
The influence of trapping agents on the antitumor efficacy of irinotecan liposomes: head-to-head comparison of ammonium sulfate, sulfobutylether-β-cyclodextrin and sucrose octasulfate
Wenqian
Yang
a,
Zimeng
Yang
a,
Jingru
Fu
a,
Mengran
Guo
a,
Bingjun
Sun
a,
Wei
Wei
a,
Dan
Liu
*b and
Hongzhuo
Liu
 *a
*a
aSchool of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China. E-mail: liuhongzhuo@syphu.edu.cn; Fax: +86-24-43520586; Tel: +86-24-4352586
bKey Laboratory of Structure-Based Drugs Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China. E-mail: sammyld@163.com
First published on 15th November 2018
Abstract
Remote loading technology is an outstanding achievement in liposome-based drug delivery systems. Compared with conventional passive loading, remote loading technology exhibits unique superiority in terms of high drug loading efficiency, low leakage rate and adequate drug accumulation. In the intra-liposome aqueous phase, the counterion of the trapping agent can control the state of aggregation/crystallization of the drug-counterion salt, and thereby contribute to control the efficiency of remote loading. Herein, irinotecan (CPT-11)-loaded liposomes were developed using three trapping agents: ammonium sulfate (AS), sulfobutylether-β-cyclodextrin (SBE-β-CD) and sucrose octasulfate (SOS). The corresponding formulations were named as AS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and TEA-SOS liposomal CPT-11, respectively. Cryo-transmission electron micrographs showed that bundles of CPT-11 fibers were gathered inside TEA-SOS liposomal CPT-11. Furthermore, compared with AS liposomal CPT-11 and TEA-SBE-β-CD liposomal CPT-11, TEA-SOS liposomal CPT-11 demonstrated slower drug release, prolonged circulation time and significantly improved antitumor efficiency. To avoid the protection of ONIVYDE®-related patents, a number of other liposomal CPT-11 formulations are under preclinical investigation or even in clinical trials. Our study gives new insights into the impact of the trapping agent on remote loading, and provides valuable information to evaluate the development of CPT-11 loaded liposomes.
Introduction
In 1972, Deamer and coworkers first introduced a remote loading liposomes system.1,2 Analyses of current loading approaches have revealed that the remote loading approach is the only option to achieve liposomal formulation with high drug encapsulation, enhanced safety and increased efficacy.3–5Inside liposomes, drugs can form an aggregation state with a specific trapping agent such as ammonium sulfate (AS), chloride ammonium, calcium acetate or sodium acetate.6–9 Moreover, polyanions such as polyphosphate, sulfobutylether-β-cyclodextrin (SBE-β-CD) or sucrose octasulfate (SOS), in conjunction with a transmembrane triethylammonium (TEA) gradient, could also mediate effective drug loading.10,11
It is noteworthy that encapsulated drugs are themselves not bioactive; only when the drugs are released from the liposomes, delivered to the disease site and exceed the minimum effective concentration, can optimal therapeutic activity be anticipated.12,13 The drug-trapping-agent complex, which determines the drug loading ability and encapsulated drug release kinetics,6,14 influences the therapeutic activity and toxicity of liposomal drug delivery systems.15,16 The solubility of the drug-trapping-agent complex is important,17,18 with very low solubility leading to slow drug release. In some cases, slow release of the encapsulated drug is advantageous, with a low leakage rate in vivo.19 However, slow release and a low leakage rate in turn cause low drug availability in inflammatory tissues.20–22 This is a clear indication of the importance of the drug-trapping agent complex. However, the influence of the trapping agent on drug loading, release and retention properties, as well as toxicity and efficacy, still needs to be evaluated.
To see how the encapsulation method can influence drug retention properties, we used irinotecan (CPT-11) as the model drug. CPT-11 is a water-soluble derivative of camptothecin.23 However, the therapeutic efficiency of CPT-11 is severely restricted by its stability and non-specific toxicity. The terminal lactone ring of CPT-11 is easily hydrolyzed in physiological conditions and then converted to the inactive carboxylate form.24 To overcome these problems, ONIVYDE®, which was developed by encapsulating CPT-11 into liposomes using sucrose octasulfate triethylammonium as a trapping agent, was approved by the Food and Drug Administration (FDA) in 2015 for the clinical treatment of advanced pancreatic cancer.25 In order to bypass ONIVYDE®-related patents, other liposomal CPT-11 formulations prepared with different trapping agents are currently under investigation. Therefore, it is necessary to directly compare and evaluate the effectiveness of these formulations in order to direct the development of liposomal CPT-11 formulations. In this study, three trapping agents containing different numbers of sulfate groups were selected. AS is a widely used trapping agent in remote loading techniques. SBE-β-CD is one of the most popular β-CD derivatives, with a lipophilic cavity containing a mean of 6.5 sulfates per cyclodextrin (CD). Due to its improved toxicity profile, SBE-β-CD has been widely used to solubilize poorly water-soluble drugs. Moreover, as one SBE-β-CD molecular could bind multiple drug molecules,11 SBE-β-CD could improve drug retention. Finally, SOS is a highly charged polyanionic moiety with eight sulfonic groups. As expected, the number of sulfates has a profound impact on the physicochemical properties of the trapping agent. In this study, the influence of the trapping agent on the retention properties and kinetics of the encapsulated drug in a remote loading system were evaluated.
