
Topology-assisted, photo-strengthened DNA/siRNA delivery mediated by branched poly(β-amino ester)s via synchronized intracellular kinetics†
Shanzhou
Duan
a,
Desheng
Cao
b,
Xudong
Li
a,
Huifang
Zhu
*c,
Min
Lan
a,
Zhengzhong
Tan
d,
Ziyuan
Song
 d,
Rongying
Zhu
a,
Lichen
Yin
*b and
Yongbing
Chen
*a
d,
Rongying
Zhu
a,
Lichen
Yin
*b and
Yongbing
Chen
*a
aDepartment of Thoracic Surgery, the Second Affiliated Hospital of Soochow University, Suzhou 215004, P.R. China. E-mail: chentongt@sina.com
bJiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Institute of Functional Nano and Soft Materials (FUNSOM), Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou 215123, P.R. China. E-mail: lcyin@suda.edu.cn
cAnalysis and Testing Center, Soochow University, Suzhou, 215123, P.R. China. E-mail: hfzhu@suda.edu.cn
dDepartment of Materials Science and Engineering, University of Illinois at Urbana-Champaign, 1304 W Green St, Urbana 61801, IL, USA
First published on 4th October 2019
Abstract
The performance of non-viral gene delivery vehicles, especially cationic polymers, is often challenged by the multiple cellular barriers that pose inconsistent requirements for material properties. A most pronounced inconsistency is exemplified by the molecular weight (MW)-related transfection efficiency and cytotoxicity. In this study, we report the development of photo-degradable, branched poly(β-amino ester)s (BPAE-NB) to realize efficient and photo-controlled DNA and siRNA delivery. BPAE-NB possessing built-in light-responsive 2-nitrobenzene moieties in the polymer backbone was synthesized via the A2 (amine) + B3 (triacrylate) + C2 (diacrylate) polycondensation reaction from 4-amino-1-butanol (A2), trimethylolpropane triacrylate (B3), and (2-nitro-1,3-phenylene)bis(methylene) diacrylate (NPBMDA, C2). The highly branched BPAE-NB with the multivalent arrangement of cationic groups provides stronger nucleic acid binding capacity than its linear analogue LPAE-NB, and thus features stronger trans-membrane gene delivery capabilities and higher transfection efficiencies. Upon UV light irradiation, the backbone of BPAE-NB can quickly degrade into low-MW fragments as a consequence of the cleavage of the light-responsive 2-nitrobenzene, thus promoting intracellular gene release and diminishing the toxicity of materials at the post-transfection state. As such, in multiple mammalian cells, BPAE-NB exhibited remarkably higher DNA/siRNA transfection efficiency yet lower cytotoxicity than its non-responsive analogue BPAE-CC upon light irradiation, notably outperforming commercial reagents PEI 25k and Lipofectamine 2000. This study therefore provides an effective topology- and photo-controlled approach to precisely manipulate the transfection efficiency and toxicity of polycationic gene vectors, and may also provide promising additions to the existing non-viral gene delivery vectors.
Introduction
Gene therapy is an effective paradigm for the treatment of various genopathies, and rectifies cellular functions by delivering therapeutic nucleic acids into the target cells.1–8 In the past two decades, a variety of gene delivery materials have been developed to realize effective gene transfection. Among them, non-viral gene vectors have attracted broad attention in both fundamental research and clinical application, because of their low immunogenicity, low oncogenicity, desired biocompatibility, and simple synthesis.9–13 Cationic polymers (also termed polycations) are widely explored non-viral gene delivery materials, because they can condense the negatively charged nucleic acids into nano-sized polyplexes via electrostatic interactions to enable trans-membrane gene delivery.14–17 However, the full performance of polycationic vectors is often hampered by the multiple cellular barriers that pose inconsistent requirements for material properties. A most pronounced inconsistency is exemplified by the molecular weight (MW)-related transfection efficiency and cytotoxicity.18–23 Particularly, polycations with a higher MW normally feature higher nucleic acid condensation capability and trans-membrane delivery efficiency, while in the meantime, impede intracellular nucleic acid release to inhibit effective gene transfection. Additionally, polycations with a higher MW induce stronger cytotoxicity than the low-MW analogues.24–27 Thus, the development of polycations that can synchronize these inconsistencies is of great importance towards effective gene therapy.Linear poly(β-amino ester)s (LPAEs) are an important category of polycationic gene delivery materials with hydrolyzable ester bonds in their main chains, first reported by Lynn and Langer in 2000.28 They can be synthesized via a facile yet versatile polycondensation reaction between diacrylates and amine molecules. By tailoring the monomer structures and end groups, researchers have constructed a huge library of structurally diverse LPAEs, and upon high-throughput screening, optimal structures are identified to enable efficient gene delivery to varieties of mammalian cells.29–31 Hydrolysis of the ester bonds allows the LPAEs to degrade and release gene cargoes in the cells, and meanwhile, contributes to the desired biocompatibility.32–34 However, such hydrolysis is non-specific and uncontrollable, making it unlikely to manipulate the gene release profile in a spatiotemporal manner. For instance, the pre-release of gene cargoes outside the cells will hamper the efficient cellular internalization, while retarded release in the cytoplasm will also impair the transfection process.35 Another shortcoming of LPAEs is their relatively low cationic charge density that mainly originates from a tertiary amine on the backbone. Thus, they suffer from relatively low binding affinity for nucleic acids, especially small interfering RNA (siRNA) with a linear, short, and rigid structure.32 In order to achieve sufficient siRNA condensation and efficient siRNA transfection, high polymer/siRNA weight ratios up to 150 are often required, which may induce additional material toxicity.36 Therefore, it is highly necessary to design poly(β-amino ester)s with potent gene condensation capacity and precise gene release profiles.
To address such a dilemma, we herein designed and synthesized a new type of photo-degradable, branched poly(β-amino ester)s (BPAE-NB) for efficient and photo-controlled DNA and siRNA delivery. BPAE-NB containing built-in light-responsive 2-nitrobenzene moieties in the polymer backbone were synthesized via the A2 (amine) + B3 (triacrylate) + C2 (diacrylate) polycondensation reaction from 4-amino-1-butanol (A2), trimethylolpropane triacrylate (B3), and (2-nitro-1,3-phenylene)bis(methylene) diacrylate (NPBMDA, C2).37–43 We hypothesize that the highly branched polymer with the multivalent arrangement of cationic groups would provide stronger nucleic acid binding capacity, thus forming compact polyplexes to promote trans-membrane gene delivery. In addition, the backbone of BPAE can quickly degrade into low-MW fragments upon UV light irradiation as a consequence of the cleavage of the light-responsive 2-nitrobenzene, thus promoting intracellular gene release to potentiate gene transfection and diminishing the toxicity of materials at the post-transfection state.44,45 Such a photo-manipulating strategy of gene delivery processes features easy maneuverability and high spatiotemporal precision, which can be realized by controlling the light irradiation site, dose, and time. The critical roles of branched topology in mediating DNA/siRNA delivery were demonstrated by comparing branched BPAE-NB with the linear LPAE-NB, and the photo-strengthened DNA/siRNA transfection was mechanistically explored by comparing the responsive BPAE-NB with the non-responsive analogue BPAE-CC in terms of polymer degradation, intracellular DNA/siRNA release, transfection efficiencies in multiple mammalian cell types, and material toxicity.
Experimental
Materials and cell lines
All chemicals and solvents were purchased from Energy Chemical (Shanghai, China). DNA plasmids encoding luciferase (pLuc) or enhanced green fluorescence protein (pEGFP) were purchased from Elim Biopharm (Hayward, CA, USA). Lipofectamine 2000 (LPF), YOYO-1, Hoechst 33258, and Lysotracker Red were purchased from Invitrogen (Carlsbad, CA, USA). Survivin siRNA (siSur) and negative control siRNA (siNC) were purchased from GenePharma (Shanghai, China), and their sequences are shown in Table S1.† Primers were purchased from Sangon Biotech (Shanghai, China), and their sequences are shown in Table S2.†HeLa (human cervix adenocarcinoma cells), COS-7 (African green monkey kidney cells), and 3T3-L1 (mouse embryonic fibroblast) cells were purchased from the American Type Culture Collection (Rockville, MD, USA) and were cultured in DMEM containing 10% fetal bovine serum (for HeLa and COS-7 cells) or 10% bovine calf serum (for 3T3-L1 cells).
