
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyaluronan derived nanoparticle for simvastatin delivery: evaluation of simvastatin induced myotoxicity in tissue engineered skeletal muscle†
Julia M.
Jones
ab,
Darren J.
Player
 a,
Sumanta
Samanta
c,
Vignesh K.
Rangasami
c,
Jöns
Hilborn
d,
Mark P.
Lewis
b,
Oommen P.
Oommen
*c and
Vivek
Mudera
a
a,
Sumanta
Samanta
c,
Vignesh K.
Rangasami
c,
Jöns
Hilborn
d,
Mark P.
Lewis
b,
Oommen P.
Oommen
*c and
Vivek
Mudera
a
aDivision of Surgery and Interventional Science, University College London, London, UK
bSchool of Sport, Exercise and Health Sciences, Loughborough University, Leicestershire, UK
cBioengineering and Nanomedicine Lab, Faculty of Medicine and Health Technology & BioMediTech Institute, Tampere University, 33720 Tampere, Finland. E-mail: oommen.oommen@tuni.fi
dDepartment of Chemistry, Ångström Laboratory, Uppsala University, 75121 Uppsala, Sweden
First published on 1st November 2019
Abstract
Statins are currently the most prescribed hypercholesterolemia-lowering drugs worldwide, with estimated usage approaching one-sixth of the population. However, statins are known to cause pleiotropic skeletal myopathies in 1.5% to 10% of patients and the mechanisms by which statins induce this response, are not fully understood. In this study, a 3D collagen-based tissue-engineered skeletal muscle construct is utilised as a screening platform to test the efficacy and toxicity of a new delivery system. A hyaluronic acid derived nanoparticle loaded with simvastatin (HA-SIM-NPs) is designed and the effect of free simvastatin and HA-SIM-NPs on cellular, molecular and tissue response is investigated. Morphological ablation of myotubes and lack of de novo myotube formation (regeneration) was evident at the highest concentrations (333.33 μM), independent of delivery vehicle (SIM or HA-SIM-NP). A dose-dependent disruption of the cytoskeleton, reductions in metabolic activity and tissue engineered (TE) construct tissue relaxation was evident in the free drug condition (SIM, 3.33 μM and 33.33 nM). However, most of these changes were ameliorated when SIM was delivered via HA-SIM-NPs. Significantly, homogeneous expressions of MMP2, MMP9, and myogenin in HA-SIM-NPs outlined enhanced regenerative responses compared to SIM. Together, these results outline statin delivery via HA-SIM-NP as an effective delivery mechanism to inhibit deleterious myotoxic side-effects.
1. Introduction
Due to poor solubility and toxic side effects, various drugs fail to deliver their full therapeutic potential.1 Drugs are formulated using surfactants, synthetic polymers or other amphiphilic molecules that indirectly hamper the efficacy and potency of the compound. In addition, several synthetic polymers and excipients used for drug delivery applications elicit undesired immune activation, significantly limiting their clinical translation.2 Therefore, delivery systems that could enhance the solubility, bioavailability and mitigate potentially toxic side effects are required.3Statins are the most prescribed hypercholesterolemia-lowering drug worldwide.4–7 According to the prediction of cardiovascular risk factors more than one-sixth of the worldwide population (a billion) are now estimated to use statins.8 Simvastatin (SIM) is a subtype of lipid-lowering drugs from the statin family that has beneficial cholesterol lowering effects, through the prevention of the enzyme activity of hydroxyl-methyl glutaryl coenzyme A (HMG-CoA) reductase.5 Specifically, SIM molecules occupy the HMG binding site of the enzyme, inhibiting the synthesis of cholesterol.9 Out of the eight statins, SIM has the third highest relative lipid lowing potency and second greatest half-life in plasma for a particular potency. As such, SIM is one of the most prescribed drugs for the prevention of cardiovascular diseases.10
Although statins have positive effects on cardiovascular health, some statins are known to have adverse effects and are reported to cause myopathies that range from fatigue and muscle weakness to rhabdomyolysis; a life-threatening condition of skeletal muscle (SkM) proteolysis (also referred to as myotoxicity). The mechanisms and pathways that govern the myotoxicity related to any statin are poorly defined, however such myopathies affect nearly 1.5% to 10% of patients causing significant pain and discomfort.11 Despite high potency and low solubility,10 delivery of SIM is typically in tablet form via oral administration.12 To this end, it is necessary to develop and test new mechanisms for the delivery of SIM, which would minimise the myopathic effects of the drug.
Nanomedicine strategies allow efficient drug loading, prevent premature drug elimination by the reticuloendothelial system (RES) and block interactions with immune stimulatory cells, thereby reducing side effects.13 Engineering nanocarriers using glycosaminoglycans (GAGs) are advantageous due to being the main component of extracellular matrix (ECM), while possessing natural biocompatibility and biodegradability.14 The therapeutic potential of nanocarriers is well documented as a targeted drug delivery agent for anticancer studies.15,16 Tailoring nanocarriers using GAGs offers significant promise for cellular delivery of toxic drugs as they provide effective stealth properties and mitigate the toxic side effects mediated by the drug.17
HA is known to play an important role in muscle repair by upregulating myoblast migration, differentiation18 and enhancing the recruitment of muscle progenitor cells.19 Furthermore, HA has been well known to regulate multiple aspects of tissue repair and regeneration by modulating the activation of inflammatory cells.20 Therefore, tailoring HA-based nanocarriers offers a promising strategy for SkM applications. This inspired us to study the efficacy of HA derived nanoparticles for the delivery of SIM.
In order to study the effects of certain drugs and carriers, in vitro 3D tissue engineered (TE) constructs have been proposed as a suitable model platform, as they accurately recapitulate in vivo structures compared to traditional monolayer cell cultures.21 Designing 3D TE SkM by encapsulating muscle precursor cells (or myoblasts) within an ECM mimetic gel would provide physiologically relevant tissue model for evaluating the drug responses to human tissue.22 3D TE SkM constructs have previously been used to examine the force response to SIM treatment, using image-based motion detection technology of silicone posts.23 Although these models are very useful, they were not used to study the drug response at the molecular level. The aim of the current investigation was to study the effect of SIM on myotoxicity and evaluate the efficacy of ECM mimetic nanocarriers for SIM delivery. We successfully tailored HA derived nanocarriers for SIM delivery and demonstrated its osteoinductive efficacy in pre-osteoblast cells and investigated the effect of SIM and SIM loaded nanocarriers on inducing myotoxicity in 3D TE SkM constructs at the tissue, cellular and molecular levels.
2. Experimental section
2.1 Materials
Hyaluronic acid (HA, MW 130 kDa) was purchased from LifeCore Biomedical (Chaska, USA). Dopamine hydrochloride (DA), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), 1-hydroxybenzotriazole hydrate (HOBt), fluorescein-5-thiosemicarbazide (FTSC) were purchased from Sigma-Aldrich. Simvastatin (SIM) was purchased from Tocris bioscience (Bristol, UK). Dialysis membranes used for purification were purchased from Spectra Por-6 (MWCO 3500). All other chemicals were purchased from Sigma-Aldrich. All solvents were of analytical quality. The NMR experiments (δ scale) were recorded with Varian Mercury 300 MHz or JEOL ECZR 500 instruments, operating at 500 MHz for 1H. Spectra for all HA conjugates were recorded in D2O at 293 K.2.2 Synthesis of dopamine modified hyaluronic acid (HA-DA)
HA (1 mmol, 400 mg, 1 equivalent) was dissolved in 60 mL deionised water, to which 1 mmol (153 mg, 1 equivalent) HOBt and 1.5 mmol (285 mg, 1.5 equivalent) DA was then added. The pH of the reaction solution was adjusted to 5.5 with 1 M HCl and 1 M NaOH. Then 1 mmol EDC (192 mg, 1 equivalent) was added in 4 batches at 30 minutes interval. pH of the solution was maintained at 5.5 for 6 hours, and then allowed to stir overnight. The reaction mixture was loaded into a dialysis bag (Spectra Por-6, MWCO 3500 g mol−1) and dialysed against dilute HCl (pH = 3.5) containing 100 mM NaCl (4 × 2 L, 48 hours) and then dialysed against deionised water (2 × 2 L, 24 hours). The solution was lyophilised to obtain fluffy material. Degree of dopamine conjugation was 4.1% (with respect to the disaccharide units of HA) which was estimated by UV measurement (at pH 7.4 in PBS buffer) using the dopamine extinction coefficient of 2.67 mM−1 cm−1 at 280 nm.24 The HA-DA polymer was further characterized by 1H NMR spectroscopy (Fig. S1 in ESI†).2.3 Synthesis of dopamine modified hyaluronic acid nanoparticle (HA-D-NPs)
The conjugation of FTSC on dopamine modified hyaluronic acid (HA-DA) was carried out by carbodiimide coupling chemistry. Briefly, 0.625 mmol of HA-DA (250 mg, 1 equivalent) was dissolved in 60 mL of deionised water. Thereafter, 0.125 mmol FTSC (53 mg, 0.2 equivalent) and 0.625 mmol HOBt (95.6 mg, 1 equivalent) was dissolved in 30 mL of DMSO and added dropwise to the aqueous HA-DA solution. The reaction mixture was stirred for 30 minutes (until it becomes homogeneous, no turbidity), and the pH of the reaction mixture was adjusted to 4.7 by careful addition of 1 M NaOH. Finally, 0.315 mmol EDC·HCl (60.4 mg, 0.5 equivalent) was added in 3 batches at 30 minutes interval, and the mixture was stirred overnight. The reaction mixture was loaded into a dialysis bag (Spectra Por-6, MWCO 3500 g mol−1) and dialysed against dilute HCl (pH = 3.5) containing 100 mM NaCl (4 × 2 L, 48 hours) and then dialysed against deionised water (2 × 2 L, 24 hours). The solution was lyophilised to obtain as yellow fluffy material. This product was finally washed with methanol to remove any traces of unreacted FTSC. Degree of FTSC conjugation was 2.4% (with respect to the disaccharide units) which was estimated by UV measurement (at pH 7.4 in water) using the FTSC extinction coefficient of 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 492 nm.25 The HA-D-FTSC polymer was further characterized by 1H NMR spectroscopy (Fig. S2 in ESI†).