Materials and methods
Materials
Irinotecan was kindly provided by Jiangsu Hengrui Medicine Co., Ltd (China). SOS was obtained from Wuhan Hengruikang Reagent Co., Ltd (China). Methanol, acetonitrile and dichloromethane (high-performance liquid chromatography [HPLC] grade) were purchased from Tianjin Kemer Chemical Reagent Co., Ltd (Tianjin, China). Disaturated phosphatidylcholine (DSPS), cholesterol (Chol) and 2-distearoyl-snglycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (mPEG2000-DSPE) were purchased from Shanghai Advanced Vehicle Technology Ltd Co. Sephadex G-75 size exclusion resins were procured from Sigma Chemical Company (St Louis, USA). All other reagents and solvents described were of analytical grade.The HT-29 tumor cell line was originally obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai, China). Nude mice (8–10 weeks old), Kunming (KM) mice (20 ± 2 g) and Sprague Dawley (SD) rats (250 ± 20 g) were obtained from Shenyang Pharmaceutical University.
Preparation of SBE-β-CD and SOS salts
TEA salt solutions of SBE-β-CD and SOS were prepared from commercially acquired sodium salts by ion-exchange chromatography on 732 cation exchange resin, immediately followed by titration with TEA. The sodium salt was then determined by an Na+ selective electrode; residual sodium salt in either solution was less than 1% of the cation content. The concentration was adjusted to 0.65 mol L−1 for the TEA-SOS solution and 0.2 mol L−1 for the TEA-SBE-β-CD solution. For both solutions, the final pH was 5.5 to 6.0. AS solution (0.25 mol L−1) was prepared by dissolving commercial ammonium sulfate salt in water.Preparation of liposomes
A mixture including DSPC (3 mol. parts), Chol (2 mol. parts) and mPEG2000-DSPE (0.015 mol. parts) was dissolved and mixed in dichloromethane, dried to a thin lipid film, and then subsequently hydrated with TEA-SOS solution, TEA-SBE-β-CD solution and AS solution, respectively. Hydration was performed under continuous shaking at 65 °C for 30 min to form multilamellar vesicles. To obtain large unilamellar vesicles, lipid suspensions were then gradually extruded 20 times through polycarbonate membranes with 0.8 μm, 0.4 μm and 0.2 μm pore sizes at 65 °C. The corresponding formulations were named as AS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and TEA-SOS liposomal CPT-11, respectively.Drug loading
Unentrapped salts of TEA-SOS, TEA-SBE-β-CD and AS were removed by column chromatography using Sephadex G-75 columns. Sephadex G-75 columns were equilibrated in pH 6.5 HEPES/NaCl (17/144, mM/mM) buffer to exchange the unentrapped TEA-SOS and AS. pH 6.5 sucrose/histidine (300/20, mM/mM) buffer was used to exchange the unentrapped TEA-SBE-β-CD. Upon buffer exchange, CPT-11 HCl was then added to the empty preformed liposomes at the desired ratio. The loading process was then optimized and performed above the phase transition temperature (Tm) of the lipids. The resulting mixture was incubated at 65 ± 5 °C for 1 h to realize drug loading, and then the mixture was chilled on ice for 20 min. In the TEA-SBE-β-CD gradient remote loading process, drug loading was performed in the presence of nigericin (20 ng mg−1 DSPC), and empty liposomes were incubated with nigericin for 15 min before drug loading.The entrapment efficiency (EE) of the preformed liposomes was determined by a centrifugal ultrafiltration method. Briefly, liposomes (100 μL) were added to an ultrafiltration centrifuge tube (10 kDa), then centrifuged (3000 rpm) for 30 min to allow the free drug to flow to the bottom. To determine the CPT-11 concentration of the liposomes, 100 μL liposomes were solubilized in 10% Triton X-100, followed by a mixture of methanol and 0.001 M citric acid. Drug concentration was determined by HPLC on a reverse Agilent 5 TC-C18 (2) column (250 mm × 4.6 mm, 4.5 μm). Ultraviolet (UV) detection was set at 255 nm, with a mixture of anhydrous sodium dihydrogen phosphate and sodium octane sulfonate/methanol/acetonitrile (57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:18) as the eluent at a flow rate of 1.5 ml min−1. The EE was calculated using the following equation: EE% = liposomes fraction/(liposomes fraction + free drug fraction) × 100.
:25:18) as the eluent at a flow rate of 1.5 ml min−1. The EE was calculated using the following equation: EE% = liposomes fraction/(liposomes fraction + free drug fraction) × 100.
