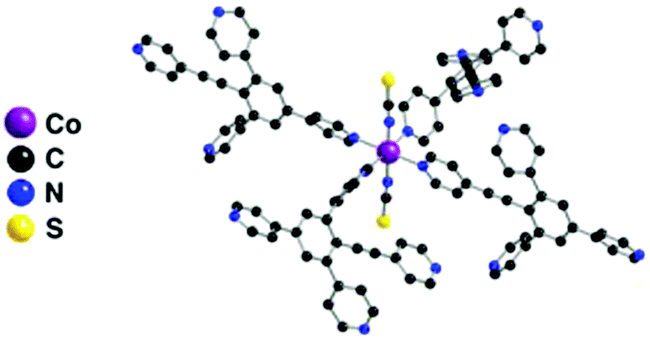 | ||
| Fig. 1 Portion of the X-ray structure of 1·Co(SCN)2 showing the Oh coordination geometry adopted by the Co2+ cation. | ||
The formation of the 3D network results from the interconnection of three identical but differently oriented 1D directional linear chains (Fig. 2a). Indeed, the organic tecton 1 forms a 1D linear coordination network through coordination of Co2+ cations by its 4-ethynylpyridyl unit and the 4-pyridyl moiety in the trans position. The remaining two appended pyridyl moieties of each tecton 1 behave as bridging units interconnecting consecutive 1D chains in the other two directions of space, thus leading to the 3D architecture. Within the 1D chains (red, blue or green, Fig. 2a), consecutive Co2+ cations are distant by 18.3 Å, whereas along the bridging axis (Fig. 2b), they are distant by 8.16 Å.
 | ||
| Fig. 2 Portion of the X-ray structure of 1·Co(SCN)2 showing the bridging of consecutive directional 1D linear networks (a) and view along the z axis (b). | ||
The 3D network described above is chiral (Fig. 3). Its chirality arises from the formation of double stranded helical motifs along the z-axis (Fig. 4a and b). Each double stranded helix is formed by two helices of the same M handedness interconnected by coordination bonds (Fig. 4b). The pitch of each helix is 30.57 Å. For the generation of these helical motifs, three out of the four coordination sites on the Co2+ cation and three pyridyl units out of the four belonging to the organic coordinating tecton 1 are involved (Fig. 4a top).
 | ||
| Fig. 3 Portion of the X-ray structure along the z axis showing the 3D structure of 1·Co(SCN)2. | ||
 | ||
| Fig. 4 Portion of the X-ray structure of 1·Co(SCN)2 showing the double stranded helical repeating motifs. | ||
The remaining coordination site on the Co2+ cation and the pyridyl unit of tecton 1 are involved in the interconnection of consecutive double stranded helical motifs in the xy plane (Fig. 3). As a result, each double stranded helical network is linked to six surrounding similar motifs, yielding a hexagonal arrangement (Fig. 3).
The 3D chiral 1·Co(SCN)2 network displays 1D chiral channels along the z axis (Fig. 3) with a solvent accessible void of 63% (Platon software18). Unfortunately, the crystals are not stable and collapse upon removal of the solvent molecules. Indeed, Powder X-Ray Diffraction (PXRD) performed on the powdered sample revealed the presence of an amorphous phase. Fe(II) based orange/red single crystals of 1·Fe(SCN)2 were obtained after few days in a similar fashion by slow diffusion of a deoxygenated MeOH solution of Fe(SCN)2(py)4 into a deoxygenated CHCl3 solution of tecton 1.
X-ray analysis of the single crystals revealed the formation of a 3D chiral network iso-structural to 1·Co(SCN)2 discussed above.
1·Fe(SCN)2 crystallizes in the chiral space group P3121. The crystal is composed of the organic tecton 1, Fe2+ cation, SCN− anion and solvent molecules. Again, owing to the disorder of the latter, the squeeze command was applied to solve the structure.
Within the coordination network, each Fe2+ metallic node adopts a distorted Oh geometry as observed for the Co2+ cation in 1·Co(SCN)2. The two apical positions of the octahedron are occupied by SCN− anions with N–Fe distances of 2.13 Å and a N–Fe–N angle of 180.0°.
The four corners of the square base of the octahedron are occupied by four tectons 1 through N–Fe coordination bonds (dN–Fe in the 2.19–2.23 Å range). As in 1·Co(SCN)2, 1D directional linear networks are formed (purple, blue and green, Fig. 5) and are further bridged along the z axis by the two appended pyridyl units belonging to each tecton 1. Within each 1D chain, the Fe–Fe distance is 18.37 Å, whereas it is 8.12 Å along the bridging z axis (Fig. 5a and b).
 | ||
| Fig. 5 Portion of the X-ray structure of 1·Fe(SCN)2 showing the bridging of consecutive directional 1D linear networks (a) and view along the helical axis (b). | ||
Similarly to 1·Co(SCN)2, 1·Fe(SCN)2 is a 3D chiral network (see the ESI†). Again, the double stranded helical motifs are of the same M handedness (see the ESI†). The crystal displays a solvent accessible void of 63% identical to the one calculated for 1·Co(SCN)2. Unfortunately, crystals of 1·Fe(SCN)2 are also unstable once out of the mother liquor, leading to an amorphous phase after a few minutes.