Synthesis of LPAE-NB
The UV-responsive monomer, (2-nitro-1,3-phenylene)bis(methylene) diacrylate (NPBMDA, C2), was synthesized as reported previously (Scheme S1†),42 and was obtained as a white crystal. NPBMBA (174.6 mg, 0.6 mmol) and 4-amino-1-butanol (44.5 mg, 0.5 mmol, A2) were then mixed and stirred at 60 °C for 5 h. Then 3-(4-methylpiperazin-1-yl)propan-1-amine (MPZ) (94.2 mg, 0.6 mmol) in dichloromethane (DCM, 1 mL) was added and stirred at RT overnight. Finally, the polymer was purified by precipitation with diethyl ether (3 × 50 mL) and isolated as a yellowish liquid (0.15 g, yield 80%, Scheme 1). | ||
| Scheme 1 Synthetic route of LPAE-NB. | ||
Synthesis of BPAE-NB
NPBMBA (242 mg, 0.83 mmol), trimethylolpropane triacrylate (TMPTA, 74 mg, 0.25 mmol), and 4-amino-1-butanol (AB, 89 mg, 1 mmol) were polymerized without a solvent at 60 °C for 4.5 h. Termination was achieved by diluting the polymer with DCM (1 mL) and end-capping with 1-(3-aminopropyl)-4-methylpiperazine (MPZ, 157 mg, 1 mmol) at RT overnight. The product was purified by precipitation in diethyl ether, and the solvent was removed under vacuum to obtain BPAE-NB as a yellowish liquid (0.19 g, yield 72%, Scheme 2). Rhodamine B isothiocyanate (RBITC)-labeled BPAE-NB was prepared by dissolving BPAE-NB (10 mg) and RBITC (2 mg) in DCM (1 mL), stirring overnight at room temperature, precipitating with ether, and removing the solvent under vacuum. The degree of branching (DB) was calculated as illustrated in Fig. S5.†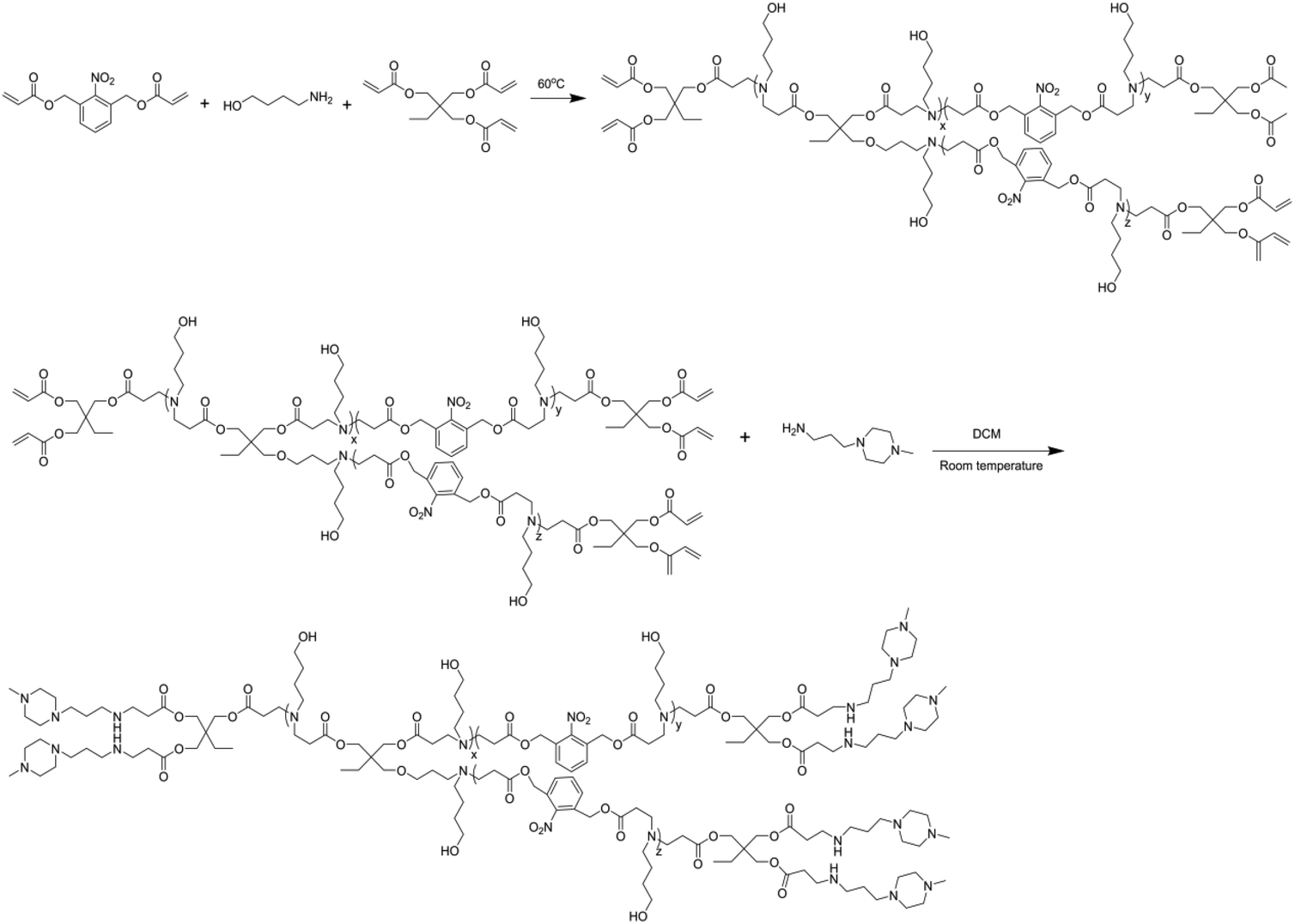 | ||
| Scheme 2 Synthetic route of BPAE-NB. | ||
Synthesis of BPAE-CC
The non-degradable BPAE (BPAE-CC) was polymerized from 1,6-hexanediol diacrylate (HD, 188 mg, 0.83 mmol), TMPTA (74 mg, 0.25 mmol), and AB (89 mg, 1 mmol) using the same method as described for BPAE-NB (Scheme 3). | ||
| Scheme 3 Synthetic route of BPAE-CC. | ||
UV-triggered polymer degradation
BPAE-NB or BPAE-CC in DMF (1 mg mL−1) was irradiated with UV light (λ = 365 nm, 20 mW cm−2) for different times. The wavelength and optical density of UV light were maintained constant for all the following assays. The UV-Vis spectra of the UV-irradiated polymers were recorded, and the MWs of polymers were determined by GPC.Preparation and characterization of polymer/DNA polyplexes
Polymers were dissolved in sodium acetate buffer (20 mM, pH 5.2) at 1 mg mL−1, and were mixed with DNA solution (0.2 mg mL−1) at various weight ratios to form the polyplexes. The DNA condensation was qualitatively evaluated using agarose (1%) gel electrophoresis and quantitatively explored using the ethidium bromide (EB) exclusion assay.34 The size and zeta potential of the polyplexes were monitored by dynamic laser scattering (DLS).UV-triggered polyplex dissociation and DNA release
In order to investigate the alteration of the DNA condensation capacity of BPAE-NB upon UV irradiation, the BPAE-NB/DNA polyplexes were irradiated with UV light for 5 min before being subjected to gel electrophoresis and EB exclusion assay as described above. Additionally, freshly prepared polyplexes (polymer/DNA weight ratio of 5) were UV-irradiated for different times and thereafter subjected to particle size analysis. The UV-triggered DNA release was further explored by incubating the UV-irradiated or non-irradiated polyplexes with heparin at various final concentrations prior to the EB exclusion assay.46In vitro DNA transfection
HeLa cells on 96-well plates (70% confluence) were incubated with the polymer/pLuc polyplexes in Opti-MEM (100 μL per well) at 1 μg DNA mL−1 for 4 h. The cells were then incubated in fresh serum-containing medium for another 20 h, and the transfection efficiency was determined and calculated as described previously.16 To explore photo-strengthened DNA transfection, the same transfection protocol was adopted except that the cells were irradiated with UV light for different times (1, 2, 5, and 10 min) after 4 h of incubation with polyplexes.pEGFP was also used to determine the transfection efficiency. The cells on 24-well plates (70% confluence) were incubated with the polymer/pEGFP polyplexes in Opti-MEM (500 μL per well) at 1 μg DNA mL−1 for 4 h. After refreshment with the serum-containing medium, the cells were irradiated with UV light for 5 min, further incubated for 48 h, and analyzed in terms of the EGFP expression by both flow cytometry and fluorescence microscopy.44
Intracellular kinetics of polymer/DNA polyplexes
HeLa cells on 96-well plates (70% confluence) were incubated with the polymer/YOYO-1-DNA polyplexes in Opti-MEM (100 μL per well) at 1 μg DNA mL−1 for 4 h at 37 °C. The cellular uptake level of polyplexes was determined by lysing the cells and quantifying the YOYO-1-DNA content in the lysate using spectrofluorimetry as described before.16To elucidate the cellular internalization mechanism of polyplexes, the cells were incubated with the BPAE-NB/YOYO-1-DNA polyplexes (w/w = 15) for 4 h at 4 °C or at 37 °C but with various endocytic inhibitors (chlorpromazine, CPZ, 10 μg mL−1; genistein, GNT, 100 μg mL−1; methyl-β-cyclodextrin, mβCD, 50 μM; wortmannin, WTM, 50 nM). The uptake level was normalized to that at 37 °C without inhibitors.44
To observe the intracellular distribution of polyplexes, HeLa cells on a culture dish (Φ = 20 mm, 30% confluence) were incubated with YOYO-1-DNA-containing polyplexes (1 μg DNA mL−1) for 4 h. The cells were then washed with cold PBS, stained with Hoechst 33258 (5 μg mL−1, 30 min, for nuclei) and Lysotracker Red (200 nM, 1 h, for endolysosomes), and visualized by confocal laser scanning microscopy (CLSM).