Characterization of CPT-11 liposome
Cryo-TEM
An aliquot of 3.5 μL liposomes was added to negatively glow discharged R1.2/1.3 100 holey carbon films grids (Cu 200 mesh) (Quantifoil, Jena, Germany), blotted to remove the excess, and plunge-frozen in a liquid ethane–propane mixture in a chamber at 100% humidity with a Vitrobot Mark IV (FEI, Hillsboro, OR). Data were acquired using a FEI Talos F200C electron microscope at an operating voltage of 200 kV. Images were collected with a charge-coupled device operating at an absolute magnification of 36000×. The dose rate of 35 e Å−2 s−1 was used during data collection. Data were collected using Serial EM software with a nominal defocus value of −5 μm.
In vitro drug release study
The release behavior of CPT-11 liposomes formulations was investigated at 37 °C with a solution of glucose–histidine–NH4Cl (250 mm/10 mm/20 mm, pH 7.5). Typically, liposomes containing 0.4 mg CPT-11 were dispersed in 50 mL of release media with shaking (100 rpm). At 1, 2, 4, 8, 12, 48, 96 and 144 h, 2 mL of solution was withdrawn. The sample was then passed through a 0.22 μm membrane filter. The CPT-11 concentration was determined by HPLC with Waters e2695 Separations Module and Waters 2489 UV/Visible Detector on a reverse Agilent 5 TC-C18 (2) column (250 mm × 4.6 mm, 4.5 μm). The column was maintained at 40 °C, and UV detection was set at 255 nm. A mixture of anhydrous sodium dihydrogen phosphate and sodium octane sulfonate/methanol/acetonitrile (57:25:18) was used as the eluent at a flow rate of 1.5 mL min−1. The results were expressed as mean ± standard deviation (SD).
Pharmacokinetics study
The animal experiments were approved by the Animal Ethics Committee of Shenyang Pharmaceutical University (Shenyang, China), in accordance with the “Guideline for the Care and Use of Laboratory Animals”. Male SD rats (250 ± 20 g) were randomly assigned to three groups (n = 5). Each rat was injected with a liposomes formulation (5 mg kg−1 CPT-11 equivalent) via the tail vein. At 5, 15 and 30 min, and 1, 2, 4, 6, 8, 12 and 48 h, blood samples (0.2 mL to 0.3 mL) were drawn from the suborbital venous plexus and immediately transferred to heparinized tubes. Blood samples were then centrifuged at 10000 rpm for 5 min to separate the plasma, and the stored at −20 °C until further analysis.
To access the pharmacokinetic profiles of the CPT-11 liposomes formulations, released CPT-11 (F-CPT-11) and total CPT-11 (T-CPT-11) (released CPT-11 + encapsulated CPT-11) in plasma were separated and quantified.26
Bio-distribution study
Bio-distribution experiments were performed on male KM mice. Mice (22 ± 2 g) were randomly assigned to three groups (n = 3) and administered with CPT-11 liposomes at 5 mg kg−1via the tail vein. At 6, 24 and 48 h after dosing, mice were sacrificed, and the main tissues (heart, liver, spleen, lung and kidney) were immediately harvested. Tissues were then rinsed in saline, dried, weighed and diluted 1:1 by saline, and the homogenized in an ice bath. Samples were then centrifuged at 3000 rpm and supernatants were stored at −20 °C until analysis. To access tissue distribution profiles of CPT-11 liposome formulations, released CPT-11 (F-CPT-11) and total CPT-11 (T-CPT-11) (released CPT-11 + encapsulated CPT-11) in liver and spleen were separated and quantified.26
In vivo antitumor activity
HT-29 cells bearing male nude mice were used to evaluate the in vivo antitumor efficacy of preformed CPT-11 liposomes. Briefly, HT-29 cells (approximately 2 × 106 cells per 100 μL) were inoculated subcutaneously in the right flank region of the mice. When the tumor size reached approximately 200 mm3, HT-29 xenografts nude mice were randomly divided into six groups (n = 5): untreated control (Saline), 25 mg kg−1 TEA-SOS liposomal CPT-11, 25 mg kg−1 TEA-SBE-β-CD liposomal CPT-11, 10 mg kg−1 TEA-SOS liposomal CPT-11, 10 mg kg−1 TEA-SBE-β-CD liposomal CPT-11 and 10 mg kg−1 AS liposomal CPT-11. These formulations were administrated every 6 days through the tail veins for a total of five injections. Tumor volume and body weight were measured every 2 or 4 days after the first injection. Mice were sacrificed 4 days after the final injection, then the tumor and major organs were collected and fixed in formalin at room temperature for hematoxylin and eosin (H&E) analysis. In addition, tumors were weighed for the calculation of tumor burden. 0.2 mL serum was also collected to determine the activities of aspartate transaminase (AST), alanine transaminase (ALT), blood urea nitrogen (BUN) and creatinine.Data analysis
In this research, all data were expressed as mean ± SD. Statistical differences were verified by Student's t-test or one-way analysis of variance (ANOVA).Results and discussion
Preparation of CPT-11 liposomes
