
Boosting the triplet activity of heavy-atom-free difluoroboron dibenzoylmethane via sp3 oxygen-bridged electron donors†
Wenhuan
Huang
a,
Xuepeng
Zhang
a,
Biao
Chen
 *a,
Hui
Miao
a,
Carl O.
Trindle
b,
Yucai
Wang
a,
Yi
Luo
a and
Guoqing
Zhang
*a
*a,
Hui
Miao
a,
Carl O.
Trindle
b,
Yucai
Wang
a,
Yi
Luo
a and
Guoqing
Zhang
*a
aHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: gzhang@ustc.edu.cn; biaochen@ustc.edu.cn
bDepartment of Chemistry, University of Virginia, Charlottesville, VA 22904, USA
First published on 24th November 2018
Abstract
Purely organic phosphors have important applications in imaging, sensing, informatics and illumination. Methoxy-substituted difluoroboron dibenzoylmethane (BF2dbm) complexes exhibit intense fluorescence with an almost unity quantum yield. Here we show that by simply introducing an sp3 oxygen-bridged methoxylphenyl group as a pendant to BF2dbm, the boron complex exhibits a triplet quantum yield of 0.16, a more than 100-fold increase compared to that of BF2dbm.
Photoluminescent organoboron molecules are routinely investigated for their exceptional photophysical properties and have found applications in optical sensing1,2 and light-emitting devices.3–7 Among these reports on organoboron materials, difluoroboron diketonates (BF2bdks)8–10 have received tremendous attention due to their strong interaction with visible light, high fluorescence quantum yields, tunable emission features, long-lived phosphorescence in polymer matrices, and mechanochromic luminescence (ML) in the solid state.11–15 In order to increase the relative quantum yield of the oxygen-sensitive, emissive triplet excited state of BF2bdks, halogen-substitution has been used in various applications such as tumour oxygen imaging,16–20 wound recovery monitoring21,22 and ML quenching23–27 due to the enhanced heavy-atom effect with boosted triplet activity. Otherwise, the nearly unity fluorescence from BF2bdks would have precluded most applications based on a balanced ratio between fluorescence and phosphorescence. In addition, the halogen atom can be photochemically unstable, toxic and unfriendly to the environment, which makes heavy-atom free BF2dbm with high triplet activity an attractive substitute. Here we show a new methodology, where the fluorescent species can be transformed into phosphorescent ones by minor alterations in the molecular structure. The strategy employs no heavy atoms, nor does it require functional groups strongly enhancing spin–orbit coupling (SOC) such as carbonyl, nitro or amine substitution. Instead, the sp3 oxygen-bridged electron donor moiety significantly boosts the triplet state yield via a combination of SOC related to transition angular momentum and charge-transfer (CT) mediated intersystem crossing (ISC).28,29
Three BF2dbm model complexes 1–3 (Fig. 1a) were readily synthesized by Claisen condensation and subsequent coordination with boron trifluoride in high yield (overall yields >50%, Scheme S1; for synthetic procedure and characterization, see Fig. S10–S18 in ESI†), where the oxygen atom is linked to methyl, phenyl and p-methoxylphenyl to produce 1, 2 and 3, respectively, and provides a twist angle between the BF2dbm core and the pendant group. The absorption and emission spectra in dilute solutions (2-methyl-tetrahydrofuran, m-THF, 4.95 μM) are presented in Fig. 1b, where complex 1 has been investigated by many research groups including us, both in solutions and in the solid state.13,30 All three compounds have almost the same absorption between 350 and 420 nm (λmax = 396, 393 and 395 nm, respectively); the slight blue shift for 2 is likely due to the electronic conjugation between the oxygen lone pair and the phenyl group, which reduces the donating ability of the lone pair. Both 1 and 2 emit strong blue photoluminescence (PL, λem = 420 nm, ΦF = 0.97 and 0.81) at room temperature under UV irradiation at 372 nm, whereas, interestingly, 3 exhibits extremely attenuated blue PL as shown in the inset (ΦF < 0.001, Fig. 1b and Table 1). It is not uncommon for PL molecules to show reduced fluorescence yield with more substituent groups due to extra vibrational and rotational modalities that could dissipate the energy in the excited state. In this case, however, the reduction in PL quantum yield from 2 to 3 (from nearly unity to nothing) is far too significant to attribute to nuclear motions. A more reasonable explanation would be fluorescence quenching from a dark, CT-mediated ISC process, as has been noted from other donor-sp3-acceptor systems.28 As shown in Fig. 1c, 3 exhibited strong photoluminescence at 77 K with a strong phosphorescence peak at ∼500 nm (τP = 1.41 s) and a calculated phosphorescence yield of 0.16, while the value for 1 was almost negligible.