The light-promoted intracellular DNA release was also explored by CLSM. HeLa cells on a culture dish (Φ = 20 mm, 30% confluence) were treated with RBITC-BPAE-NB/YOYO-1-DNA polyplexes (w/w = 15) for 4 h, UV-irradiated for 5 min, and further cultured in fresh medium for 1 h before staining with Hoechst 33258 and observation by CLSM. The non-responsive RBITC-BPAE-CC/YOYO-1-DNA polyplexes were used as a control. The colocalization ratio between RBITC-BPAE and YOYO-1-DNA was calculated from 50 individual cells using Image-J software.47
Material cytotoxicity
The cells on 96-well plates (70% confluence) were incubated with LPAE-NB or BPAEs (with or without pre-treatment with 5 min UV irradiation) at various concentrations in Opti-MEM (100 μL per well) for 4 h. The cells were further incubated in serum-containing medium for 20 h, and the cell viability was determined by the MTT assay. The cytotoxicity of polyplexes was similarly monitored, wherein the cells were incubated with polyplexes at 1 μg DNA mL−1 for 4 h, irradiated with UV light for 2, 5, and 10 min, further cultured in fresh serum-containing medium for another 48 h, and subjected to the MTT assay. The cells without UV irradiation served as the control.Preparation, characterization, and UV-responsiveness of polymer/siRNA polyplexes
The polymer/siRNA polyplexes were prepared from a polymer and siRNA (5 and 0.2 mg mL−1 in DEPC water, respectively) using the same method as described for polymer/DNA polyplexes. siRNA condensation was evaluated by electrophoresis in 2% agarose gel at 90 mV, and the particle size as well as zeta potential of polyplexes was determined by DLS. The UV light-promoted siRNA release was similarly explored in terms of siRNA migration upon gel electrophoresis and size alteration of polyplexes.Intracellular kinetics of polymer/siRNA polyplexes
FAM-siRNA was used to complex with polymers, and the obtained polyplexes were incubated with HeLa cells on 96-well plates (70% confluence) at 2 μg siRNA mL−1 for 4 h. The uptake level of FAM-siRNA was determined using the same method as described above using spectrofluorimetry, and the results were expressed as ng FAM-siRNA per mg cellular protein. The intracellular distribution of polyplexes was similarly observed by CLSM after incubation of cells (30% confluence in culture dish) with BPAE-NB/FAM-siRNA or LPAE-NB/FAM-siRNA polyplexes (w/w = 30) for 4 h at 2 μg siRNA mL−1. The light-triggered intracellular siRNA release was explored by incubating the cells with RITC-BPAE/FAM-siRNA polyplexes (w/w = 30) for 4 h, light irradiation for 5 min, and further incubation for 1 h followed by CLSM observation.In vitro survivin silencing and anti-cancer efficiencies
HeLa cells on 6-well plates (70% confluence) were treated with polymer/siSur polyplexes at 2 μg siRNA mL−1 in Opti-MEM for 4 h. After refreshment with serum-containing medium, the cells treated with BPAE/siSur polyplexes were irradiated with UV light for 5 min. The cells were further incubated for 20 h, and the survivin mRNA level was determined by real-time PCR.To further evaluate the viability of HeLa cells after survivin silencing, the cells on 96-well plates (70% confluence) were incubated with BPAE-NB/siSur or BPAE-NB/siNC polyplexes (w/w = 30) at various siRNA concentrations for 4 h, UV light-irradiated for 5 min, and further cultured in serum-containing medium for 48 h before assessment of the cell viability using the MTT assay.
Statistical analysis
Statistical analysis was performed using Student's t-test. The differences between the test and control groups were judged to be significant at * p < 0.05 and very significant at ** p < 0.01 and *** p < 0.001.Results and discussion
Synthesis and characterization of UV-responsive BPAE-NB
The branched BPAE-NB was synthesized via the A2 (amine) + B3 (triacrylate) + C2 (diacrylate) Michael addition reaction as shown in Scheme 3, wherein the branching monomer (B3) was utilized to form the highly branched architecture and the nitrobenzene-containing monomer (NPBMDA, A2) was selected to endow the polymer with UV-responsive degradability. The obtained polymer was further end-capped with 1-(3-aminopropyl)-4-methylpiperazine (MPZ) to enhance the cationic charge density, thus yielding the UV-responsive, branched poly(β-amino ester) (BPAE-NB). To allow direct comparison on UV responsiveness, the non-degradable analogue of BPAE-NB, named BPAE-CC, was similarly synthesized using 1,6-hexanediol diacrylate as A2. The linear counterpart (LPAE-NB) was also prepared from A2 and C2 but without B3. The chemical structures of polymers were verified by 1H NMR (Fig. S1–S4†), and the degree of branching (DB) of BPAE-NB was calculated to be 0.55 as illustrated in Fig. S5.† The MW of BPAE-NB was determined using gel permeation chromatography (GPC), which showed an Mn of 18.1 kDa and a PDI of 1.83 (Table S3†).UV-triggered degradation of BPAE-NB
We first studied light-triggered polymer degradation using both UV-Vis spectrometry and GPC. When a DMF solution of BPAE-NB was irradiated with UV light (λ = 365 nm, 20 mW cm−2) for up to 2 min, a notable increase of the absorbance at 280 nm (OD280) was observed (Fig. 1A), which indicated the generation of the nitrosobenzene derivatives due to cleavage of the photo-labile ester bond. When the irradiation time was further prolonged, the OD280 did not greatly change, indicating that the majority of the polymer was degraded within 2 min irradiation. In support of such observation, BPAE-CC, a non-responsive analogue of BPAE-NB, showed an unappreciable alteration of OD280 upon UV irradiation (Fig. S6†). The degradation of BPAE-NB was further confirmed by GPC analysis. As illustrated in Fig. 1B, the MWs of BPAE-NB were dramatically decreased upon UV irradiation (λ = 365 nm, 20 mW cm−2, 10 min), and the chromatographic peak almost disappeared. In comparison, the non-responsive BPAE-CC cannot be degraded by UV irradiation and thus its MW remained unchanged. Such results indicated that the photo-responsive BPAE-NB can be degraded into low-MW fragments upon UV light irradiation.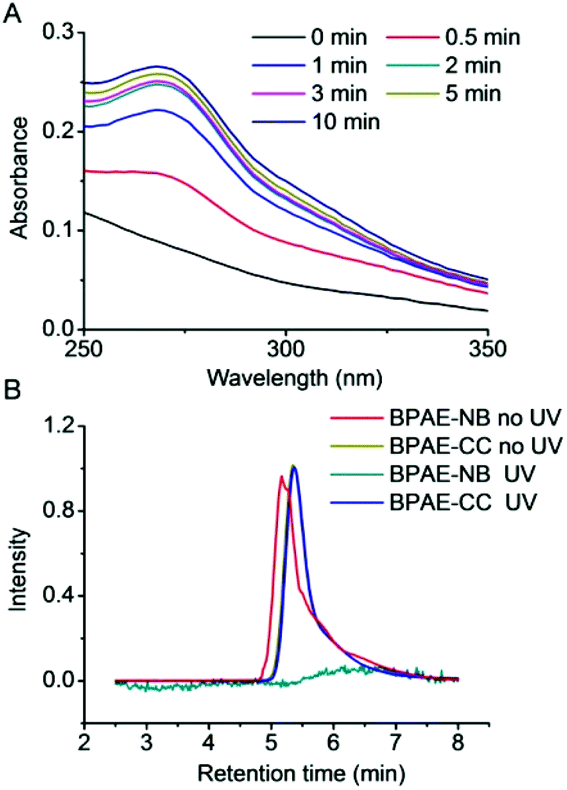 | ||