Fig. 1 and 2 describe the overall mechanism of the loading process. In AS liposomal CPT-11 (Fig. 2A), liposomes were prepared in 250 mM ammonium sulfate, then the ammonium sulfate was removed and replaced by HEPES/NaCl solution. The higher ammonium concentration inside AS liposomal CPT-11 induced the diffusion of neutral NH3, and thus a pH gradient was formed. The magnitude of this AS gradient is derived by the ratio [NH4+] extra/[NH4+]inter. Next, the protonated NH4+ leads to an increase of pH, promoting the internalization of CPT-11 molecules. This effect accelerates the flocculation of CPT-11, further improving the drug encapsulation. As TEA is a weak base (pKa of TEA is 10.75), in the case of TEA-SOS liposomal CPT-11 and TEA-SBE-β-CD liposomal CPT-11 (Fig. 2B and C), they dissociate to produce protons, and neutral TEA can freely permeate lipid bilayer, resulting in the acidification of intra-liposomal media and further improving the accumulation and encapsulation of CPT-11. Nevertheless, the drug-to-lipid (D/L) ratio for the different trapping agents differed, with the different D/L ratios possibly ascribed to different physicochemical properties of the drug-trapping agent complex. In the AS gradient, AS could mediate stable encapsulation with a D/L ratio of 0.1. In contrast, each of SBE-β-CD and SOS could bind multiple CPT-11 molecules via electrostatic effects, such that D/L ratios of 0.40 and 0.63 were obtained. In the TEA-SBE-β-CD gradient remote loading process, nigericin was added to further improve the loading efficacy of the TEA-SBE-β-CD gradient. Nigericin, as a proton ionophore, was used to exchange Na+ with proton to facilitate the pH gradient.27,28 Accordingly, CPT-11 could be effectively loaded into liposomes by the TEA-SBE-β-CD gradient, and >99% drug encapsulation was obtained in the presence of nigericin. | ||
| Fig. 1 Remote loading mechanism of AS liposomal CPT-11 (A), TEA-SBE-β-CD liposomal CPT-11 (B), (empty liposomes was incubated with nigericin) (C), Cryo-TEM of AS liposomal CPT-11 (D), TEA-SBE-β-CD liposomal CPT-11 (E) and TEA-SOS liposomal CPT-11 (F). | ||
 | ||
| Fig. 2 In vitro release of CPT-11 liposomes with release buffer (250 mm glucose, 10 mm histidine, 20 mm NH4Cl, pH 7.5); incubation temperatures were 37 °C. | ||
Characterization of CPT-11 liposomes
After preparation of the CPT-11 liposomes, the particle size, zeta potential and encapsulation efficiency were investigated (Table 1). The particle sizes of the three formulations were approximately 130 nm. For the three formulations, >99% CPT-11 encapsulation was obtained.| Formulation | Particle size (nm) | PDI | Zeta potential (mV) | EE (%) | D/L |
|---|---|---|---|---|---|
| TEA-SOS liposomal CPT-11 | 137.8 ± 3.477 | 0.096 ± 0.038 | −11.30 ± −0.884 | 99.40 | 0.63 |
| TEA-SBE-β-CD liposomal CPT-11 | 132.4 ± 2.460 | 0.045 ± 0.022 | −17.30 ± −1.356 | 99.16 | 0.40 |
| AS liposomal CPT-11 | 133.3 ± 1.484 | 0.030 ± 0.010 | −18.80 ± −1.474 | 99.00 | 0.10 |
Cryo-TEM was applied to visualize the physical state of the encapsulated CPT-11; representative images are shown in Fig. 1. The results confirm the size distribution determined in dynamic light scattering. Further, the interior of the liposomes appear highly electron-dense, indicating the existence of entrapped CPT-11. Bundles of fibers are clearly shown in the TEA-SOS liposomal CPT-11. In comparison, intra-liposomal CPT-11 gathers into an amorphous/gel precipitate, with no clearly defined structural organization in TEA-SBE-β-CD liposomal CPT-11 or AS liposomal CPT-11.
Intra-liposomal precipitation results from the drug self-association and interaction between the drug and counterion. Indeed, nano-crystal formation occurs only at drug concentrations in excess of its aqueous solubility limit. Furthermore, the property of precipitation (crystalline or non-crystalline form) is affected by many factors including the physicochemical properties of the drug, intra-liposomal pH and the counterion of the gradient-forming ion. In a previous study, the physical state of doxorubicin inside the liposomes was examined. Doxorubicin loaded via an AS gradient existed in straight rods, while doxorubicin loaded via a citrate gradient existed as curved and circular bundles of fibers. However, doxorubicin aggregates appeared as uncondensed fibers in a liposomal formulation containing lactobionic acid. This suggests that the physical state of doxorubicin is related to the trapping agent.29 In this study, AS could only interact with two CPT-11 molecules. In comparison, the polyanionic SBE-β-CD molecule (with a mean 6.5 sulfates) could bind more CPT-11 molecules. Moreover, SOS (with eight sulfates) carries multiple negative charges, and could interact with multiple CPT-11 molecules and facilitate inter-fiber crosslinking via an electrostatic effect. Hence, bundles of CPT-11 fibers could be observed in TEA-SOS liposomal CPT-11.