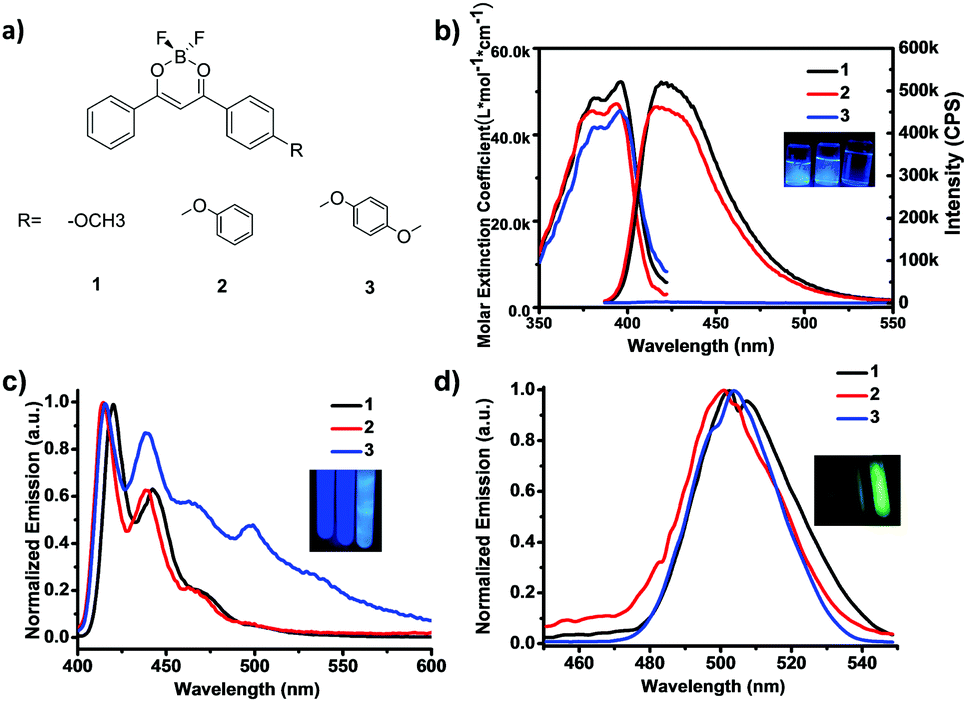 | ||
| Fig. 1 (a) Chemical structures of BF2dbm model complexes 1–3; (b) UV/Vis absorbance and steady-state emission (λex = 372 nm) spectra of model complexes 1–3 in m-THF at room temperature, inset: image showing PL emission in m-THF at RT at an identical concentration of 4.95 μM; (c) steady-state emission spectra of model complexes 1–3 in m-THF at low temperature (77 K, LT) (λex = 372 nm), inset: image showing steady-state PL emission in m-THF at 77 K; (d) delayed (Δt = 100 ms) emission spectra of model complexes 1–3 in m-THF at 77 K, inset: image showing afterglow 100 ms after excitation (using a 365 nm handheld lamp) had ceased with the aid of a mechanical shutter. | ||
| λ F (nm) | λ P (nm) | τ F (ns) | τ LP (s) | Φ PL | Φ PL | Φ P | |
|---|---|---|---|---|---|---|---|
| a Fluorescence emission maximum from steady-state emission spectra (λex = 372 nm) in m-THF at 77 K. b LP maximum at 77 K (λex = 372 nm). c FL lifetime fitted from single-exponential decay (λex = 374 nm, nanoLED) at 77 K d Phosphorescence lifetime fitted from single-exponential decay (λex = 372 nm, spectraLED) at 77 K. e Absolute quantum yield from 400–600 nm at RT. f Absolute quantum yield from 400–600 nm at 77 K. g Calculated phosphorescence quantum yield at 77 K by spectral subtraction. h Signal too weak for accurate measurement. | |||||||
| 1 | 420 | 502 | 1.75 | 1.25 | 0.97 | 0.97 | <0.01 |
| 2 | 414 | 497 | 1.60 | 1.30 | 0.81 | 0.82 | 0.045 |
| 3 | 415 | 539 | —h | 1.41 | <0.001 | 0.30 | 0.16 |
Purely organic, heavy-atom-free RTP molecules have wide applications in optical sensing, bioimaging and illumination.31–41 Many applications require that small molecular phosphors be embedded into rigid polymer matrices as an efficient approach to achieve RTP due to suppression of molecular motions in these systems (restriction of non-radiative relaxations).42,43 All three molecules were dissolved in PMMA films (film thickness ∼200 μm, solute 0.1% by