| Fig. 1 (A) Light-triggered degradation of BPAE-NB. (A) UV-Vis spectra of BPAE-NB after UV irradiation (λ = 365 nm, 20 mW cm−2) for various times. (B) GPC diagrams of BPAE-NB and BPAE-CC before and after UV irradiation (λ = 365 nm, 20 mW cm−2, 5 min). | ||
UV-triggered polyplex dissociation and DNA release
The capacity of BPAE-NB to condense DNA was first characterized by the gel retardation assay. As shown in Fig. 2A, DNA migration in the 1% agarose gel was completely restricted to the loading well at the polymer/DNA weight ratio higher than 0.5, indicating the complete condensation of DNA by BPAE-NB with a branched architecture. Comparatively, the linear LPAE-NB showed weaker DNA condensation capability, and it required a notably higher polymer/DNA weight ratio of 15 to completely condense DNA. Such results were further verified by the quantitative EB exclusion assay, wherein more than 85% of the DNA was condensed by BPAEs at polymer/DNA weight ratios higher than 1, while LPAE-NB afforded the DNA condensation efficiency of 85% at a higher polymer/DNA weight ratio of 10 (Fig. 2B). Such discrepancy thus highlighted the critical role of the branched structure in potentiating DNA condensation, which was mainly attributed to its multivalent structure that provided a higher local positive charge density to promote DNA condensation and polyplex formation through electrostatic interactions. Consistently, DLS measurement revealed that polyplexes with a diameter of ∼180 nm and a positive surface charge of ∼30 mV were formed at the BPAE/DNA weight ratio ≥5 (Fig. 2C and S7†).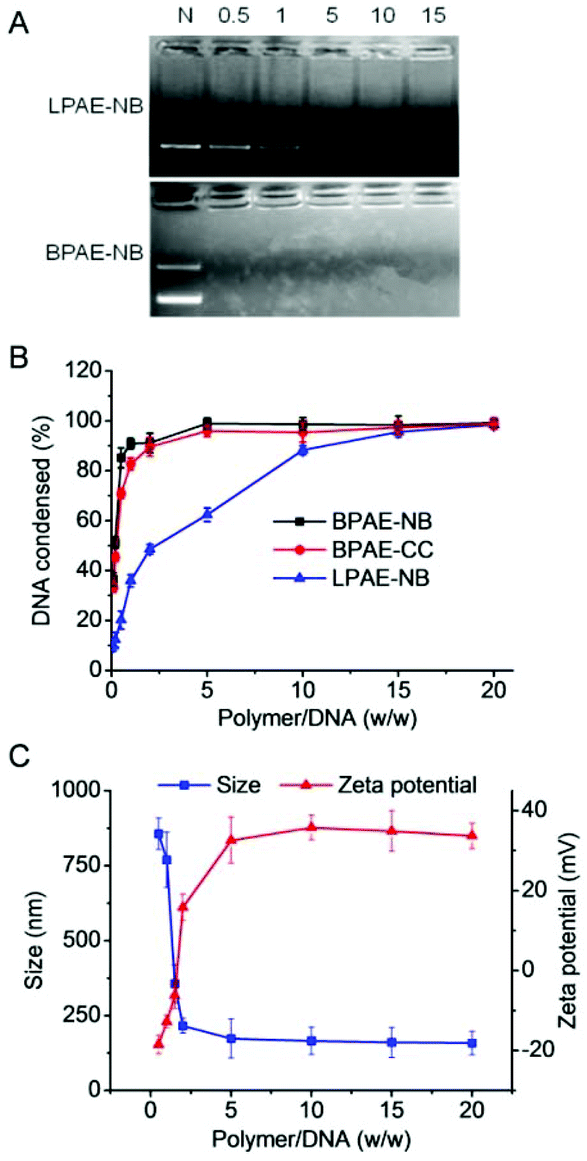 | ||
| Fig. 2 Characterization of polymer/DNA polyplexes. (A) DNA condensation by BPAE-NB and LPAE-NB at various polymer/DNA weight ratios as evaluated by the gel retardation assay. N represents naked DNA. (B) DNA condensation efficiencies of BPAE-NB, BPAE-CC, and LPAE-NB at various polymer/DNA weight ratios as evaluated by the EB exclusion assay (n = 3). (C) Size and zeta potential of BPAE-NB/DNA polyplexes at various polymer/DNA weight ratios. | ||
The UV-triggered DNA release was first investigated by gel electrophoresis and the EB exclusion assay. Upon UV light irradiation (λ = 365 nm, 20 mW cm−2, 10 min) of the BPAE-NB/DNA polyplexes, DNA completely migrated in the agarose gel at the polymer/DNA weight ratio of 2 (Fig. 3A), and the quantitative EB exclusion assay showed that DNA condensation efficiency notably dropped from 99.0 to 41.2% (Fig. 3B), indicating that the capability of BPAE-NB to condense DNA was weakened by UV light irradiation. In accordance with the polymer degradation profile, a longer UV irradiation time correlated to a higher DNA unpackaging rate within the observation period of 48 h (Fig. 3C). Furthermore, the particle size of the BPAE-NB/DNA polyplexes was greatly enhanced upon UV treatment (Fig. 3D and S8†), suggesting the dissociation of the polyplexes as a result of polymer degradation and reduced binding affinity toward DNA. In support of the above assessments on the UV-responsiveness of BPAE-NB, the DNA condensation by the non-responsive BPAE-CC was not affected by UV treatment (Fig. 3A and B), and the particle size of the BPAE-CC/DNA polyplexes also revealed an unappreciable change (Fig. 3D and S8†), further demonstrating that UV-triggered polymer degradation contributed to the promoted DNA release.
 | ||
| Fig. 3 UV light irradiation (λ = 365 nm, 20 mW cm−2) triggered polyplex dissociation and DNA release. (A) DNA condensation by BPAE-NB and BPAE-CC at various polymer/DNA weight ratios after UV irradiation for 5 min. (B) DNA condensation efficiencies of BPAE-NB, BPAE-CC, and LPAE-NB at the polymer/DNA weight ratio of 5 following UV irradiation for 5 min (n = 3). (C) DNA release from BPAE-NB/DNA polyplexes (w/w = 5) irradiated with UV light for different times followed by incubation for up to 48 h (n = 3). (D) Size alteration of BPAE-NB/DNA and BPAE-CC/DNA polyplexes (w/w = 5) following UV irradiation for different times. (E) DNA release from BPAE-NB/DNA polyplexes (w/w = 5) in the presence of heparin at various concentrations before and after UV irradiation for 5 min (n = 3). | ||
Light-facilitated DNA release was further probed using the heparin replacement assay. As shown in Fig. 3E, DNA was almost completely released from UV light-irradiated BPAE-NB/DNA polyplexes (λ = 365 nm, 20 mW cm−2, 10 min) in the presence of 0.2 mg mL−1 heparin. Comparatively, a higher heparin concentration of 0.5 mg mL−1 was required to dissociate DNA from the non-irradiated polyplexes. Such discrepancy clearly demonstrated that the DNA binding affinity of the UV-responsive BPAE-NB was notably reduced after UV irradiation. As a control, the non-responsive BPAE-CC showed unappreciable alteration in terms of heparin-induced DNA release profiles upon UV treatment (Fig. S9†). These results collectively substantiated our design strategy to promote “on-demand” DNA release by cascading instantaneous polymer degradation using external light triggers.