000 M−1 cm−1 at 492 nm.25 The HA-D-FTSC polymer was further characterized by 1H NMR spectroscopy (Fig. S2 in ESI†).
2.4 Preparation of SIM loaded nanoparticles (HA-SIM-NP)
SIM was loaded on the HA-D-NPs by reverse emulsion method. Briefly, 60 mg of HA-D-NPs was dissolved in 30 mL of DMSO. Thereafter, 6 mg of SIM was added under magnetic stirring (850 rpm) and the reaction mixture was stirred overnight. Thereafter, 60 mL of deionised water was carefully added dropwise to the DMSO solution to avoid excess heat generation and assemble the nanoparticles. The reaction mixture was subsequently loaded into a dialysis bag (Spectra Por-6, MWCO 3500) and dialysed against deionised water (5 × 2 L, 24 hours). The unloaded drug (insoluble) was recovered by filtering through a 0.45 μm filter and the filtrate was lyophilised yielding 50 mg of orange-yellow fluffy product. The drug trapped in the 0.45 μm filter was recovered by washing with methanol–water solution (50:50) (till no further increment in UV absorbance) to estimate the unloaded drug. The unloaded drug was found to be 19.6% of the total drug, which was quantified by UV measurement using SIM extinction coefficient of 15068 M−1 cm−1 at 230 nm.26
2.5 Determination of encapsulation of SIM in the NP and drug loading efficiency
For calculating the percentage of drug loading, the HA-SIM-NPs were disrupted by incubating the sample in DMSO at 1 mg mL−1 concentration at 37 °C for 24 hours. Thereafter, the soluble drug was isolated from the polymer by filtering through 0.2 μm membrane filter. The DMSO solution was freeze dried and subsequently dissolved in methanol–water solution (50:50) to estimate the percentage loading using UV measurements as described above.
2.6 Particle size and surface zeta potential measurement
The particle size distribution and surface zeta potential measurement were carried out using Zetasizer Nano ZS, Malvern. HA-SIM-NPs and HA-D-NPs were dispersed in deionised water at 0.33 mg mL−1 concentration at 25 °C and the hydrodynamic size of the nanoparticles were recorded using 10 × 10 × 45 mm disposable polystyrene cuvette at 25 °C. The surface zeta potential was subsequently measured at 25 °C using disposable folded capillary DTS1070 cells.2.7 Surface topography analysis by atomic force microscopy
Atomic Force Microscopy (AFM) topography of surface was taken using XE10 Park systems at room temperature. The AFM images were recorded by scanning in non-contact mode under air. The probe was supported on an APPNANOTM AFM cantilever (Applied NanoStructures Inc., USA, type: ACTA). The spring constant was 25–75 N m h. 10 μL of 0.1 mg mL−1 SIM loaded nanoparticles (HA-SIM-NP) in water were drop-casted on a thin glass sheet and dried in air for 2 hours. The surface roughness of the samples was determined from the data by XEI 1.7 image processing software (Park Systems, Santa Clara, USA). Different areas on the surfaces were analysed to check the consistency of the surface roughness data.2.8 Cell culture conditions for alkaline phosphatase (ALP) assay
In order to test the osteoinductive property of SIM, clonal pre-osteoblastic cell lines (MC3T3-E1) derived from new born mouse calvaria were cultured in α-MEM (Gibco, Thermo-scientific) with nucleosides and without ascorbic acid along with 10% foetal bovine serum at 37 °C in 5% CO2.2.9 Measurement of ALP assay
MC3T3-E1 cells were counted using Countess II cell counter (Life technologies) and 40000 cells were seeded in each well of a 24-well plate. The plates were incubated at 37 °C in 5% CO2 for 18 hours after which the medium was changed and fresh medium along with SIM (80 μg mL−1) was added to the wells. The nanoparticles loaded with SIM (HA-SIM-NP) were also added to the wells such that the final concentration of the drug is 80 μg mL−1. Nanoparticles without SIM (HA-D-NPs) and untreated cells were used as control. The cells were then incubated for 24 hours after which medium was changed and fresh medium added. The ALP activity was measured with the alkaline phosphatase colorimetric assay kit (ab83369, Abcam) at day 2, 4, 6, 10 and 14. Briefly, the cells were washed with PBS, trypsinised and re-suspended in 200 μl assay buffer. The cell lysates were then centrifuged at high speed for 15 minutes. The supernatant was collected and 40 μL of it was added to 96-well plate. The samples were incubated for 60 minutes at 25 °C after the addition of 40 μL of assay buffer and 50 μL of 5 mM p-nitrophenyl phosphate. The absorbance was then measured at 405 nm using an Envision PerkinElmer system.
2.10 Cell culture conditions for 3D TE muscle constructs
C2C12 myoblasts (Public Health England, sourced from the European Collection of Authenticated Cell Cultures (ECACC)) at passages 3–12 were maintained in growth medium (GM) consisting of: Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich, UK) supplemented with 20% v/v fetal calf serum (First Link Ltd, United Kingdom) and 1% v/v penicillin–streptomycin (Gibco Life Technologies, UK). All cell cultures were kept in a humidified incubator at 37 °C and 5% CO2 for the duration of the experiment.2.11 Chamber configuration
Manufactured chambers were fabricated from poly(ether ether ketone) (PEEK), a polymer that is biocompatible for use with cell cultures.41 The custom manufactured chamber has inbuilt cylindrical attachment/anchor points that are posts set within the wells (Fig. S3 in ESI†). The chamber dimensions are 10 mm × 21.5 mm × 5 mm, with the volumetric capacity of 0.5 mL. As in previous studies, these chambers were designed and tested for use within 6-multi-well plates.272.12 Engineering skeletal muscle tissue scaffolds using 3D collagen matrix
SkM TE constructs were engineered for evaluating the activity of SIM and HA-SIM-NP on differentiated aligned myotubes as seen in vivo. For this purpose, type-1 collagen hydrogels were prepared following a previously published protocol.28–31Collagen hydrogels were composed of 85% v/v Type 1 collagen (2.05 mg mL−1) (First Link, UK), 10% of 10× minimal essential media (MEM) (Gibco, Life Technologies, UK) and 5% v/v GM containing C2C12 cells at cellular density of 4 × 106 per mL. To prepare the hydrogels, collagen-MEM solution was neutralised via dropwise addition of NaOH at 5 M and then 1 M concentrations, prior to the addition of cell suspensions. Once neutralised the collagen solution containing cells remained on ice, until casting into the PEEK chambers. The homogeneous mixed seeded constructs were cast into the 0.5 mL PEEK chambers and placed in a humidified incubator at 37 °C and 5% CO2 for 15 minutes. Following polymerisation, 6 mL of GM was added to each construct. GM was replenished every 24 hours, for 4 days, at which point the medium was removed and replaced with differentiation medium (DM) consisting of DMEM supplemented with 2% v/v horse serum (Sigma-Aldrich, UK) and 1% v/v penicillin–streptomycin (Gibco Life Technologies, UK). DM was replenished at 24-hour intervals for the remaining 17 days of culture. The experimental duration was 21 days; allowing 7 days post tissue maturation for assessment of SIM on SkM gene expression and myotube morphology. Eighteen hydrogels were prepared for each concentration, derived from n = 3 experimental repeats; totalling nine hydrogels per analysis per SIM concentration/condition.