In vitro drug release
To quantitatively compare the role of the trapping agent in drug retention, in vitro release was performed in NH4Cl-containing release media. NH3 could freely permeate the lipid bilayer and elevate intra-liposomal pH, and thus induce CPT-11 release.30 The in vitro profiles are shown in Fig. 2. Based on these results, TEA-SOS mediated the most stable CPT-11 encapsulation and showed reduced CPT-11 release. In TEA-SOS liposomal CPT-11, the amount of CPT-11 released was 60.96% over 96 h, which was lower than the amount released in both TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11.The difference between the release rates may be attributed to the difference in the extent of precipitation with different trapping agents. In other words, the higher the fraction precipitated, the slower the release rate. In TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11, CPT-11 exists in a non-crystalline form. However, in TEA-SOS liposomal CPT-11, this is not the case, as CPT-11 exists in a crystalline form. Under this condition, the release property is governed by Fick's law relationship.31,32
| d[C]toto/dt = −pA([C]o − [C]i)/Vo |
Based on this formula, the release rate is proportional to the membrane area of the liposomes and the concentration of CPT-11 between the inner and outer liposome membrane. As the outer volume of the liposome is infinitely larger than the inner volume, thus [C]i ≫ [C]o. The formula was then converted to d[C]toto/dt = pA[C]i, which suggests that the rapid release of CPT-11 relies on higher CPT-11 accumulation in the intra-liposome aqueous phase. Inside TEA-SOS liposomal CPT-11, the crystalline form of CPT-11 was in equilibrium with the small amount of the soluble and neutral form; therefore, solubility of the crystal aggregates is limited. Accordingly, if the dissolution of encapsulated CPT-11 crystals is a rate-limiting step, TEA-SOS liposomal CPT-11 would lead to a slower CPT-11 release than TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11.
Pharmacokinetics study
The pharmacokinetics of TEA-SOS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11 after intravenous injection were investigated in SD rats. The plasma concentration-time curves of total CPT-11 (T-CPT-11), released CPT-11 (F-CPT-11) and the main metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) are shown in Fig. 3, and the main pharmacokinetic parameters are presented in Tables 2–4. | ||
| Fig. 3 Plasma concentrations of total CPT-11 levels (A), free CPT-11 levels (B), and main metabolite SN-38 levels (C) in SD rats injected with AS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and TEA-TEA-SOS liposomal CPT-11. The CPT-11 dose was 5 mg kg−1, CPT-11 levels and SN-38 levels were assayed using the ultra-performance liquid chromatography – tandem mass (UPLC-MS-MS) method. Data are shown as mean ± SD (n = 5). | ||
| Pharmacokinetic parameters | AS liposomal CPT-11 | TEA-SBE-β-CD liposomal CPT-11 | TEA-SOS liposomal CPT-11 |
|---|---|---|---|
| Pharmacokinetic parameters were calculated for T-CPT-11 after the intravenous injection of CPT-11 liposomes at 5 mg kg−1. Values represent the mean ± SD (n = 5). p < 0.05 (★), p < 0.01 (★★) and p < 0.001 (★★★) versus AS liposomal CPT-11 as the control; p < 0.05 (*) and p < 0.01 (**) and p < 0.001 (***) versus TEA-SBE-β-CD liposomal CPT-11 as the control. | |||
| AUC(0–t) (ng L−1 h−1) | 278006.44 ± 22516.72 |
468465.57 ± 46623.90★★★ |
645543.45 ± 98561.19★★★* |
| AUC(0–∞) (ng L−1 h−1) | 287201.85 ± 25973.99 |
503789.36 ± 54035.14★★★ |
682368.64 ± 127510.22★★★ |
| MRT(0–t) (h) | 2.99 ± 0.18 | 6.60 ± 0.39★★★ | 6.45 ± 0.52★★★ |
| CLz (L h−1 kg−1) | 0.058 ± 0.012 | 0.01 ± 0.001★★★ | 0.007 ± 0.001★★★** |
| t 1/2 (h) | 2.33 ± 0.53 | 6.32 ± 1.33★★★ | 5.52 ± 1.38★★ |
| Pharmacokinetic parameters | AS liposomal CPT-11 | TEA-SBE-β-CD liposomal CPT-11 | TEA-SOS liposomal CPT-11 LPs |
|---|---|---|---|