In vitro DNA transfection
The transfection efficiencies of various polyplexes were first evaluated in HeLa cells using pLuc as the reporter plasmid. As shown in Fig. 4A, at the optimal weight ratio of 15, BPAE-NB/DNA or BPAE-CC/DNA polyplexes showed remarkably higher transfection efficiency than LPAE-NB/DNA polyplexes, further substantiating the essential role of the branched architecture toward gene transfection. In direct comparison with PEI as the benchmark positive control, BPAE-NB also demonstrated a 15-fold higher transfection efficiency. UV irradiation (λ = 365 nm, 20 mW cm−2) was performed after the cells were incubated with polyplexes for 4 h, wherein effective cellular internalization of the polyplexes had been achieved. As illustrated in Fig. 4B, UV irradiation significantly promoted the transfection efficiencies of the BPAE-NB/DNA polyplexes but not the BPAE-CC/DNA polyplexes, demonstrating that UV-triggered backbone cleavage potentiated gene transfection by facilitating intracellular DNA release. It was further noted that prolonging the UV irradiation time up to 5 min led to greatly increased transfection efficiency, while it decreased when the irradiation time was further prolonged to 10 min. It therefore indicated that 5 min irradiation was sufficient to provoke intracellular DNA release towards maximal transfection, and excessively long-time irradiation may impair cell integrity. Similarly, in other mammalian cell types, including COS-7 and 3T3-L1 cells, the photo-strengthened transfection efficiencies were also noted, which confirmed the generality of the proposed strategy (Fig. 4C). Furthermore, pEGFP was adopted as another reporter plasmid to evaluate the transfection efficiency, and UV light irradiation (5 min) again augmented the GFP expression level of BPAE-NB but not the non-responsive BPAE-CC and commercial reagent LPF (Fig. 4D). | ||
| Fig. 4 UV light irradiation (λ = 365 nm, 20 mW cm−2) promoted the DNA transfection capabilities of polyplexes. (A) Comparison of the transfection efficiencies of BPAE-NB/pLuc, BPAE-CC/pLuc, LPAE-NB/pLuc, and PEI/pLuc polyplexes in HeLa cells (n = 3). (B) Effect of UV irradiation time on the transfection efficiencies of BPAE-NB/pLuc or BPAE-CC/pLuc polyplexes (w/w = 15) in HeLa cells (n = 3). (C) Transfection efficiencies of LPAE-NB/pLuc polyplexes (w/w = 15) and BPAE-NB/pLuc polyplexes (w/w = 15) in HeLa, COS-7, and 3T3-L1 cells with or without UV irradiation for 5 min (n = 3). (D) Fluorescence images of HeLa cells transfected with LPF/pEGFP lipoplexes, BPAE-NB/pEGFP polyplexes (w/w = 15), and BPAE-CC/pEGFP polyplexes (w/w = 15) with or without UV irradiation (5 min). The scale bar represents 20 μm. | ||
Intracellular kinetics of polymer/DNA polyplexes
The gene transfection performance of non-viral vectors is closely related to their internalization pathways and intracellular fate.32 We then studied the cellular uptake level and internalization mechanisms of the polyplexes formed by various polymers and YOYO-1-DNA. As shown in Fig. 5A, BPAE-NB remarkably promoted the internalization of DNA in HeLa cells at polymer/DNA weight ratios of 10–30, outperforming LPAE-NB and PEI by 2–4 fold. The non-responsive BPAE-CC showed a similar DNA uptake level to BPAE-NB, because of their similar cationic charge density and molecular structure. Notably a lower uptake level was noted for LPAE-NB, because of its weaker ability to condense DNA and form a compact nanostructure. In consistence, CLSM images also revealed a higher cellular uptake level of BPAE-NB/DNA polyplexes than LPAE-NB/DNA polyplexes (Fig. 5C). | ||
| Fig. 5 Intracellular kinetics of polyplexes in HeLa cells. (A) The uptake level of various polymer/YOYO-1-DNA polyplexes following incubation at 37 °C for 4 h (n = 3). (B) The uptake level of BPAE-NB/YOYO-1-DNA polyplexes (w/w = 15) at 4 °C or in the presence of various endocytic inhibitors (n = 3). (C) CLSM images of HeLa cells following incubation with LPAE-NB/YOYO-1-DNA or BPAE-NB/YOYO-1-DNA polyplexes (w/w = 15) for 4 h. Cell nuclei were stained with Hoechst 33258 and the endolysosomes were stained with Lysotracker Red. The scale bar represents 10 μm. (D) CLSM images of HeLa cells incubated with RBITC-BPAE-NB/YOYO-1-DNA polyplexes (w/w = 15) for 4 h, UV light irradiated for 5 min (λ = 365 nm, 20 mW cm−2), and further incubated for 1 h. The colocalization ratios between YOYO-1-DNA and RBITC-BPAE-NB were calculated from 50 cells and are listed. The scale bar represents 10 μm. | ||
The internalization pathway of the polyplexes was then probed by performing the uptake study at 4 °C when energy-dependent endocytosis is blocked or using different endocytic inhibitors including CPZ as the inhibitor for clathrin-mediated endocytosis (CME), GNT and mβCD as caveolae inhibitors, and WTM as the macropinocytosis inhibitor.10,48 As shown in Fig. 5B, the cell uptake was notably decreased by more than 70% at 4 °C, suggesting that the majority of the BPAE-NB/DNA polyplexes was internalized via energy-dependent endocytosis. The cell uptake level was also significantly inhibited by CPZ, GNT, and mβCD but not WTM, indicating that the polyplexes were internalized mainly via caveolae and CME rather than macropinocytosis.
Endosomal entrapment and lysosomal degradation of the gene cargoes serve as critical barriers against effective non-viral gene delivery. Therefore, the capability of BPAE-NB polyplexes to avoid endolysosomal entrapment was further studied. CLSM images revealed that BPAE-NB/YOYO-1-DNA polyplexes were effectively internalized into HeLa cells as evidenced by the extensive cytoplasmic distribution of YOYO-1-DNA post 4 h incubation (Fig. 5C), and green fluorescence (YOYO-1-DNA) greatly separated from red fluorescence (Lysotracker Red-stained endolysosomes), which demonstrated that the BPAE-NB/DNA polyplexes were able to effectively avoid endosomal/lysosomal entrapment (Fig. 5C).
CLSM was further utilized to evaluate the intracellular DNA release from polyplexes composed of RBITC-labeled BPAE-NB and YOYO-1-DNA. As shown in Fig. 5D, after exposure to UV light (λ = 365 nm, 20 mW cm−2) for 5 min, green fluorescence (YOYO-1-DNA) largely separated from red fluorescence (RBITC-BPAE-NB) in HeLa cells following 4 h incubation, conferring a significantly decreased co-localization ratio from 78.6% to 25.2%. Thus the UV-triggered degradation of BPAE-NB to facilitate the intracellular DNA release was evidenced. YOYO-1-DNA was also noted to be distributed in Hoechst 33258-stained nuclei, which indicated that it can be effectively transported into the nuclei where gene transcription was initiated (Fig. 5D).
Material cytotoxicity
Cytotoxicity and transfection efficiency of polycations are often conversely related.43 Polycations with a higher MW often afford higher transfection efficiencies yet stronger cytotoxicities than their low-MW analogues. As shown in Fig. 6A, BPAE-NB and BPAE-CC with a branched structure showed greatly higher and concentration-dependent cytotoxicity than the linear LPAE-NB after 24 h incubation, which could be attributed to their higher cationic charge densities. In comparison, UV light pre-treated BPAE-NB showed minimal cytotoxicity at the highest concentration tested (0.2 mg mL−1), substantiating that UV irradiation degraded the BPAE-NB into small segments to eliminate the MW-dependent material toxicity. As a control, UV light irradiation showed no effect on the cytotoxicity of the non-responsive BPAE-CC, due to its high MW and inability to degrade into low-MW segments upon UV treatment. | ||
| Fig. 6 Cytotoxicity of free polymers and polyplexes. (A) Viabilities of HeLa cells following 24 h incubation with polymers at various concentrations (n = 3). BPAE-NB and BPAE-CC were alternatively irradiated with UV light (λ = 365 nm, 20 mW cm−2, 5 min) before incubation with cells. (B) Viabilities of HeLa cells transfected with polyplexes at various polymer/DNA weight ratios for 4 h (1 μg DNA mL−1), irradiated with UV light (λ = 365 nm, 20 mW cm−2) for different times, and further cultured for 48 h (n = 3). | ||
A similar trend was noted when the cytotoxicities of polyplexes were tested following the transfection protocol. Consistently, UV irradiation significantly reduced the cytotoxicity of BPAE-NB/DNA polyplexes but not the non-responsive BPAE-CC/DNA polyplexes, and longer irradiation time further reduced the cytotoxicity (Fig. 6B). These findings again demonstrated our proposed strategy to eliminate the long-term cytotoxicity of BPAEs via light-triggered polymer degradation at the post-transfection state.