2.13 SIM & HA-SIM-NP administration and incubation
SIM, in both aqueous and HA-SIM-NP forms, was reconstituted in PBS and briefly vortexed to ensure a homogeneous solution prior to administration. The use of PBS for dilution of aqueous SIM was intended to match the delivery vehicle of HA-SIM-NP, with typical agents; DMSO and ethanol eliciting deleterious effects to the HA-NP. Concentrations of aqueous SIM and HA-SIM-NP were categorized as high (333.33 μM), intermediate (3.33 μM) and low (33.3 nM) with respect to the total delivered drug content. To obtain the above listed concentrations, SIM (5 mg) was diluted in 1.195 mL to obtain a 10 mM stock prior to being serially diluted 1 in 10 to obtain further stocks of 1 mM, 100 μM, 10 μM and 1 μM. Final concentrations were then obtained by adding 200 μL of each stock to 6 mL of DM to simulate bolus drug delivery. Drug-free nanoparticles (HA-D-NPs), were used as a negative control. A further control of standard HA (333.33 μM) was included, to account for the effect of this naturally occurring in the ECM. Constructs were administered with drug conditions for 24 hours on day 14, prior to a further 6 days in culture investigate the efficacy of SIM delivery mechanisms. 21 day no treatment controls were also included for separately both SIM and HA-SIM-NP, to account for increased SkM construct maturation across time.2.14 Cell viability alamarBlue® assay
Cellular viability, indicative of myotoxicity, was measured using an alamarBlue® assay. A 10% (v/v) alamarBlue® solution within un-supplemented medium (89% DMEM and 1% penicillin–streptomycin) was added to each well at day 21 and incubated for 4 hours. 100 μl of solution was added in triplicate to black-well 96 multi-well plates (Nunc Ltd, United States of America). Absorbance was measured using a Varioskan Fluorscan (Thermo-Scientific, United State of America) with excitation at 540–570 nm and emission at 580–610 nm. Cell metabolic activity for all conditions were normalised to their internal control for that experiment. Extrapolated data of different concentrations were normalised to their non-drug control by subtraction of mean control value to remove possible background interference.2.15 Fluorescent staining
At experimental termination, constructs were fixed using 3.7% paraformaldehyde for ≥1 hour. Constructs were washed three times in 1× tris-buffered saline (TBS), prior to being permeabilised via addition of 0.2% v/v Triton X-100 (Thermo Fisher Scientific, United Kingdom) solution diluted in TBS for 2 hours. Constructs were then incubated overnight within rhodamine-phalloidin (Life Technologies, United States of America) diluted 1:200 v/v in TBS. The following day, constructs were washed three times with 1× TBS before addition of 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, United States of America) diluted 1:2000 v/v in TBS for ≥30 minutes. Following a final three washes with 1× TBS, constructs were placed on polylysine-coated microscope slides (VWR, United Kingdom) and mounted to a coverslip using Fluoromount™ (Sigma-Aldrich, USA) mounting medium.
2.16 Microscopic & macroscopic images
Images of fluorescently stained TE SkM constructs were obtained using a Confocal Microscope (Zeiss LSM 880, Carl Zeiss, Germany) using a 40× oil immersion objective. Sets of 12 random images were taken of myotubes within the constructs of each concentration for each condition. Macroscopic images of whole constructs within their chambers were taken throughout the experiment to determine hydrogel deformation (Fig. 6).2.17 Image analysis of seeded collagen skeletal muscle construct
All images (micro and macroscopic) were processed using FIJI Software by Image J (NIH, Bethesda, MD). The following list of measurements were taken (known in this paper as the myotube index): myotube width, length, fusion index, number of myotubes, cell density and the number of nuclei per myotube. Only three are presented and discussed below, as these measures were determined to be most relevant: myotube width, fusion index and number of myotubes. Myotube classification was determined when a single elongated membrane structure contained 3 or more nuclei.32,33 Irregular masses, clumps (myo-sacs) or multi-branched aggregate conformations (dysmorphic myotubes), with three or more nuclei were not counted as myotubes. Most myotubes were aligned, to the uniaxial isometric lines of strain within the gel. An average of 10 measurements enabled an average myotube diameter to be calculated.34,35 The fusion index was calculated as the number of nuclei residing within myotubes expressed as a percentage value for the total number of nuclei per image frame.362.18 RNA extraction and real-time polymerase chain reaction (RT-qPCR)