weight) and the steady-state PL emissions in air and under vacuum were measured as shown in Fig. 2a and b, respectively. The PL emissions of 1 and 2 are similar with a single fluorescence peak at 433–434 nm (τF = 2.2–2.3 ns, Table 2) in PMMA films while that of 3 is significantly broadened in the lower energy region (450–650 nm). We ascribed the broadening to the enhanced ISC and subsequently boosted RTP with polymer protection. Indeed, a lifetime of 250 μs (Fig. 2c inset) was recorded for 3 in the low energy shoulder band area (500–550 nm). Under vacuum, the relative intensity of the broad shoulder band is further increased, which confirms the oxygen-sensitive nature of the RTP from 3. The RTP lifetime exhibits a concomitant increase by more than 1000 fold to 310 ms (Fig. 2c). Although the relative RTP intensity is much lower for 1 and 2, their lifetimes are nonetheless much longer, which can be explained by Fig. 2d and e. The delayed emission spectra recorded not only show the main RTP band at 510 nm but also the thermally activated delayed fluorescence (TADF)44–47 band at around 430 nm, which disappears at 77 K (Fig. 2e). The RTP bands for all three complexes 1–3 are almost super-imposable, indicating the identical origin from the localized π–π* state from the BF2dbm core structure, as has been previously reported.13,30,48 The fact that 3 alone exhibits the most intense TADF peak strongly suggests multiple energy pathways for ISC, most likely a combination of localized (∼430 nm) and other mediating states (>440 nm), such as CT, in between 1π–π* and 3π–π*.44,45 Higher TADF ratios generally promise shorter RTP lifetimes, irrespective of thermal activation or triplet–triplet annihilation. In addition, the similarity in TADF peak intensity of 1 and 2 also points to the fact that vibrational coupling caused by the additional phenyl ring plays a minor role in effectuating the ISC process. Fig. 2f–h compare the steady-state and delayed emission spectra of 