Characterization and UV-responsiveness of polymer/siRNA polyplexes
After identifying the photo-controlled DNA delivery performance, we further explored the potential of BPAE-NB in mediating photo-strengthened siRNA delivery. The siRNA condensation capability of BPAE-NB was first characterized by the gel retardation assay. As shown in Fig. 7A, compared to LPAE-NB which was unable to completely condense siRNA even at high weight ratio up to 150, BPAE-NB could condense siRNA at the polymer/siRNA weight ratios ≥15, which was similar to the non-responsive BPAE-CC (Fig. 7D). Such discrepancy thus highlighted the pronounced roles of the branched architecture in enabling effective siRNA condensation. DLS measurements showed that at the BPAE-NB (or BPAE-CC)/siRNA weight ratios higher than 20, nanoscale polyplexes with a diameter of ∼180 nm and a positive surface charge of ∼20 mV could be obtained (Fig. 7B and C).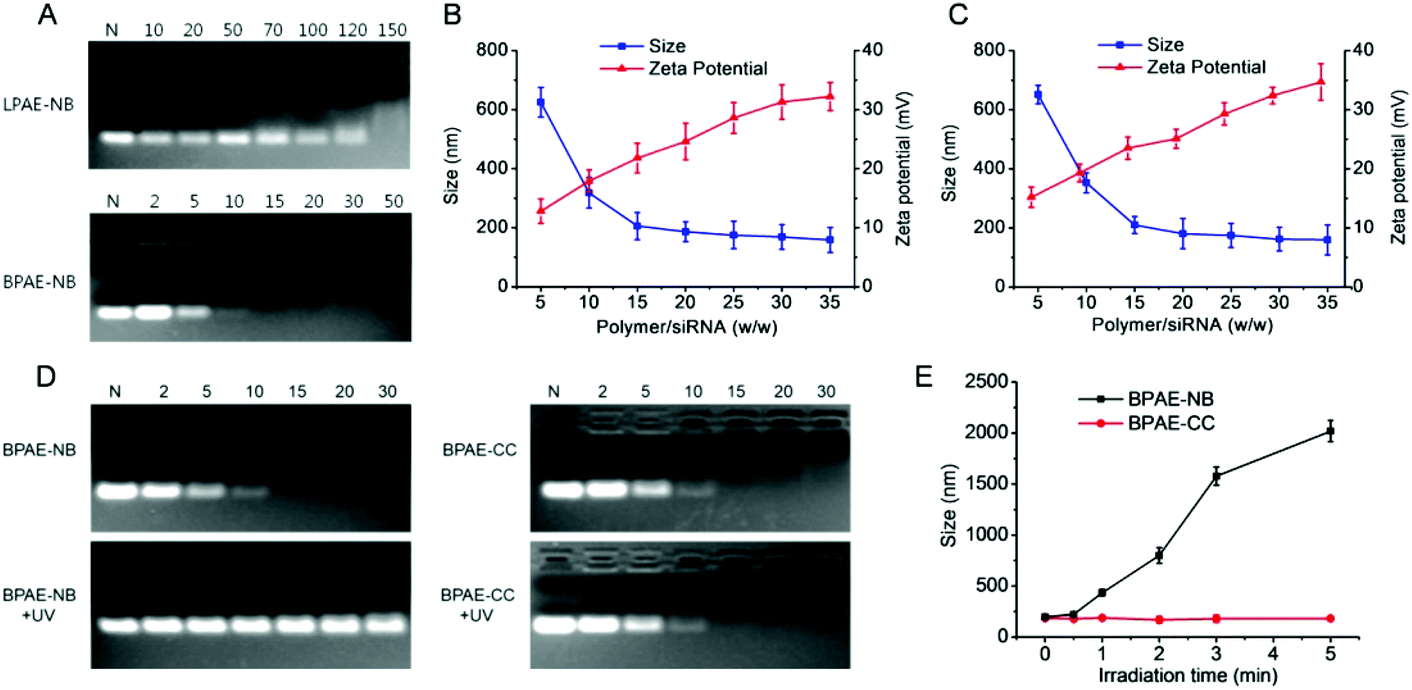 | ||
| Fig. 7 Characterization and light responsiveness of polymer/siRNA polyplexes. (A) siRNA condensation by BPAE-NB and LPAE-NB at various polymer/siRNA weight ratios as evaluated by the gel retardation assay. N represents naked siRNA. Size and zeta potential of BPAE-NB/siRNA (B) and BPAE-CC/siRNA (C) polyplexes at various polymer/siRNA weight ratios. (D) siRNA release from BPAE-NB/siRNA and BPAE-CC/siRNA polyplexes at various polymer/siRNA weight ratios following UV light irradiation (λ = 365 nm, 20 mW cm−2, 5 min). N represents naked siRNA. (E) Size alteration of BPAE-NB/siRNA and BPAE-CC/siRNA polyplexes (w/w = 30) following UV irradiation for different times. | ||
Upon treatment with UV light (λ = 365 nm, 20 mW cm−2, 5 min), siRNA completely migrated in the agarose gel at the BAPE-NB/DNA weight ratio of 30 (Fig. 7D), in comparison to the retarded siRNA in the loading well before UV irradiation. The particle size of the BPAE-NB/siRNA polyplexes also dramatically increased upon UV irradiation (Fig. 7E), which demonstrated the dissociation of polyplexes as a result of UV-triggered polymer degradation. As expected, the siRNA condensation level and the particle size of the non-responsive BPAE-CC/siRNA polyplexes did not alter upon UV light irradiation (Fig. 7D and E).
Intracellular fate of polymer/siRNA polyplexes
Similar to the results obtained for DNA delivery, BPAE-NB showed a dramatically enhanced cellular uptake level of FAM-siRNA compared to the linear LPAE-NB by ∼10 fold, and it outperformed the commercial reagent PEI (Fig. 8A). CLSM observation also revealed the capability of BPAE-NB to mediate the endolysosomal escape of siRNA cargoes, as evidenced by the clear separation between Lysotracker Red-stained endosomes/lysosomes and FAM-siRNA after 4 h incubation (Fig. 8B). When HeLa cells were incubated with RBITC-BPAE-NB/FAM-siRNA polyplexes for 4 h followed by UV light irradiation (λ = 365 nm, 20 mW cm−2) for 5 min, green fluorescence (FAM-siRNA) notably separated from red fluorescence (RBITC-BPAE-NB) at a decreased co-localization ratio from 67.2% to 20.6% (Fig. 8C). It therefore clearly evidenced the promoted intracellular siRNA in response to light-promoted BPAE-NB degradation.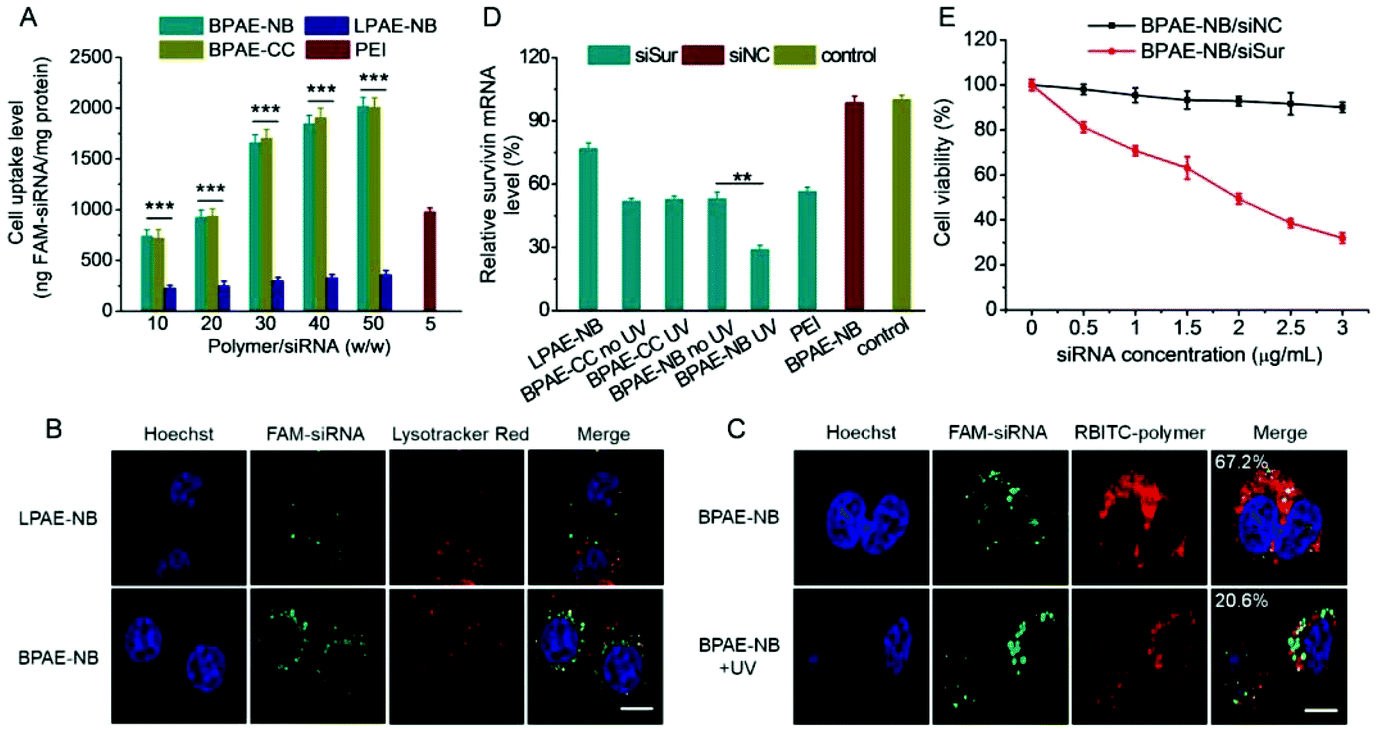 | ||
| Fig. 8 Light-enhanced intracellular delivery efficiency of siSur and anti-cancer efficiency in HeLa cells. (A) Uptake level of various polymer/FAM-siRNA polyplexes following incubation at 37 °C for 4 h (n = 3). (B) CLSM images of HeLa cells incubated with LPAE-NB/FAM-siRNA or BPAE-NB/FAM-siRNA polyplexes (w/w = 30) for 4 h. The scale bar represents 10 μm. (C) CLSM images of HeLa cells incubated with RBITC-BPAE-NB/FAM-siRNA polyplexes (w/w = 30) for 4 h, UV light irradiated (λ = 365 nm, 20 mW cm−2) for 5 min, and further incubated for 1 h. The colocalization ratios between FAM-siRNA and RBITC-BPAE-NB were calculated from 50 cells and are listed. The scale bar represents 10 μm. (D) Relative survivin mRNA levels in HeLa cells transfected with various polyplexes (w/w = 30 for LPAE-NB, BPAE-CC, and BPAE-NB, and w/w = 5 for PEI). Cells treated with BPAE-CC and BPAE-NB polyplexes were irradiated with UV light (λ = 365 nm, 20 mW cm−2, 5 min) following 4 h incubation (n = 3). (E) Viabilities of HeLa cells transfected with BPAE-NB/siNC and BPAE-NB/siSur polyplexes (w/w = 30) for 4 h, irradiated with UV light (λ = 365 nm, 20 mW cm−2) for 5 min, and further cultured for 48 h (n = 3). | ||
In vitro survivin knockdown efficiency and anti-cancer efficacy