3D TE SkM constructs for all treatment types were detached from their anchor points and transferred to sterile 1.5 mL microcentrifuge tubes containing 500 μl of TRI Reagent (Sigma-Aldrich, United States of America) prior to being snap frozen in liquid nitrogen. The homogenization process (maximal shear) was achieved using agitation via needle (23/21G) and syringe. RNA was extracted using the TRIzol method, according to manufacturer's instructions (Sigma). RNA concentration and purity were obtained by UV spectroscopy at ODs of 260 and 280 nm using a Nanodrop 2000 (Fisher, Rosklide, Denmark). All RNA samples were analysed in duplicate. Twenty nanograms of RNA were used per RT-PCR reaction for RPII-β, Myogenin, MMP2 and MMP9 (the primers used for the estimation of mRNA expression is provided in Table S1 in ESI†).RT-qPCR amplifications were carried out using Power SYBR Green RNA-to-CT 1 step kit (Qiagen) on a ViiATM Real-Time PCR System (Applied bio-systems, Life Technologies), analysed using ViiATM 7RUO Software. RT-qPCR procedure was as follows: 50 °C, 10 min (for cDNA synthesis), 95 °C, 5 min (transcriptase inactivation), followed by 95 °C, 10 s (denaturation), 60 °C, 30 s (annealing/extension) for 40 cycles. Relative gene expressions were calculated using the comparative CT (ΔΔCT) equation for normalised expression ratios; relative expression calculated as 2-ΔΔCT, where CT is representative of the threshold cycle. RPII β was used as the housekeeping gene in all RT-PCR assays. To compare conditions, one SkM control sample at day 21 was used as the calibrator condition in the CT (ΔΔCT) equation. RT-PCR data is presented as relative gene expression level, determined by the ΔΔCT equation.52
2.19 Statistical analysis
Mauchly's test of sphericity and Shapiro–Wilk tests were used to confirm homogeneity of variance and normal distribution of data respectively. Where parametric assumptions were met, one-way factorial analysis of variance (ANOVA) were performed for myotube Index. Two-way ANOVA were used for construct deformation and alamarBlue® cellular viability using GraphPad Prism software V6 (GraphPad Software Inc., United States of America). These tests were used to determine if statistical differences existed between the two different delivery methods of aqueous and HA-SIM-NPs delivery, where significance between individual conditions were determined using Bonferroni post-hoc analysis. All data is reported as mean ± standard deviation (SD) per condition at day 21 unless otherwise stated. Significance was assumed at P ≤ 0.05.3. Results and discussion
3.1 Synthesis of hyaluronan derived nanoparticles
It is well characterised that SIM has low solubility,12 a bioavailability of less than 5%,37 and is also known to exhibit adverse myotoxicity.38 Therefore, we engineered an ECM derived nanocarrier that could enhance the solubility and preserve bioactivity, allowing us to examine the role of the drug and the delivery mechanism on myotoxicity. For this purpose, polymeric HA-based nanoparticles were synthesised by inducing HA amphiphilicity via conjugating non-toxic aromatic fluorescein and dopamine molecule using carbodiimide chemistry (Scheme 1). We hypothesised that hydrophobic fluorescein and dopamine molecules would synergistically stabilise the hydrophobic SIM drug, via van der Waals interactions. Such formulations (HA-DA-FTSC or HA-conjugate NPs) have previously been shown to contribute to enhanced cell adhesion, proliferation and viability, demonstrating suitable ability in biological applications.39–41 | ||
| Scheme 1 Schematic representation of the synthesis and self-assembly of SIM laoded HA nanoparticles. | ||
The degree of chemical modification of fluorescein and dopamine in HA-D-FTSC were 2.4% and 4.1% respectively, as determined by ultraviolet (UV) spectroscopy at pH 7.4 within PBS buffer (Fig. 1a). The percentage of SIM loading on the HA-SIM-NP was estimated to be 8% by weight (corresponding to 80% drug loading with respect to the feed ratio) as determined by UV spectroscopy. The loading was further confirmed by recovering the unloaded insoluble SIM (∼20%) by filtering the dialysed reaction mixture with 0.45 μm filters. The HA-SIM-NP morphology and size were analysed via AFM (Fig. 1b). The AFM surface topology assessment outlined that HA-SIM-NPs were spherical in nature, at 200–300 nm in size. Subsequently, we estimated the hydrodynamic size of HA-D-NPs and HA-SIM-NP by dynamic light scattering (DLS) experimentation, found to be 661 nm and 280 nm respectively (Fig. 2a and b). The addition of the hydrophobic SIM drug into the HA-D-NPs augmented the amphiphilicity, resulting in shrinkage and decreases in size (Fig. 2b). Thereafter, an estimation of the net surface charge of the particle was measured using the zeta potential (δ). The δ of HA-D-NPs and HA-SIM-NP were found to be −28.4 mV and −25.6 mV respectively (Fig. 2c and d), suggesting that high net negative charge on the particles promoted high electrostatic repulsion between nanoparticles. This ensures efficient stabilisation and prevents aggregation of the NPs upon lyophilisation.39
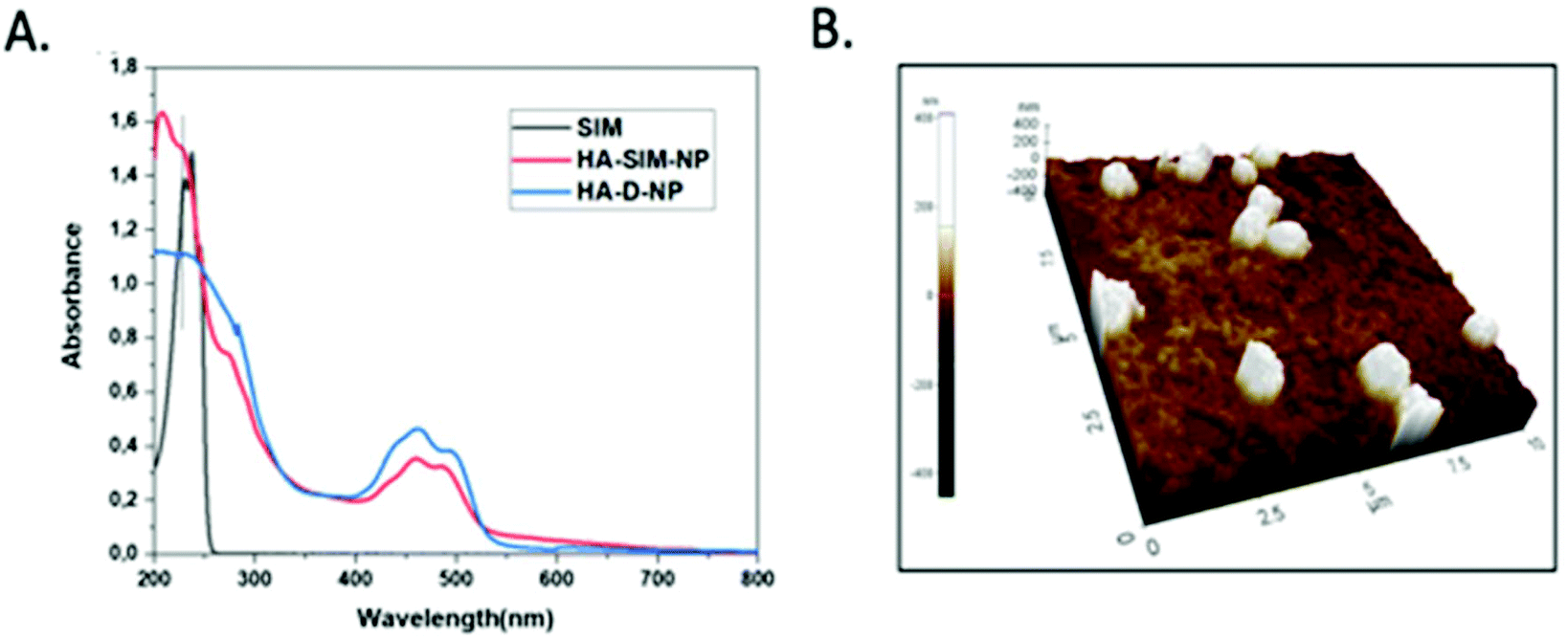 | ||
| Fig. 1 (a) UV-VIS spectrum of SIM, HA-D-NPs and HA-SIM-NPs recorded in water at 25 °C. (b) AFM image showing the spherical topology of HA-SIM-NP in the range of 200–300 nm. | ||
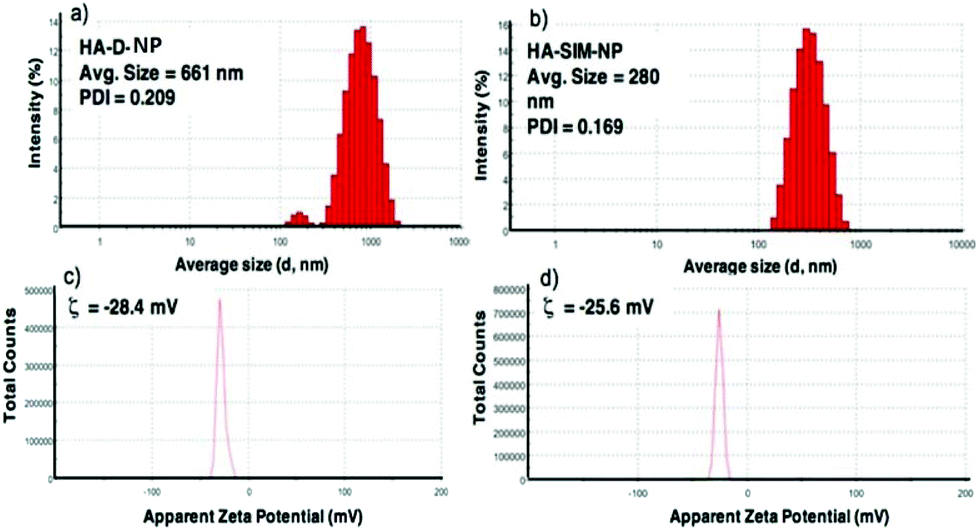 | ||
| Fig. 2 The hydrodynamic size of (a) HA-D-FTSC and (b) HA-SIM-NPs and zeta potential of (c) HA-D-FTSC and (d) HA-SIM-NP as determined at 25 °C in water by dynamic light scattering measurements using Malvern's Zetasizer NanoZS. | ||
3.2 Hyaluronan derived nanoparticles delay osteogenic differentiation of MC3T3-E1 cells