Both 1·Co(SCN)2 and 1·Fe(SCN)2 discussed above are enantiomerically pure chiral crystals resulting from the interconnection of double stranded helical motifs with the same M handedness. This behavior was investigated in both cases by repeating the crystallization process several times. Crystals of two different batches have been randomly selected and structurally investigated by X-ray diffraction on single crystals. In both cases enantiomerically pure crystals based on helical strands of the same M handedness have been obtained. However, one cannot exclude the formation of enantiomerically pure crystals with the opposite chirality resulting from interconnection of double helical networks with P handedness, thus leading to a racemic conglomerate in the bulk. Further investigations in that direction are being pursued.
In conclusion, the synthesis of tecton 1 based on a central aryl moiety and bearing four monodentate coordinating sites of the pyridyl type was achieved using two distinct synthetic strategies. Although the first strategy based on a [4+2] cycloaddition reaction was original, owing to its rather low yield, a sequential approach based on coupling reactions was developed. The combination of the achiral tecton with Co(SCN)2 and Fe(SCN)2 afforded crystalline materials. Their structural investigation by X-ray diffraction on single crystals revealed the formation of iso-structural chiral 3D coordination networks. The chirality of the networks arises from the formation of double stranded helical motifs with the same M handedness. The two helical motifs are interconnected by N–M coordination bonds. The linking of consecutive helices leads to the formation of a 3D coordination network displaying 1D chiral channels with a solvent accessible void of 63% arranged into a honeycomb type arrangement. These two unprecedented architectures in the solid state are novel examples of 3D chiral networks formed upon combination of achiral components. The combination of tecton 1 with other metallic cations is under investigation.
We thank the University of Strasbourg, the Institut Universitaire de France, the C.N.R.S., the International Centre for Frontier Research in Chemistry (icFRC), and the LabEx CSC (ANR-10-LABX-0026 CSC) within the Investissement d’Avenir program ANR-10-IDEX-0002-02 for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Basak, I. Singh, A. Ferranco, J. Syed and H.-B. Kraatz, Angew. Chem., Int. Ed., 2017, 56, 13288 CrossRef CAS PubMed.
- (a) B. F. Abrahams, B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1991, 113, 3607 CrossRef; (b) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- (a) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084 CrossRef CAS PubMed; (b) W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163 CrossRef CAS PubMed; (c) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC; (d) T. Hang, D. W. Fu, Q. Ye and R. G. Xiong, Cryst. Growth Des., 2009, 9, 2026 CrossRef CAS; (e) X. M. Jiang, M. J. Zhang, H. Y. Zeng, G. C. Guo and J. S. Huang, J. Am. Chem. Soc., 2011, 133, 3410 CrossRef CAS PubMed.
- (a) X.-H. Bu, W. Chen, M. Du, K. Biradha, W.-Z. Wang and R.-H. Zhang, Inorg. Chem., 2002, 41, 437 CrossRef CAS PubMed; (b) T. Ezuhara, K. Endo and Y. Aoyama, J. Am. Chem. Soc., 1999, 121, 3279 CrossRef CAS; (c) A. Erxleben, Inorg. Chem., 2001, 40, 412 CrossRef CAS PubMed; (d) L. Han and M. Hong, Inorg. Chem. Commun., 2005, 8, 406 CrossRef CAS; (e) K. Biradha, C. Seward and M. J. Zaworotko, Angew. Chem., Int. Ed., 1999, 15, 492 CrossRef.
- (a) T. J. Prior and M. J. Rosseinsky, Inorg. Chem., 2003, 42, 1564 CrossRef CAS; (b) Y.-Q. Chen, G.-R. Li, Z. Chang, Y.-K. Qu, Y.-H. Zhang and X.-H. Bu, Chem. Sci., 2013, 4, 3678 RSC; (c) M. C. Alonso, A. Arca, F. Iaia, R. Lai, V. Lippolis, S. K. Callear, M. Caricato, D. Pasini, S. J. Coles and M. C. Aragoni, CrystEngComm, 2014, 16, 8582 RSC.