siRNA against survivin, a member of the inhibitors of apoptosis protein (IAP) family that is involved in both cell division and apoptosis inhibition,49 was selected to further evaluate the gene knockdown efficiency and siRNA-mediated anti-cancer efficacy mediated by BPAE-NB. As shown in Fig. 8D, compared with the LPAE-NB/siSur polyplexes that only afforded ∼20% survivin knockdown in HeLa cells, the highly branched BPAE-NB/siSur polyplexes showed a notably higher knockdown efficiency (∼50%), and UV light irradiation further enhanced the silencing efficiency to ∼70%. The non-responsive BPAE-CC/siSur polyplexes showed a comparable knockdown efficiency to the BPAE-NB/siSur polyplexes, which was negligibly affected by UV irradiation. It was therefore substantiated that UV light irradiation greatly potentiated the BPAE-NB-mediated gene silencing as a consequence of light-triggered intracellular siRNA liberation.As a consequence of the effective survivin knockdown, BPAE-NB/siSur polyplexes coupled with post-transfection UV light irradiation led to an appreciable and siRNA concentration-dependent anti-cancer efficacy in HeLa cells, while the BPAE-NB/siNC polyplexes showed almost no toxicity at all tested siRNA concentrations up to 3 μg mL−1 (Fig. 8E). This thus confirmed that the successful knockdown of survivin mediated by BPAE-NB can significantly kill HeLa cells.
Conclusions
In summary, we developed a strategy to synchronistically overcome the multiple cellular barriers against non-viral gene delivery by tailoring the molecular topology of polycations and photo-controlling the degradation of materials. With this strategy, we designed and developed BPAE-NB with built-in photo-degradable domains in the backbone. BPAE-NB afforded strong DNA/siRNA condensation capabilities due to its multivalent structure, and thus featured a high trans-membrane gene delivery efficiency. With a UV light trigger, BPAE-NB underwent instantaneous degradation, which triggered intracellular DNA/siRNA unpackaging to potentiate the gene transfection efficiency and in the meantime self-diminished the toxicity of materials at the post-transfection state. By provoking these multiple delivery responses, transfection efficiencies of BPAE-NB were greatly improved to outperform the linear LPAE-NB, and it also outperformed the non-responsive BPAE-CC with higher transfection efficiency yet lower cytotoxicity. This study therefore provides an effective approach to address the efficiency–toxicity inconsistency of polycation-based gene vectors, and would provide promising insight into the rational design of non-viral gene delivery materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Ministry of Science and Technology of China (2016YFA0201200), the National Natural Science Foundation of China (51873142, 51573123 and 51722305), the Natural Science Foundation of Jiangsu Province (BK20161224), the Science and Technology Projects for The Youth of Suzhou Health and Family Planning Commission (KJXW2016016), and the Suzhou Key Laboratory of Thoracic Oncology (SZS201907).References
- U. Ben-David, B. Siranosian, G. Ha, H. Tang, Y. Oren, K. Hinohara, C. A. Strathdee, J. Dempster, N. J. Lyons, R. Burns, A. Nag, G. Kugener, B. Cimini, P. Tsvetkov, Y. E. Maruvka, R. O'Rourke, A. Garrity, A. A. Tubelli, P. Bandopadhayay, A. Tsherniak, F. Vazquez, B. Wong, C. Birger, M. Ghandi, A. R. Thorner, J. A. Bittker, M. Meyerson, G. Getz, R. Beroukhim and T. R. Golub, Nature, 2018, 560, 325–330 CrossRef CAS.
- J. J. Nie, B. Qiao, S. Duan, C. Xu, B. Chen, W. Hao, B. Yu, Y. Li, J. Du and F. J. Xu, Adv. Mater., 2018, 30, e1801570 CrossRef.
- X. T. Sun, M. H. Li, Y. Yang, H. Z. Jia and W. G. Liu, Biomater. Sci., 2018, 6, 3300–3308 RSC.
- C. Xu, Y. Zhang, K. Xu, J. J. Nie, B. Yu, S. Li, G. Cheng, Y. Li, J. Du and F. J. Xu, Nat. Commun., 2019, 10, 3184 CrossRef PubMed.
- J. B. Liu, L. L. Song, S. L. Liu, Q. Jiang, Q. Liu, N. Li, Z. G. Wang and B. Q. Ding, Nano Lett., 2018, 18, 3328–3334 CrossRef CAS.
- R. Q. Li, Y. Wu, Y. Zhi, X. Yang, Y. Li, F. J. Xua and J. Du, Adv. Mater., 2016, 28, 7204–7212 CrossRef CAS.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS.
- J. J. Cui, L. F. Qin, J. W. Zhang, P. Abrahimi, H. Li, G. X. Li, G. T. Tietjen, G. Tellides, J. S. Pober and W. M. Saltzman, Nat. Commun., 2017, 8, 191 CrossRef.
- S. Raisin, M. Morille, C. Bony, D. Noel, J. M. Devoisselle and E. Belamie, Biomater. Sci., 2017, 5, 1910–1921 RSC.
- M. M. Wang, H. M. Liu, L. Li and Y. Y. Cheng, Nat. Commun., 2014, 5, 3053 CrossRef.