SIM has previously been demonstrated to stimulate osteogenic differentiation of stem cells.42 Therefore, to test the functional activity of SIM loaded in HA-SIM-NPs, osteogenic differentiation experiments using clonal pre-osteoblastic cell lines (MC3T3-E1) were conducted. MC3T3-E1 cells were incubated with 80 μg mL−1 (∼190 μM) of the aqueous SIM drug (dissolved in 70% ethanol and reconstituted in cell culture medium) or the drug equivalent of HA-SIM-NPs (dissolved in medium). The rate of differentiation was evaluated by measuring the early osteoblast marker, alkaline phosphatase (ALP) at different time-points.43 No differences were observed at the early time points (day 2 and 6) however, the aqueous drug delivery promoted higher ALP expression than HA-SIM-NPs (Fig. 3) after 14 days in culture. As the differentiation of MC3T3-E1 cells to osteoblasts proceeds slowly, it is reasonable to speculate that the lower expression of ALP by HA-SIM-NP may be due to slower release of the drug when compared with free SIM.44 Nevertheless, the osteoblastic differentiation of MC3T3-E1 by HA-SIM-NPs demonstrates release of active drug from the nanoparticles.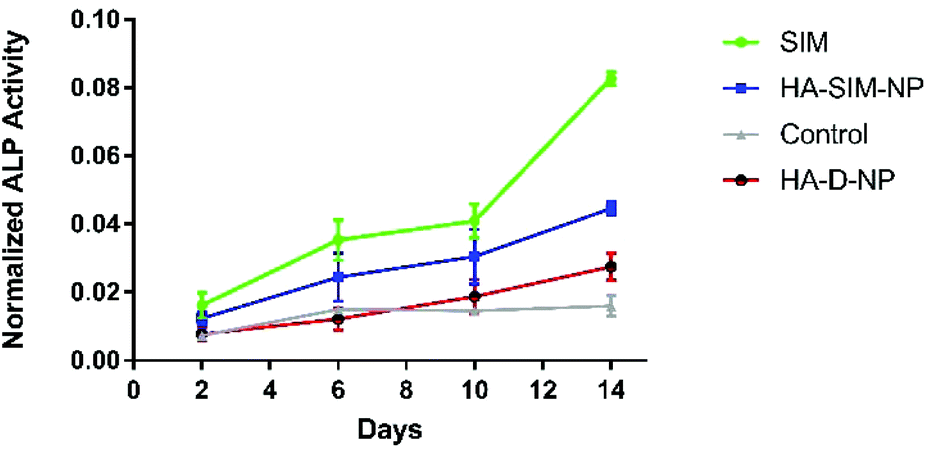 | ||
| Fig. 3 ALP activity: The figure depicts the ALP activity due to effect of the free aqueous drug (SIM), nanoparticles loaded with drug at equivalent concentration (HA-SIM-NPs) and nanoparticles without SIM (HA-D-NP), compared with respect to untreated control MC3T3-E1 cells. | ||
3.3 Hyaluronan derived nanoparticles inhibit simvastatin induced myopathy
After establishing the functional activity of SIM in HA-SIM-NPs via osteoblastic differentiation of MC3T3-E1 cells, the efficacy of HA-SIM-NP as a delivery mechanism for SIM was evaluated within a TE SkM model, compared to standard aqueous SIM administration. SIM administration altered the morphological appearance of myotubes cultured in a 3D collagen matrix in a dose response fashion, regardless of delivery mechanism at high concentrations (Fig. 4). The sensitive nature of the dose dependent morphological disruption observed within this work (following SIM and/or HA-SIM-NP treatment), demonstrates that the TE SkM model utilised is a viable method for the investigation of mechanisms that contribute toward stain induced myopathy.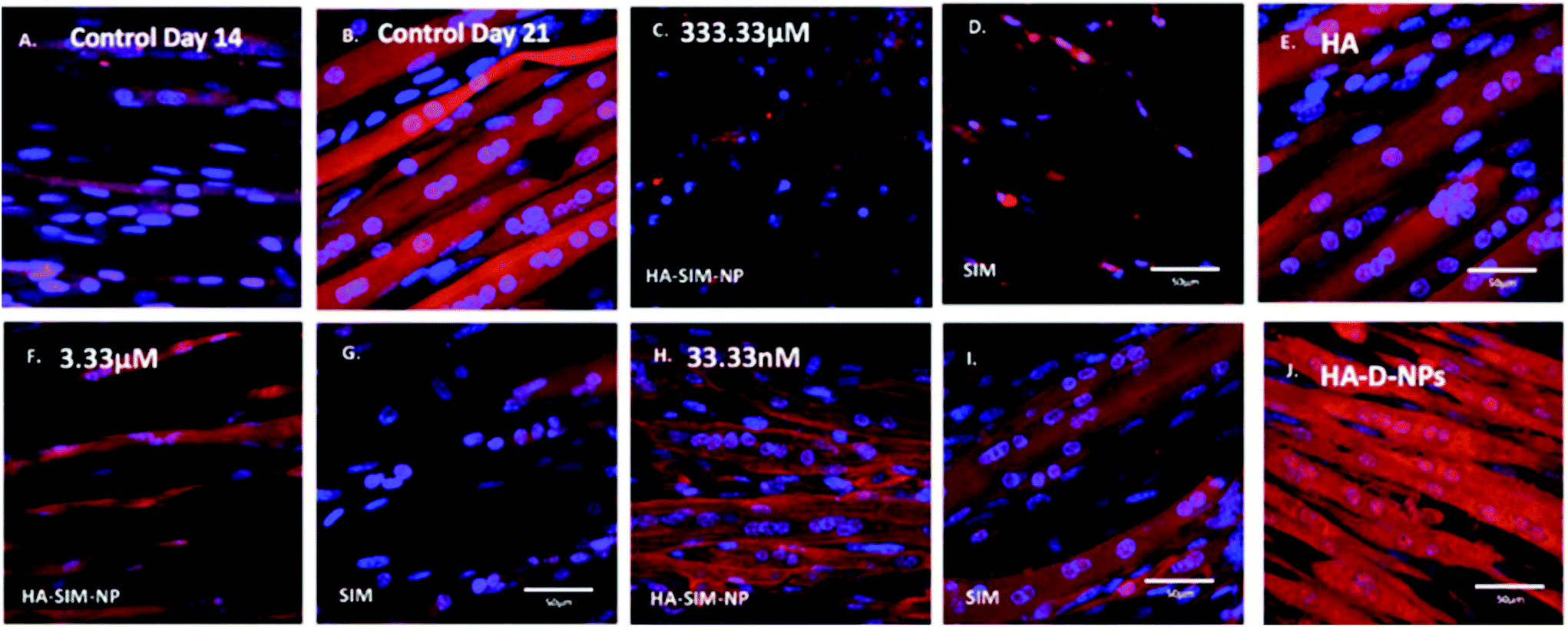 | ||
| Fig. 4 Morphological staining of actin cytoskeleton (red) and nucleic DNA (blue) of tissue engineered skeletal muscle 6 days after aqueous (SIM) or nanoparticle (HA-SIM-NP) delivery of simvastatin. A.–B. controls at days 14 and 21 respectively. C., F., H., are HA-SIM-NPs. D., G., I., are the free aqueous SIM constructs. Doses: C.–D. 333.33 μM. F.–G. 3.33 μM. H.–I. 33.33 nM. E.–J. are positive (HA) and neutral (HA-D-NPs) controls respectively, as explained in the methods delivered at the highest dose. Images show clear distinction of myotube degradation at the higher doses 333.33 μM to 3.33 μM, with progressive myotube preservation towards the lower doses (33.33 nM). Scale bars = 50 μm. | ||
The highest concentration (333.33 μM) in both conditions resulted in degradation of myotubes and physical myotube disruption (i.e. 6 membrane fragments and compromised nuclei). This was outlined with significant decreases in morphological parameters of fusion, myotube width and number compared to control conditions (Fig. 6, P ≤ 0.0005). The definitive mechanism for statin-related myotube disruption is yet to be fully elucidated, however, contributing factors are hypothesised to include apoptosis and/or necrosis, that result in the disruption of the cytoskeleton.45,46 The dose response observed in this study further coincides with monolayer studies that found high statin concentrations elicited impaired morphological myotube phenotypes (shorter or thinner myotubes). In addition, cellular degradation, changes in cell/myotube contour, cytoplasmic vacuolation, or disruption and/or loss of myotubes were also noted,4,47 in a concentration dependent manner. Two independent monolayer studies,48,49 further support this assertion, using fluorescent based cellular morphology change to ascertain apoptosis of statin treated L6 myoblasts (including SIM). Here, nuclei fragmentation was observed after treatment with high doses of 10 mM fluvastatin or SIM for 2 days. Utilising a 3D TE SkM, further work demonstrated decreases in active force with 1–3 days incubation at high atorvastatin (stronger in its effect than SIM) concentrations (2.5–25 μM) and 3–5 days at lower levels (0.01 μM).23
Significantly, reductions in both width and myotube number observed in 3.33 μM doses were negated when SIM was delivered via hyaluronan nanoparticles (HA-SIM-NPS, P ≥ 0.05), however aqueous SIM delivery continued to inhibit fusion and impair morphological indices of width and number 6 days post drug administration. This data would appear to outline a protective capacity of drug administration within HA-SIM-NPs. HA has been reported amongst the literature as a positive promoter of myotube development,18 which correlates with the significant increases in myotube width (P ≤ 0.0005) and comparative fusion of nuclei to control conditions. As such, it is evident that HA-SIM-NP, as novel nanoparticle drug carriers, contribute to a HA induced hypertrophic myotube phenotype and/or slower drug release kinetics that are sufficient to counteract SIM induced myopathy at physiologically significant intermediate doses (3.33 μM).