| Pharmacokinetic parameters were calculated for F-CPT-11 after the intravenous injection of CPT-11 liposomes at 5 mg kg−1. Values represent the mean ± SD (n = 5). p < 0.05 (★), p < 0.01 (★★) and p < 0.001 (★★★) versus AS liposomal CPT-11 as the control. | |||
| AUC(0–t) (ng L−1 h−1) | 44968.31 ± 11775.20 |
96506.50 ± 22702.21★★ |
67177.53 ± 32943.54 |
| AUC(0–∞) (ng L−1 h−1) | 45152.20 ± 12006.30 |
104630.83 ± 28683.36★★ |
67788.94 ± 32956.49 |
| MRT(0–t) (h) | 5.03 ± 0.63 | 11.28 ± 1.31★★★ | 9.72 ± 3.18★ |
| CLz (L h−1 kg−1) | 0.12 ± 0.031 | 0.27 ± 0.41★★ | 0.089 ± 0.039 |
| t 1/2 (h) | 4.29 ± 1.46 | 8.67 ± 2.72★ | 6.73 ± 1.86★ |
| Pharmacokinetic parameters | AS liposomal CPT-11 | TEA-SBE-β-CD liposomal CPT-11 | TEA-SOS liposomal CPT-11 |
|---|---|---|---|
| Pharmacokinetic parameters were calculated for SN-38 after the intravenous injection of CPT-11 liposomes at 5 mg kg−1. Values represent the mean ± SD (n = 5). p < 0.05 (★), p < 0.01 (★★) and p < 0.001 (★★★) versus AS liposomal CPT-11 as the control. | |||
| AUC(0–t) (ng L−1 h−1) | 49950.66 ± 14203.54 |
72878.63 ± 17371.80 |
59743.30 ± 7,925.85 |
| AUC(0–∞) (ng L−1 h−1) | 61337.22 ± 22224.52 |
191025.44 ± 140791.81 |
252664.79 ± 80033.28★★★ |
| MRT(0–t) (h) | 7.84 ± 1.86 | 10.91 ± 1.40★ | 11.08 ± 0.42★★ |
| CLz (L h−1 kg−1) | 0.092 ± 0.038 | 0.036 ± 0.019★ | 0.022 ± 0.008★★ |
| t 1/2 (h) | 9.57 ± 3.00 | 28.33 ± 14.35★★★ | 63.02 ± 24.52★★★ |
After liposomes were injected into the systemic circulation, the clearance of the liposomal drug was dependent on: (i) the clearance of the liposomal nanocarrier due to interaction with plasma proteins or phagocytosis of reticuloendothelial system; (ii) the disassociation of the entrapped drug in the liposomes inner; (iii) the clearance and metabolism of the released drug.31,33,34 Despite having the same lipid components, TEA-SOS was more supportive in constructing a sterically stabilized liposomes system, as shown in Fig. 4 and Tables 2–4. The half-lives of T-CPT-11 for TEA-SOS liposomal CPT-11 and TEA-SBE-β-CD liposomal CPT-11 were 5.52 and 6.32 h; significantly longer than that of AS liposomal CPT-11 (2.328 h) (p < 0.01 and p < 0.001). Moreover, the AUC(0–t) values were 645543.45, 468465.57 and 278006.44 ng L−1 h−1 for TEA-SOS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11, respectively.
 | ||
| Fig. 4 Bio-distribution of T-CPT-11 levels in the main tissues including the heart (A), spleen (B), liver (C), lung (D) and kidney (E), and metabolite SN-38 levels in the liver (F) in KM mice injected with AS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and TEA-SOS liposomal CPT-11 at 5 mg kg−1, and F-CPT-11/T-CPT-11 ratio, namely F in the liver (G) and spleen (H). Data are shown as mean ± SD (n = 3). | ||
To fully understand the therapeutic efficacy and side effects of the liposomal formulation, the pharmacokinetics of the released CPT-11 were further evaluated. The ratio of F-CPT-11 concentration to T-CPT-11 concentration in plasma was then calculated. We observed that the percentage of F-CPT-11 between the three formulations were different. In the case of TEA-SOS liposomal CPT-11, 95.33% of detected CPT-11 was encapsulated in liposomes, and only 4.67% was present in F-CPT-11 at 6 h after intravenous administration. In contrast, CPT-11 cannot form a precipitate with SBE-β-CD and AS inside liposomes, and the amount of free drug outside the liposomes was relatively high, with 19.09% of detected CPT-11 present in F-CPT-11 in TEA-SBE-β-CD liposomal CPT-11 and 20.92% in AS liposomal CPT-11. Accordingly, TEA-SOS liposomal CPT-11 prepared by a remote loading method with TEA-SOS gradient showed different in vivo release behavior. TEA-SOS gradient resulted in a close interaction between the SOS and CPT-11 and a lower release rate in the systemic circulation. Moreover, TEA-SOS liposomal CPT-11 showed the lowest F-CPT-11 levels, which could reduce the gastrointestinal toxicity.