- H. Wei, L. R. Volpatti, D. L. Sellers, D. O. Maris, I. W. Andrews, A. S. Hemphill, L. W. Chan, D. S. H. Chu, P. J. Horner and S. H. Pun, Angew. Chem., Int. Ed., 2013, 52, 5377–5381 CrossRef CAS.
- B. P. Mead, N. Kim, G. W. Miller, D. Hodges, P. Mastorakos, A. L. Klibanov, J. W. Mandell, J. Hirsh, J. S. Suk, J. Hanes and R. J. Price, Nano Lett., 2017, 17, 3533–3542 CrossRef CAS.
- H. Wang, Y. T. Wang, Y. Wang, J. J. Hu, T. F. Li, H. M. Liu, Q. Zhang and Y. Y. Cheng, Angew. Chem., Int. Ed., 2015, 54, 11647–11651 CrossRef CAS.
- H. P. Fang, Z. P. Guo, L. Lin, J. Chen, P. J. Sun, J. Y. Wu, C. N. Xu, H. Y. Tian and X. S. Chen, J. Am. Chem. Soc., 2018, 140, 11992–12000 CrossRef CAS.
- L. C. Yin, Z. Y. Song, K. H. Kim, N. Zheng, N. P. Gabrielson and J. J. Cheng, Adv. Mater., 2013, 25, 3063–3070 CrossRef CAS.
- L. C. Yin, Z. Y. Song, K. H. Kim, N. Zheng, H. Y. Tang, H. Lu, N. Gabrielson and J. J. Cheng, Biomaterials, 2013, 34, 2340–2349 CrossRef CAS.
- H. Y. Tang, L. C. Yin, K. H. Kim and J. J. Cheng, Chem. Sci., 2013, 4, 3839–3844 RSC.
- R. S. Zhu, L. C. Yin, B. Chen, X. Y. Li, T. F. Yi, M. F. Ye and A. N. Zhou, J. Mater. Eng. Perform., 2013, 22, 1744–1747 CrossRef CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS PubMed.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS.
- A. B. Cook, R. Peltier, J. L. Zhang, P. Gurnani, J. Tanaka, J. A. Burns, R. Dallmann, M. Hartlieb and S. Perrier, Polym. Chem., 2019, 10, 1202–1212 RSC.
- X. W. Guan, Z. P. Guo, L. Lin, J. Chen, H. Y. Tian and X. S. Chen, Nano Lett., 2016, 16, 6823–6831 CrossRef CAS.
- W. W. Shen, Q. W. Wang, Y. Shen, X. Gao, L. Li, Y. Yan, H. Wang and Y. Y. Cheng, ACS Cent. Sci., 2018, 4, 1326–1333 CrossRef CAS.
- Z. P. Guo, L. Lin, J. Chen, X. Z. Zhou, H. F. Chan, X. S. Chen, H. Y. Tian and M. W. Chen, Biomater. Sci., 2018, 6, 3053–3062 RSC.
- Y. Ou, K. Chen, H. Cai, H. Zhang, Q. Y. Gong, J. Wang, W. Chen and K. Luo, Biomater. Sci., 2018, 6, 1177–1188 RSC.
- M. Ahmed and R. Narain, Biomaterials, 2012, 33, 3990–4001 CrossRef CAS.
- Y. Y. Cheng, Acta Polym. Sin., 2017, 8, 1234–1245 Search PubMed.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- S. Min, Y. Jin, C. Y. Hou, J. Kim, J. J. Green, T. J. Kang and S. W. Cho, Adv. Healthcare Mater., 2018, 7, 1800052 CrossRef PubMed.
- U. C. Palmiero, J. C. Kaczmarek, O. S. Fenton and D. G. Anderson, Adv. Healthcare Mater., 2018, 7, 1800249 CrossRef.
- S. Y. Tzeng and J. J. Green, Adv. Healthcare Mater., 2013, 2, 468–480 CrossRef CAS.
- S. Tang, Q. Yin, J. H. Su, H. P. Sun, Q. S. Meng, Y. Chen, L. L. Chen, Y. Z. Huang, W. W. Gu, M. H. Xu, H. J. Yu, Z. W. Zhang and Y. P. Li, Biomaterials, 2015, 48, 1–15 CrossRef CAS.
- C. J. Bishop, T. M. Ketola, S. Y. Tzeng, J. C. Sunshine, A. Urtti, H. Lemmetyinen, E. Vuorimaa-Laukkanen, M. Yliperttula and J. J. Green, J. Am. Chem. Soc., 2013, 135, 6951–6957 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. J. Siegwart, C. A. Alabi, J. S. Rajan, R. Langer and D. G. Anderson, Biomaterials, 2012, 33, 3594–3603 CrossRef CAS.
- K. L. Kozielski, S. Y. Tzeng, B. A. H. De Mendoza and J. J. Green, ACS Nano, 2014, 8, 3232–3241 CrossRef CAS.
- S. Bai, X. L. Zhang, X. Q. Ma, J. C. Chen, Q. B. Chen, X. X. Shi, M. L. Hou, P. Xue, Y. J. Kang and Z. G. Xu, Biomater. Sci., 2018, 6, 3126–3138 RSC.
- Y. S. Gao, J. Y. Huang, J. O. Ahern, L. Cutlar, D. Z. Zhou, F. H. Lin and W. X. Wang, Biomacromolecules, 2016, 17, 3640–3647 CrossRef CAS PubMed.
- Q. Xu, L. R. Guo, A. Sigen, Y. S. Gao, D. Z. Zhou, U. Greiser, J. Creagh-Flynn, H. Zhang, Y. X. Dong, L. Cutlar, F. G. Wang, W. G. Liu, W. Wang and W. X. Wang, Chem. Sci., 2018, 9, 2179–2187 RSC.
- D. Z. Zhou, L. Cutlar, Y. S. Gao, W. Wang, J. O'Keeffe-Ahern, S. McMahon, B. Duarte, F. Larcher, B. J. Rodriguez, U. Greiser and W. X. Wang, Sci. Adv., 2016, 2, e1600102 CrossRef.
- S. Liu, Y. S. Gao, D. Z. Zhou, M. Zeng, F. Alshehri, B. Newland, J. Lyu, J. O'Keeffe-Ahern, U. Greiser, T. Y. Guo, F. Z. Zhang and W. X. Wang, Nat. Commun., 2019, 10, 3307 CrossRef.
- M. Zeng, D. Z. Zhou, F. Alshehri, I. Lara-Saez, Y. N. Lyu, J. Creagh-Flynn, Q. Xu, A. Sigen, J. Zhang and W. X. Wang, Nano Lett., 2019, 19, 381–391 CrossRef CAS.
- D. Z. Zhou, Y. S. Gao, A. Aied, L. Cutlar, O. Igoucheva, B. Newland, V. Alexeeve, U. Greiser, J. Uitto and W. X. Wang, J. Controlled Release, 2016, 244, 336–346 CrossRef CAS.
- J. Y. Huang, Y. S. Gao, L. Cutlar, J. O'Keeffe-Ahern, T. Y. Zhao, F. H. Lin, D. Z. Zhou, S. McMahon, U. Greiser, W. Wang and W. X. Wang, Chem. Commun., 2015, 51, 8473–8476 RSC.
- X. J. Deng, N. Zheng, Z. Y. Song, L. C. Yin and J. J. Cheng, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS.
- L. C. Yin, H. Y. Tang, K. H. Kim, N. Zheng, Z. Y. Song, N. P. Gabrielson, H. Lu and J. J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS.
- X. Xu, Y. J. Li, Q. J. Liang, Z. Y. Song, F. F. Li, H. He, J. H. Wang, L. P. Zhu, Z. F. Lin and L. C. Yin, ACS Appl. Mater. Interfaces, 2018, 10, 256–266 CrossRef CAS.
- Y. W. Won, A. N. McGinn, M. Lee, K. Nam, D. A. Bull and S. W. Kim, Biomaterials, 2013, 34, 6229–6238 CrossRef CAS.
- B. Newland, H. Y. Tai, Y. Zheng, D. Velasco, A. Di Luca, S. M. Howdle, C. Alexander, W. X. Wang and A. Pandit, Chem. Commun., 2010, 46, 4698–4700 RSC.
- C. Sanhueza, S. Wehinger, J. C. Bennett, M. Valenzuela, G. I. Owen and A. F. G. Quest, Mol. Cancer, 2015, 14, 198 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of instrumentation, 1H NMR spectra, and additional experimental results. See DOI: 10.1039/C9BM01452G |
| This journal is © The Royal Society of Chemistry 2020 |