3.4 Simvastatin reduces metabolic activity of murine tissue engineered skeletal muscle in a dose-dependent manner
Cell viability data (Fig. 5) outlined a significant decrease in metabolic activity (P ≤ 0.05) across all aqueous SIM concentrations compared to its respective control. However, when delivered via HA-SIM-NPs treatment, significant decreases were only observed in the high (333.33 μM) and intermediate (3.33 μM) concentrations in comparison to control (P ≤ 0.0001 and P = 0.0009 respectively). As such, the comparable levels of metabolic activity in SkM hydrogels treated with the lowest concentration (33.33 nM) of HA-SIM-NPs compared to the control appears to indicate a protective capacity of this delivery mechanism and that this concentration is non-toxic to the SkM myotubes. Furthermore, HA-SIM-NPs appeared to inhibit SIM induced reductions in metabolic activity at intermediate (3.33 μM, P = 0.333) and high (333.33 μM, P ≤ 0.0001) concentrations. Together, this would indicate that the incorporation of SIM with nanoparticle formulations that elicit the slow release of this drug, in addition to the presence of HA, may provide an effective novel alternative for the delivery of statins to reduce myopathic side effects. Cytotoxicity assays have previously been used to investigate the viability of skeletal muscle cell lines (RD and L6) after incubation with various concentrations of statins.47,50,51 Here, decreases in cell metabolism based on the redox (metabolic) activity of the simvastatin drug was observed. Further work has also outlined the significance of simvastatin's action in the reduction of cell metabolism, predominantly in a dose dependent manner.52 SIM induced myopathy is arguably linked to its accumulation in muscle tissue in vivo,53 due to low solubility in aqueous solutions or tablet form via oral administration.12 Therefore, increasing the solubility, bioavailability and release rate of this compound via HA-SIM-NPs may aid hepatic liver metabolism and consequentially reduce myotoxic side effects via reduced concentrations reaching the SkM tissue.33,38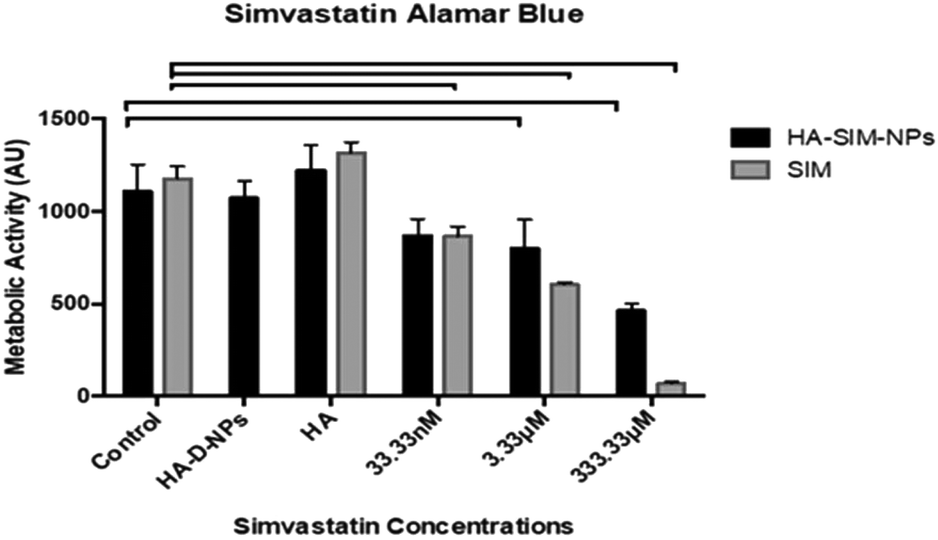 | ||
| Fig. 5 Cell viability of tissue engineered skeletal muscle constructs 6 days post aqueous (SIM) or nanoparticle (HA-SIM-NP) simvastatin delivery. Significance is indicated by notation bars that link an appropriate concentration to control (P < 0.05). All data reported as mean ± SD derived from n = 3 constructs from 3 independent repeats for each condition. | ||
 | ||
| Fig. 6 Graphical representation of the morphological analysis for simvastatin delivery in nanoparticle (HA-SIM-NP) and aqueous form (SIM) at 333 μM, 3.33 μM and 33.3 nM doses, in addition to simvastatin free nanoparticles (HA-D-NPs) and hyaluronic acid (HA) controls. All data reported as mean ± SD derived from n = 3 constructs from 3 independent repeats for each condition. Significance to control *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.0005. Asterisks donate significance in both conditions compared to respective control groups unless specifically stated (NP or SIM). | ||
3.5 Simvastatin administration elicits delivery dependent myotube/extracellular matrix interaction
Alterations in the structural integrity or biochemical environment of SkM can affect the cellular interactions that govern the surrounding ECM. As such, analysing the rate at which TE constructs reduce in size in response to longitudinal tension (deformation) via matrix remodelling, can afford an insight into the extent to which SIM treatment affects myoblast-matrix interactions. To determine whether relaxation and subsequent re-expansion of the 3D collagen matrix occurs in response to SIM administration, only the highest dose (333.33 μM) was utilised due to the significant effect on SkM morphology in this concentration. All constructs were analysed across time from day 14 (day of drug administration), with further measurements being taken at 18 days and experimental end point (day 21; Fig. 7). Construct matrix relaxation was apparent immediately post drug administration (day 14) and at day 18 when treated with SIM. Although this effect appeared reduced when delivered via HA-SIM-NPs, no statistical significance between delivery vehicles or control conditions were observed. At experimental termination time-points (day 21) delivery of aqueous SIM had, however, elicited a significant decrease in construct stiffness compared to control (P = 0.0061). Furthermore, the delivery of this statin within HA-SIM-NPs counteracted the apparent matrix relaxation (P = 0.0037), outlining a protective effect of this delivery agent in statin induced myopathy. This data coincides with the morphological responses observed, albeit at high doses. Morphological restoration was apparent at intermediate doses of 3.33 μM, however this data would indicate that HA-SIM-NP inhibits myotoxicity to some degree even at high doses (333.33 μM), preserving myotube/matrix interaction and passive construct tension. Alterations in macroscopic tissue remodelling (deformation), is not a typical measure reported amongst the literature regarding tissue specific responses to drugs. However, the tissue relaxation observed in this work at high doses that differs to morphological change outlines a simplistic, cost effective measure to analyse cell/matrix integrity when exposed to pharmaceutical agents. This data provides evidence that SIM administration at 333.33 μM doses effects cellular structure via loss of myotube integrity, which consequentially reduces the amount of longitudinal force generated, leading to reductions in tissue stiffness. This is further supported by reductions in cellular activity observed in comparable SIM concentrations. Delivery via HA-SIM-NPs does however reduce this effect, potentially via the presence of HA stimulated hypertrophy and aiding SkM regeneration as previously outlined.41,54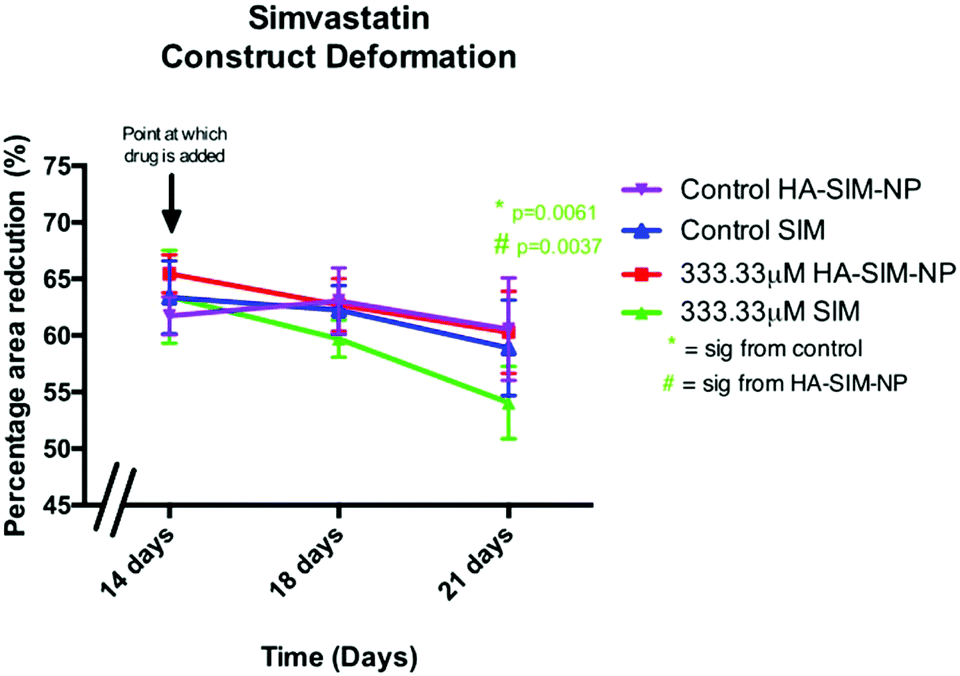 | ||
| Fig. 7 Skeletal muscle construct deformation immediately (14 days), 3 days (18 days) and 6 days (21 days) post aqueous (SIM) or nanoparticle (HA-SIM-NP) simvastatin administration at 333.33 μM doses. All data reported as mean ± SD derived from n = 3 constructs from 3 independent repeats for each condition. Significance to control *, significance between SIM and HA-SIM-NP #. | ||