Bio-distribution study
Tissue distribution (including heart, liver, spleen, lung and kidney) was evaluated at 6, 24 and 48 h after an intravenous injection of 5 mg kg−1 of TEA-SOS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11. The results are shown in Fig. 4.In the liver, CPT-11 concentration at 6 h after administration of TEA-SOS liposomal CPT-11 was approximately 3-fold higher than that of TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11. At 24 h, CPT-11 concentration in TEA-SBE-β-CD liposomal CPT-11 was about the same as that at 6 h. However, the accumulation of CPT-11 in the spleen after injection of AS liposomal CPT-11 and TEA-SOS liposomal CPT-11 was 5-fold and 9-fold higher than that of TEA-SBE-β-CD liposomal CPT-11, respectively. Similarly, the accumulation of CPT-11 in the heart also showed a significant difference between groups. To further detect the drug state (encapsulated or released), F-CPT-11 in the liver and spleen was quantified, and the F-CPT-11/T-CPT-11 ratio, namely F (Fig. 3G, H) was calculated. In the liver, 99.3% of the encapsulated CPT-11 was released from the AS liposomal CPT-11 at 24 h after intravenous administration. In contrast, 30.3% CPT-11 was released from TEA-SBE-β-CD liposomal CPT-11, and almost no CPT-11 was released from TEA-SOS liposomal CPT-11. In the spleen, the F value also showed a significant difference between the three liposomal formulations.
In this study, AS was unable to form a stable physical state to retain CPT-11, and indeed a transmembrane gradient might be generated. The encapsulated CPT-11 in AS liposomal CPT-11 underwent rapid and total release in the liver. Unlike AS, as polyanions, SOS and SBE-β-CD were able to cross-link drug molecules at the intra-liposomal phase; therefore, this could reduce the leakage of CPT-11 both in vitro and in vivo. Stable drug entrapment of TEA-SOS liposomal CPT-11 could reduce premature drug release from liposomes to normal tissues, resulting in a lower F-CPT-11 percentage and reducing the conventional hepatotoxicity associated with F-CPT-11. As mentioned above, polyanions are indispensable to improve drug retention and achieve stable drug encapsulation.
In vivo antitumor activity
Male nude mice bearing HT-29 cells were used to evaluate the in vivo antitumor efficacy of AS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and TEA-SOS liposomal CPT-11, the results are summarized in Fig. 5. Hepatorenal function parameters and H&E staining of the major organs, tumors after treatment were shown in Fig. 6. CPT-11 liposomes were administered to mice at two dosages: 10 mg kg−1 for three formulations and 25 mg kg−1 for TEA-SOS liposomal CPT-11 and TEA-SBE-β-CD liposomal CPT-11. As shown in Fig. 5A, the results revealed that (1) the therapeutic effect was dose-dependent; (2) TEA-SOS liposomal CPT-11 at 25 mg kg−1 was the most efficacious of all the formulations, and the tumor completely disappeared; (3) AS liposomal CPT-11 exhibited poor antitumor efficacy; (4) for treatment with TEA-SOS liposomal CPT-11 and TEA-SBE-β-CD liposomal CPT-11 at 10 mg kg−1, the antitumor activity was only maintained for 24 days, and re-growth of the tumor occurred after the final administration. H&E-stained pathological sections showed different levels of apoptosis in the tumor sections of TEA-SOS liposomal CPT-11, TEA-SBE-β-CD liposomal CPT-11 and AS liposomal CPT-11. Moreover, compared with mice treated with TEA-SOS liposomal CPT-11 at 25 mg kg−1, mice treated with AS liposomal CPT-11 at 10 mg kg−1 exhibited significant weight loss (p < 0.001). The weight loss may be connected with massive drug leakage of AS liposomal CPT-11 in systemic circulation, as released drug contributes significant toxicity.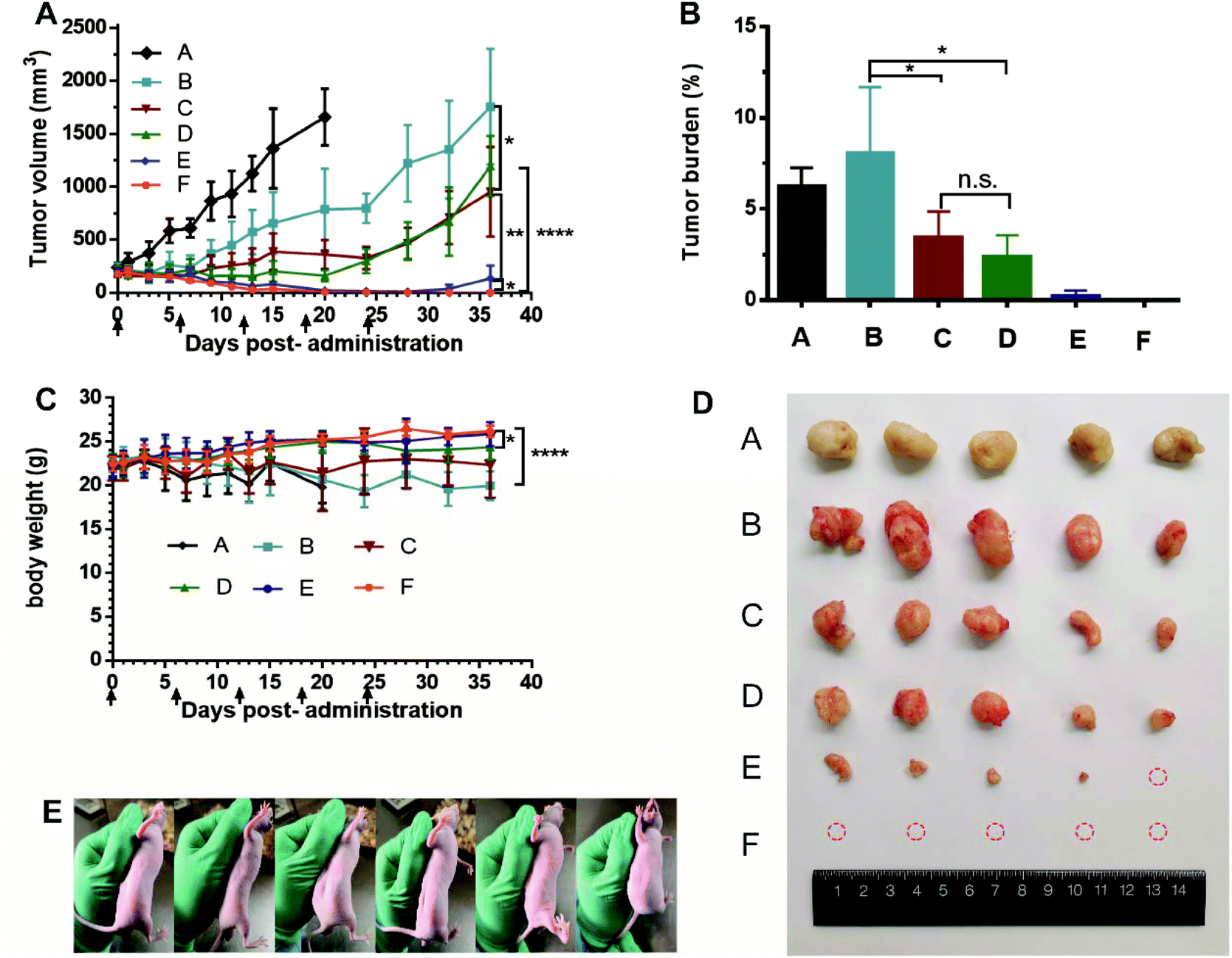 | ||