1–3, respectively. It is evident that RTP is dramatically more sensitive to oxygen quenching vs. TADF given that air quenches most RTP while TADF persists. We were also able to record the TADF spectra in different solvents (Fig. S33 and S34, ESI†); it was found that the TADF intensity is the lowest in the most polar solvent. This observation is attributed to increased non-radiative decay rate in polar solvents as has been previously reported by Adachi et al.49
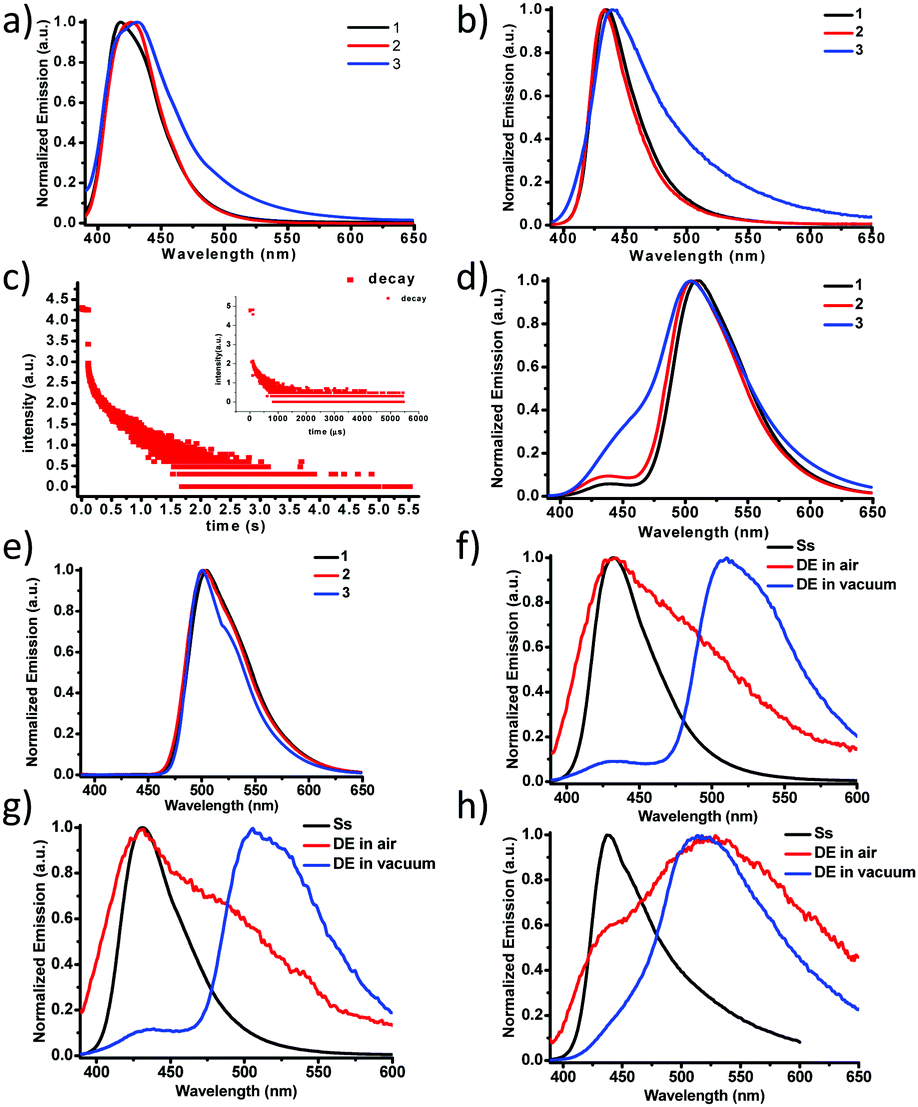 | ||
| Fig. 2 Steady-state emission spectra of model complexes 1–3 in thin PMMA films at room temperature in air (a) and under vacuum (b) (λex = 372 nm); (c) time-resolved emission decay curves of 3 at 510 nm in air and under vacuum; delayed (Δt = 100 ms) emission spectra of model complexes in PMMA at RT (d) and 77 K (e) and emission spectra of model complexes 1 (f), 2 (g), and 3 (h) in PMMA at RT (Ss: steady state emission, DE in air: delayed emission in air (Δt = 50 μs), DE in vacuum: delayed emission in vacuum (Δt = 50 μs)). | ||
| λ F (nm) | λ RTP (nm) | τ F (ns) | τ RTP (μs) | τ RTP (s) | |
|---|---|---|---|---|---|
| a Fluorescence emission maximum from steady-state spectra (λex = 372 nm) in PMMA at room temperature. b RTP maximum at RT (λex = 372 nm). c FL lifetime fitted from single-exponential decay (λex = 372 nm, nanoLED) at RT. d RTP lifetime fitted from single-exponential decay (λex = 370 nm, spectraLED) at RT in air, note that RTP emissions of 1 and 2 in air are too weak (if any) to measure. e RTP lifetime fitted from single-exponential decay (λex = 370 nm, spectraLED) at RT in vacuum. | |||||
| 1 | 434 | 510 | 2.27 | — | 0.70 |
| 2 | 433 | 510 | 2.15 | — | 0.58 |
| 3 | 440 | 510 | 2.00 | 250 | 0.31 |