3.6 Hyaluronan derived nanoparticles reduce simvastatin mediated matrix metalloproteinase gene transcription
Analysis of key myogenic and matrix metalloproteinase (MMP) remodelling mRNA further supports both the morphological and macroscopic tissue data previously presented. Being hypothesised to elicit myopathy at high doses regardless of delivery mechanism, 6 days post SIM administration was afforded to allow for SkM regeneration and matrix re-modelling. Significant increases in MMP2 mRNA at experimental termination time-points between high (333.33 μM) aqueous SIM doses and control (P ≤ 0.001) were observed. However, no difference was evident when SIM was delivered via HA-SIM-NPs. MMP2 is known within SkM to be markedly up-regulated post injury after 3 days, prior to reduced transcription at 7 days, and returning to base line by 10 days.55,56 This would indicate that high doses of SIM in both conditions likely induced myopathy, with HA-SIM-NPs eliciting a reduction in peak SkM degradation and hence returning to baseline (control) transcription levels 7 days post administration, opposed to aqueous delivery which remained elevated. Furthermore, the role of MMP2 in regeneration is centred on ECM remodelling, specifically of type IV collagen, during early phase skeletal myoblast proliferation, migration and fusion.55 This would indicate that the basal MMP2 transcription at lower doses outlines a dose dependent effect on SkM regeneration at these concentrations. Conversely, MMP9 expressions were significantly potentiated within the lowest doses only in aqueous SIM (P = 0.002) compared to control (Fig. 8). Typically, MMP9 is an early inflammation mediated modulator of ECM remodelling following injury, however has also been documented to be involved in myotube formation.57–59 This would suggest that up-regulation of this gene in the lowest doses outlines an advanced stage of regeneration. Myogenin transcription was however, enhanced only at intermediate concentrations 16.7 mM (P = 0.001). The comparable transcription in both MMP2 and MMP9, taken in combination with homogeneous myogenin expression in HA-SIM-NPs, would indicate that HA nanoparticle delivery reduced the myopathy associated with SIM and restored SkM phenotype at enhanced rates compared in aqueous SIM. This is further evident when parameters of morphological viability, tissue integrity and metabolic activity are considered, demonstrating efficacy of HA-SIM-NPs as novel delivery vehicle for cholesterol lowering statins and or other stimulants that negatively regulate SkM phenotype. Future experiments should seek to determine the physiological effects of HA-SIM-NPs delivery to SkM, through the assessment of tissue function.60 This will be fundamental to determine whether acute physiological effects are independent of observable morphological changes.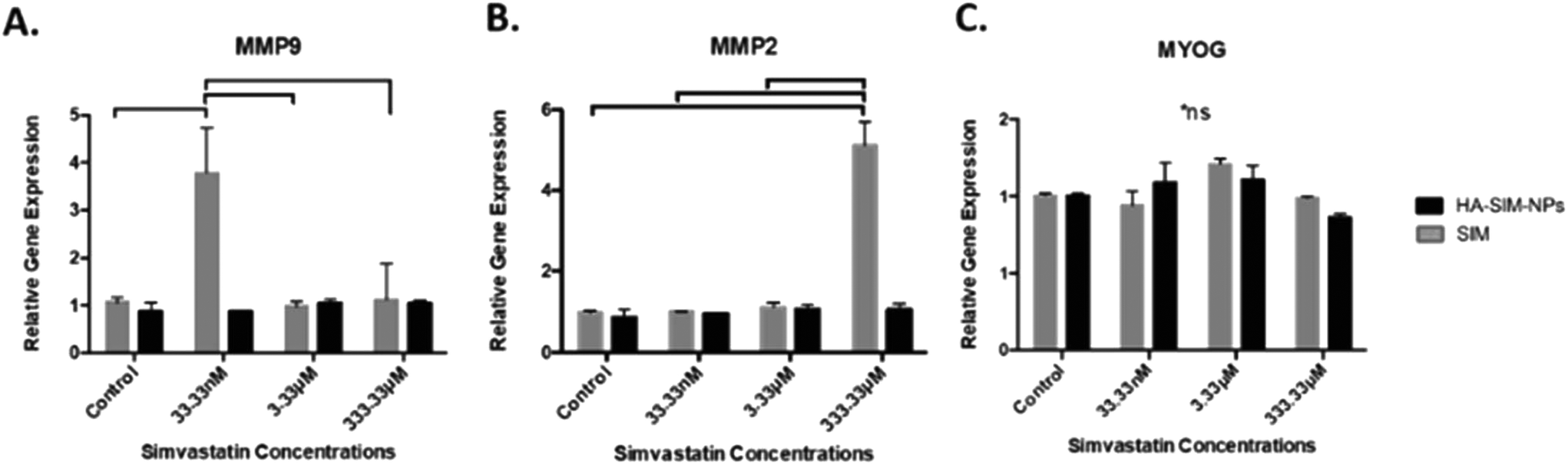 | ||
| Fig. 8 mRNA expression of matrix metalloproteinase (MMP) 2 and 9, and myogenin (MYOG) 6 days (day 21) post aqueous (SIM) or nanoparticle (HA-SIM-NP) simvastatin administration. Significance is indicated by notation bars that link an appropriate concentration to control (P < 0.05). All data reported as mean ± SD derived from n = 3 constructs from 3 independent repeats for each condition. *ns: no significance noted. | ||
4. Conclusions
Here, we have presented an in vitro 3D TE SkM screening platform, which can act as a pre-clinical model to understand the action of SIM in vitro. This was achieved using a novel delivery mechanism, whereby the toxicity of HA-SIM-NPs could be tested. In conclusion, the delivery of SIM via HA-SIM-NPs may be a more suitable way of statin administration to prevent potential side-effects, by reducing myotoxicity and loss of muscle cell integrity.Conflicts of interest
There was no conflict of interest in the execution of the experimental work nor on the submission of this manuscript.Acknowledgements
OPO conceived the project with late Prof. Robert Brown. The SkM experiments and manuscript were conducted and written by JJ; Nanoparticle characterisation and manuscript writing were VKR, SS; final editing and proof reading were conducted by DP, VM, OPO, JH and ML as well as project supervision. Authors thank EU-FP-7 BIODESIGN programme (Award No. 262948) for providing financial support to this project. SS thank EU-H2020 Marie Sklodowska-Curie BioMEP program (Award No. 713645) for financial support. Sincere gratitude to UCL's late Professor Robert Brown of whom envisaged this work long before its fruition, and of whom helped to forged shape this collaboration. Thanks to Dr Teresa Rebelo Calejo from Tampere University for assisting us with AFM. Thanks to the UCL's Dr James Phillips for his contribution of the PEEK chambers.References
- S. Stegemann, F. Leveiller, D. Franchi, H. de Jong and H. Lindén, Eur. J. Pharm. Sci., 2007, 31, 249–261 CrossRef CAS PubMed.
- S. M. Moghimi, J. Controlled Release, 2014, 190, 556–562 CrossRef CAS.
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403–416 CrossRef CAS.
- A. J. Dirks and K. M. Jones, Am. J. Physiol.: Cell Physiol., 2006, 291, C1208–C1212 CrossRef CAS.
- S. Geboers, J. Stappaerts, J. Tack, P. Annaert and P. Augustijns, Int. J. Pharm., 2016, 510, 296–303 CrossRef CAS.
- Scandinavian_simvastatin_survival_group, Lancet, 1994, 344, 1383–1389 Search PubMed.
- P. Gazzerro, M. C. Proto, G. Gangemi, A. M. Malfitano, E. Ciaglia, S. Pisanti, A. Santoro, C. Laezza and M. Bifulco, Pharmacol. Rev., 2012, 64, 102–146 CrossRef CAS PubMed.
- J. P. Ioannidis, J. Am. Med. Assoc., 2014, 311, 463–464 CrossRef CAS.
- E. Istvan, Atheroscler. Suppl., 2003, 4, 3–8 CrossRef CAS.
- J. Dulak and A. Jozkowicz, Curr. Cancer Drug Targets, 2005, 5, 579–594 CrossRef CAS.
- J. Auer, H. Sinzinger, B. Franklin and R. Berent, Eur. J. Prev. Cardiol., 2016, 23, 88–110 CrossRef.
- K. Ganesh, D. Archana and K. Preeti, Iran. J. Pharm. Res., 2015, 14, 407–415 CAS.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- V. M. Platt and F. C. Szoka, Mol. Pharm., 2008, 5, 474–486 CrossRef CAS PubMed.