- (a) Z.-G. Gu, C. Zhan, J. Zhang and X. Bu, Chem. Soc. Rev., 2016, 45, 3122 RSC; (b) L. Qin, M. X. Zheng, Z. J. Guo, H.-G. Zheng and Y. Xu, Chem. Commun., 2015, 51, 2447 RSC; (c) Z. Liao, H. Huang, G. Sun, M. Luo and F. Luo, Inorg. Chem. Commun., 2011, 14, 181 CrossRef; (d) F. Luo, Y. Ning, M. Luo and G. Huang, CrystEngComm, 2010, 12, 2769 RSC.
- Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS PubMed.
- (a) K. Kim, Nature, 2000, 404, 982 CrossRef; (b) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075 CrossRef CAS PubMed; (c) F. Song, T. Zhang, C. Wang and W. Lin, Proc. R. Soc. A, 2012, 468, 2035 CrossRef CAS; (d) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS; (e) M. Zaworotko, Z. Zhang, Y. Ji, L. Wojtas, W.-Y. Gao, S. Ma and J. Antilla, Chem. Commun., 2013, 49, 7693 RSC; (f) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS; (g) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- (a) R.-G. Xiong, X.-Z. You, B. F. Abrahams, Z. Xue and C.-M. Che, Angew. Chem., Int. Ed., 2001, 40, 4422 CrossRef CAS; (b) K. Suh, M. P. Yutkin, D. N. Dybtsev, V. P. Fedin and K. Kim, Chem. Commun., 2012, 48, 513 RSC; (c) D. Bradshaw, T. J. Prior, E. J. Cussen, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2004, 126, 6106 CrossRef CAS; (d) A. L. Nuzhdin, D. N. Dybtsev, K. P. Bryliakov, E. P. Talsi and V. P. Fedin, J. Am. Chem. Soc., 2007, 129, 12958 CrossRef CAS PubMed; (e) Y. Cui, Y. Peng and T. Gong, Chem. Commun., 2013, 49, 8253 RSC; (f) M. Zhang, Z.-J. Pu, X.-L. Chen, X.-L. Gong, A.-X. Zhu and L.-M. Yuan, Chem. Commun., 2013, 49, 5201 RSC; (g) S.-M. Xie, Z.-J. Zhang, Z.-Y. Wang and L.-M. Yuan, J. Am. Chem. Soc., 2011, 133, 11892 CrossRef CAS PubMed.
- (a) S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341 CrossRef CAS; (b) Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315 RSC.
- R. E. Morris and X. Bu, Nat. Chem., 2012, 2, 353 CrossRef.
- (a) T. Ezuhara, K. Endo and Y. Aoyama, J. Am. Chem. Soc., 1999, 121, 3279 CrossRef CAS; (b) G. Tian, G. Zhu, X. Yang, Q. Fang, M. Xue, J. Sun, Y. Wei and S. Qiu, Chem. Commun., 2005, 1396 RSC.
- (a) Z. Lin, A. M. Z. Slawin and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 4880 CrossRef CAS; (b) J. Zhang, S. Chen, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2008, 130, 12882 CrossRef CAS PubMed; (c) J. Zhang, R. Liu, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2007, 46, 8388 CrossRef CAS PubMed.
- S. Tripathi, R. Srirambalaji, N. Singh and G. Anantharaman, J. Chem. Sci., 2014, 126, 1423 CrossRef CAS.
- (a) C. Kaes, M. W. Hosseini, C. E. F. Rockard, B. W. Skelton and A. H. White, Angew. Chem., Int. Ed., 1998, 37, 920 CrossRef CAS; (b) B. Schmaltz, A. Jouaiti, M. W. Hosseini and A. De Cian, Chem. Commun., 2001, 1242 RSC; (c) A. Jouaiti, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2003, 472 RSC; (d) P. Grosshans, A. Jouaiti, V. Bulach, J.-M. Planeix, M. W. Hosseini and J.-F. Nicoud, CrystEngComm, 2003, 5, 414 RSC; (e) A. Jouaiti, V. Bulach, J.-M. Planeix, M. W. Hosseini and J.-F. Nicoud, C. R. Chimie, 2004, 7, 189 CrossRef; (f) A. Jouaiti, M. W. Hosseini, N. Kyritsakas, P. Grosshans and J.-M. Planeix, Chem. Commun., 2006, 3078 RSC; (g) M.-J. Lin, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2010, 46, 115 RSC; (h) M.-J. Lin, A. Jouaiti, P. Grosshans, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2011, 47, 7635 RSC.
- J. G. Rodríguez and C. Díaz-Oliva, Tetrahedron, 2009, 65, 2512 CrossRef.
- A. L. Spek, PLATON, The University of Utrecht, Utrecht, The Netherlands, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1413936 and 1413937. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc08877b |
| This journal is © The Royal Society of Chemistry 2019 |