To further understand the photophysics of the sp3-bridged donor effects, we performed quantum chemical calculations and modelled their molecular geometries in the ground and excited states (singlet and triplet) using a previously established method.50,51 Theoretical modeling for this sequence was accomplished with the functional B3PW91 and the basis 6-311+G(d), proved by Barlow and co-workers to be effective in describing the charge-transfer excitations.52 The medium was modeled by the polarizable continuum defined by Tomasi, with parameters for THF.53 Charge transfer plays a crucial role in populating the triplet excited states as indicated in Fig. 3a–c and Tables S1–S3 (ESI†). Briefly speaking, the energy gaps between the S1 state (localized π–π*) and the neighbouring lowest triplet state for both 1 and 2 are almost identical (∼0.22 eV), while that of 3 (1CT) exhibits a sharp decrease to 0.025 eV, readily bifurcating to T3 and T2. Specifically, MO analysis (Fig. 3a) shows little if any CT character in 1's reddest transition. There is also substantial splitting between S1 and T1 dominated by the HOMO–LUMO excitation. This is consistent with a large exchange integral K (HOMO–LUMO). Strong fluorescence emission (f = 1.04, experimentally ΦF ∼ 1) probably also helps to hinder effective passage to the nearest triplet state. When O-phenyl replaces O-methyl to form 2, the reddest (HOMO–LUMO) transition in the absorption spectrum is hardly shifted, and the MOs (Fig. 3b) involved are still confined to the core defined by 1. Little CT character is seen in the transition. The further substitution of a para MeO-fragment moves the MO on the O-phenyl-OMe up in energy; which becomes the new HOMO (Fig. 3c). The LUMO is still located at the BF2dbm core, so the HOMO–LUMO transition has a substantial CT character. This is illustrated by the small exchange splitting between the singlet and triplet states defined by those two MOs (only 0.025 eV) and the relative weakness of the lowest singlet-state transition (f = 0.016). These features of 3 are preserved in the excited S1 state (Fig. 3c, splitting 0.01 eV and f < 0.01). This defines a diradical character and implies a long lifetime for the S1 state. All these features suggest efficient passage to the triplet state and subsequent strong RTP emission of 3.