- P. Oommen Oommen, J. Garousi, M. Sloff and P. Varghese Oommen, Macromol. Biosci., 2013, 14, 327–333 CrossRef PubMed.
- O. P. Varghese, J. Liu, K. Sundaram, J. Hilborn and O. P. Oommen, Biomater. Sci., 2016, 4, 1310–1313 RSC.
- D. Gurav, O. P. Varghese, O. A. Hamad, B. Nilsson, J. Hilborn and O. P. Oommen, Chem. Commun., 2016, 52, 966–969 RSC.
- V. Krenn, B. Brand-Saberi and F. Wachtler, Am. J. Anat., 1991, 192, 400–406 CrossRef CAS.
- S. Calve, S. J. Odelberg and H. G. Simon, Dev. Biol., 2010, 344, 259–271 CrossRef CAS.
- D. Jiang, J. Liang and P. W. Noble, Physiol. Rev., 2011, 91, 221–264 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CrossRef CAS.
- J. P. Mertens, K. B. Sugg, J. D. Lee and L. M. Larkin, Regener. Med., 2014, 9, 89–100 CrossRef CAS.
- H. Vandenburgh, J. Shansky, F. Benesch-Lee, V. Barbata, J. Reid, L. Thorrez, R. Valentini and G. Crawford, Muscle Nerve, 2008, 37, 438–447 CrossRef CAS.
- J. P. Evans, K. Ahn and J. P. Klinman, J. Biol. Chem., 2003, 278, 49691–49698 CrossRef CAS.
- O. P. Oommen, C. Duehrkop, B. Nilsson, J. Hilborn and O. P. Varghese, ACS Appl. Mater. Interfaces, 2016, 8, 20614–20624 CrossRef CAS PubMed.
- V. B. Mane, S. Babar and N. Kulkarni, Int. J. PharmTech Res., 2011, 3, 1459–1466 CAS.
- J. M. Jones, D. J. Player, N. R. W. Martin, A. J. Capel, M. P. Lewis and V. Mudera, Front. Physiol., 2018, 9, 483 CrossRef.
- A. S. Smith, S. Passey, L. Greensmith, V. Mudera and M. P. Lewis, J. Cell. Biochem., 2012, 113, 1044–1053 CrossRef CAS.
- R. A. Brown, M. Wiseman, C. B. Chuo, U. Cheema and S. N. Nazhat, Adv. Funct. Mater., 2005, 15, 1762–1770 CrossRef CAS.
- U. Cheema, S. Y. Yang, V. Mudera, G. G. Goldspink and R. A. Brown, Cell Motil. Cytoskeleton, 2003, 54, 226–236 CrossRef CAS.
- V. Mudera, A. S. Smith, M. A. Brady and M. P. Lewis, J. Cell. Physiol., 2010, 225, 646–653 CrossRef CAS.
- X. Ge, Y. Zhang, S. Park, X. Cong, D. E. Gerrard and H. Jiang, PLoS One, 2014, 9, e95926 CrossRef.
- B. Cadot, V. Gache, E. Vasyutina, S. Falcone, C. Birchmeier and E. R. Gomes, EMBO Rep., 2012, 13, 741–749 CrossRef CAS.
- C. C. Agley, C. P. Velloso, N. R. Lazarus and S. D. Harridge, J. Histochem. Cytochem., 2012, 60, 428–438 CrossRef CAS PubMed.
- C. Rommel, S. C. Bodine, B. A. Clarke, R. Rossman, L. Nunez, T. N. Stitt, G. D. Yancopoulos and D. J. Glass, Nat. Cell Biol., 2001, 3, 1009–1013 CrossRef CAS.
- N. R. Martin, S. L. Passey, D. J. Player, V. Mudera, K. Baar, L. Greensmith and M. P. Lewis, Tissue Eng., Part A, 2015, 21, 2595–2604 CrossRef CAS.
- M. Kato, Drug Metab. Pharmacokinet., 2008, 23, 87–94 CrossRef CAS.
- D. A. Taha, C. H. De Moor, D. A. Barrett and P. Gershkovich, Transl. Res., 2014, 164, 85–109 CrossRef CAS.
- Y. Zhu, J. Wang, X. Li, D. Zhao, J. Sun and X. Liu, Carbohydr. Polym., 2015, 123, 72–79 CrossRef CAS.
- A. I. Neto, A. C. Cibrao, C. R. Correia, R. R. Carvalho, G. M. Luz, G. G. Ferrer, G. Botelho, C. Picart, N. M. Alves and J. F. Mano, Small, 2014, 10, 2459–2469 CrossRef CAS.
- G. Tripodo, A. Trapani, M. L. Torre, G. Giammona, G. Trapani and D. Mandracchia, Eur. J. Pharm. Biopharm., 2015, 97, 400–416 CrossRef CAS.
- J. Pagkalos, J. M. Cha, Y. Kang, M. Heliotis, E. Tsiridis and A. Mantalaris, J. Bone Miner. Res., 2010, 25, 2470–2478 CrossRef CAS.
- T. Maeda, A. Matsunuma, T. Kawane and N. Horiuchi, Biochem. Biophys. Res. Commun., 2001, 280, 874–877 CrossRef CAS.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS.
- K. Yokota, F. Miyoshi, T. Miyazaki, K. Sato, Y. Yoshida, Y. Asanuma, Y. Akiyama and T. Mimura, J. Rheumatol., 2008, 35, 193–200 CAS.
- N. Ruiz-Velasco, A. Dominguez and M. A. Vega, Biochem. Pharmacol., 2004, 67, 303–313 CrossRef CAS.
- B. A. Masters, M. J. Palmoski, O. P. Flint, R. E. Gregg, D. Wangiverson and S. K. Durham, Toxicol. Appl. Pharmacol., 1995, 131, 163–174 CrossRef CAS.
- P. Kaufmann, M. Torok, A. Zahno, K. M. Waldhauser, K. Brecht and S. Krahenbuhl, Cell. Mol. Life Sci., 2006, 63, 2415–2425 CrossRef CAS.
- J. Hanai, P. Cao, P. Tanksale, S. Imamura, E. Koshimizu, J. Zhao, S. Kishi, M. Yamashita, P. S. Phillips, V. P. Sukhatme and S. H. Lecker, J. Clin. Invest., 2007, 117, 3940–3951 CAS.
- M. Kobayashi, T. Kagawa, K. Narumi, S. Itagaki, T. Hirano and K. Iseki, J. Pharm. Pharm. Sci., 2008, 11, 1–8 CAS.
- M. Kobayashi, T. Kagawa, R. Takano, S. Itagaki, T. Hirano and K. Iseki, J. Pharm. Pharm. Sci., 2007, 10, 332–339 CAS.
- J. Sacher, L. Weigl, M. Werner, C. Szegedi and M. Hohenegger, J. Pharmacol. Exp. Ther., 2005, 314, 1032–1041 CrossRef CAS PubMed.
- L. Bjorkhem-Bergman, J. D. Lindh and P. Bergman, Br. J. Clin. Pharmacol., 2011, 72, 164–165 CrossRef.
- N. Oh and J. H. Park, Int. J. Nanomed., 2014, 9(Suppl 1), 51–63 Search PubMed.
- P. J. Ferre, L. Liaubet, D. Concordet, M. SanCristobal, E. Uro-Coste, G. Tosser-Klopp, A. Bonnet, P. L. Toutain, F. Hatey and H. P. Lefebvre, Pharm. Res., 2007, 24, 1480–1489 CrossRef CAS.
- X. Chen and Y. Li, Cell Adhes. Migr., 2009, 3, 337–341 CrossRef.
- S. Kherif, C. Lafuma, M. Dehaupas, S. Lachkar, J. G. Fournier, M. Verdiere-Sahuque, M. Fardeau and H. S. Alameddine, Dev. Biol., 1999, 205, 158–170 CrossRef CAS.
- K. Fukushima, A. Nakamura, H. Ueda, K. Yuasa, K. Yoshida, S. Takeda and S. Ikeda, BMC Musculoskeletal Disord., 2007, 8, 54 CrossRef PubMed.
- G. Lluri and D. M. Jaworski, Muscle Nerve, 2005, 32, 492–499 CrossRef CAS.
- A. J. Capel, R. P. Rimington, J. W. Fleming, D. J. Player, L. A. Baker, M. C. Turner, J. M. Jones, N. R. W. Martin, R. A. Ferguson, V. C. Mudera and M. P. Lewis, Front. Bioeng. Biotechnol., 2019, 7, 20 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00986h |
| This journal is © The Royal Society of Chemistry 2020 |