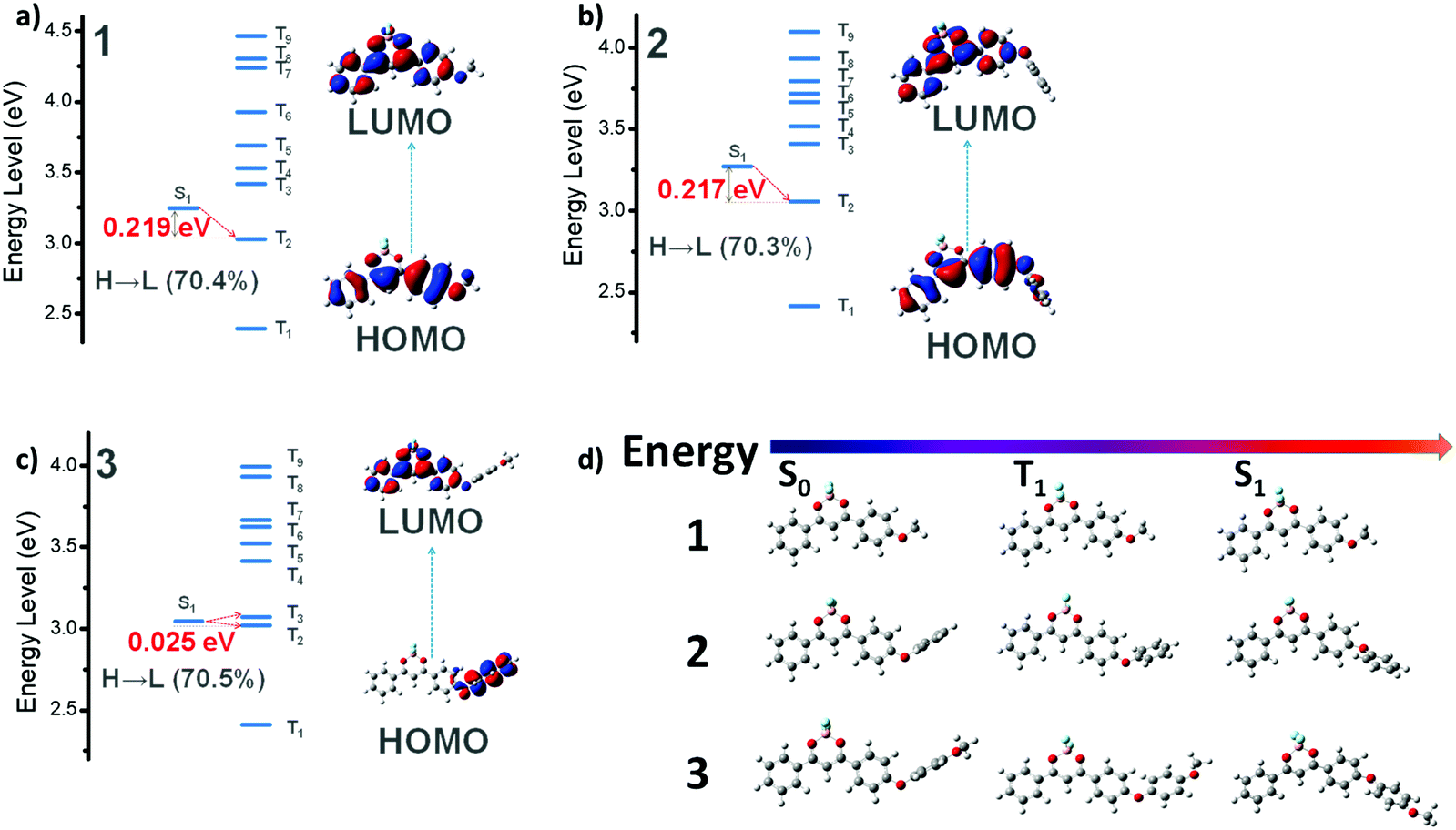 | ||
| Fig. 3 Calculated molecular orbitals for the lowest singlet-state transition for all three boron complexes. Schematic diagrams of the TD-DFT calculated energy levels and possible ISC channels of 1 (a), 2 (b) and 3 (c) at the singlet (S1) and triplet (Tn) states. H and L represent the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively. The insets are the Kohn–Sham frontier orbitals obtained from all three complexes; and (d) calculated molecular geometries at the ground state (S0), first triplet excited state (T1) and first singlet excited state (S1) for all three complexes. | ||
In summary, we have successfully synthesized three difluoroboron dibenzoylmethane derivatives with the methyl, phenyl and p-methoxylphenyl as the “pedant” groups to the BF2dbm core, respectively. Despite the subtle structural change, the molecules show substantial difference in the photophysical and photochemical properties, particularly for the p-methoxylphenyl substituted BF2dbm complex, 3, where the enhanced triplet excited state activity, including RTP and TADF, reveals that the charge-transfer mediated ISC process can also be realized in the oxygen-bridged donor-sp3-acceptor systems. The report broadens the application potential of the donor-sp3-acceptor systems and can serve as a working strategy to construct heavy-element free organic phosphors.
We thank the National Key R&D Program of China (2017YFA0303500 to G. Z. and Y. L.) and the Fundamental Research Funds for the Central Universities (WK2340000068 to G. Z.) for their financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879 CrossRef CAS PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131 CrossRef CAS PubMed.
- W. L. Ng, M. Lourenco, R. Gwilliam, S. Ledain, G. Shao and K. Homewood, Nature, 2001, 410, 192 CrossRef CAS.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416 RSC.
- X. Yin, K. Liu, Y. Ren, R. A. Lalancette, Y.-L. Loo and F. Jäkle, Chem. Sci., 2017, 8, 5497 RSC.
- B. Meng, Y. Ren, J. Liu, F. Jäkle and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 2183 CrossRef CAS PubMed.
- K. Liu, R. A. Lalancette and F. Jakle, J. Am. Chem. Soc., 2017, 139, 18170 CrossRef CAS PubMed.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2017, 56, 8173 CrossRef CAS.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS.
- K. Tanaka and Y. Chujo, NPG Asia Mater., 2015, 7, e223 CrossRef CAS.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107 CrossRef.
- S. Xu, R. E. Evans, T. Liu, G. Zhang, J. Demas, C. O. Trindle and C. L. Fraser, Inorg. Chem., 2013, 52, 3597 CrossRef CAS.
- C.-T. Poon, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 13992 CrossRef CAS.
- P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196 CrossRef CAS.
- J. Samonina-Kosicka, C. A. DeRosa, W. A. Morris, Z. Fan and C. L. Fraser, Macromolecules, 2014, 47, 3736 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed.
- A. S. Mathew, C. A. DeRosa, J. N. Demas and C. L. Fraser, Anal. Methods, 2016, 8, 3109 RSC.
- C. A. DeRosa, C. Kerr, Z. Fan, M. Kolpaczynska, A. S. Mathew, R. E. Evans, G. Zhang and C. L. Fraser, ACS Appl. Mater. Interfaces, 2015, 7, 23633 CrossRef CAS.
- J. Samonina-Kosicka, D. H. Weitzel, C. L. Hofmann, H. Hendargo, G. Hanna, M. W. Dewhirst, G. M. Palmer and C. L. Fraser, Macromol. Rapid Commun., 2015, 36, 694 CrossRef CAS.
- C. A. DeRosa, S. A. Seaman, A. S. Mathew, C. M. Gorick, Z. Fan, J. N. Demas, S. M. Peirce and C. L. Fraser, ACS Sens., 2016, 1, 1366 CrossRef CAS.
- T. Butler, A. S. Mathew, M. Sabat and C. L. Fraser, ACS Appl. Mater. Interfaces, 2017, 9, 17603 CrossRef CAS.
- W. A. Morris, M. Kolpaczynska and C. L. Fraser, J. Phys. Chem. C, 2016, 120, 22539 CrossRef CAS.
- X. Zhang, T. Xie, M. Cui, L. Yang, X. Sun, J. Jiang and G. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 2279 CrossRef CAS.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918 CrossRef CAS.
- G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295 RSC.
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242 CrossRef CAS.
- B. Chen, X. Zhang, Y. Wang, H. Miao and G. Zhang, Chem. – Asian J, 2018 DOI:10.1002/asia.201801002.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi and J. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9872 CrossRef CAS PubMed.
- G. Zhang, S. Xu, A. G. Zestos, R. E. Evans, J. Lu and C. L. Fraser, ACS Appl. Mater. Interfaces, 2010, 2, 3069 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng and X. Liu, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728 CrossRef CAS.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 2181 CrossRef CAS.
- S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989 RSC.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling and M. Gu, Angew. Chem., Int. Ed., 2018, 57, 4005 CrossRef CAS.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang and Q. Zheng, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS.
- Z. He, W. Zhao, J. W. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef.
- X.-d. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666 RSC.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916 CrossRef CAS.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589 CrossRef CAS.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177 CrossRef CAS.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe and S. R. Forrest, Nat. Commun., 2015, 6, 8947 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS.
- P. S. Rukin, A. Y. Freidzon, A. V. Scherbinin, V. A. Sazhnikov, A. A. Bagaturyants and M. V. Alfimov, Phys. Chem. Chem. Phys., 2015, 17, 16997 RSC.
- R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef.
- S. Salman, J.-L. Bredas, S. R. Marder, V. Coropceanu and S. Barlow, Organometallics, 2013, 32, 6061 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08346k |
| This journal is © The Royal Society of Chemistry 2019 |