| Fig. 5 In vivo antitumor activity of CPT-11 liposomes against HT 29 tumors (n = 5). (Group A: Saline, Group B: AS liposomal CPT-11 at 10 mg kg−1, Group C: TEA-SBE-β-CD liposomal CPT-11 at 10 mg kg−1, Group D: TEA-SOS liposomal CPT-11 at 10 mg kg−1, Group E: TEA-SBE-β-CD liposomal CPT-11 at 25 mg kg−1, Group F: TEA-SOS liposomal CPT-11 at 25 mg kg−1). Tumor growth profiles treated with different CPT-11 liposomes formulations (A), tumor burden after the last treatment (B), body weight variations during treatment (C), images of tumors at day 36 (D), images of nudes injected with TEA-SOS liposomal CPT-11 at 25 mg kg−1 at day 36 (E). | ||
 | ||
| Fig. 6 Liver and kidney functional parameters at day 36 after scarified (A). Images of the different organ sections and tumors stained with H&E after various treatments (B). | ||
As can be seen from the rapid in vitro and in vivo release profiles, the AS gradient results in a weak interaction between the AS molecule and the CPT-11 molecule, and a lower ability to retain CPT-11 inside the liposomes. Thus, more CPT-11 may escape to the blood circulation before AS liposomal CPT-11 reaches the tumor sites, so the antitumor efficacy of AS liposomal CPT-11 in vivo may be less than that of TEA-SBE-β-CD liposomal CPT-11 and TEA-SOS liposomal CPT-11. Accordingly, the superior antitumor efficacy of TEA-SOS liposomal CPT-11 was attributed to several aspects: (i) improved stability and enhanced circulation time in vivo by PEGylation, (ii) high D/L ratio and crystallization inside liposomes which further improves the benefits of liposomal formulation and produces an adequate drug concentration, (iii) stable drug entrapment which can reduce premature drug release from liposomes. These aspects cover all stages influencing the biological activity of the liposomal drug, resulting in superior chemotherapeutic efficacy of TEA-SOS liposomal CPT-11.
Conclusion
It is known that remote loading is an excellent technology for preparing sterically stabilized liposomes. In this study, intra-liposomal trapping agents including AS, SBE-β-CD and SOS were used for remote loading of CPT-11 liposomes. Bundles of crystal fibers were observed inside TEA-SOS liposomal CPT-11 using cryo-TEM. Additionally, retention properties of encapsulated CPT-11 in the three liposomes formulations were also evaluated. TEA-SOS liposomal CPT-11 showed measurably slower drug release, a longer circulation time and more potent antitumor efficacy. By contrast, AS liposomal CPT-11 resulted in a rapid release and poor antitumor efficacy. This research confirmed that CPT-11 liposome prepared using a TEA-SOS gradient, with a higher drug-lipid ratio, provides the best antitumor efficacy. To the best of our knowledge, this study is the first deep investigation into how the trapping agents influence the effectiveness of a remote loading liposomes system.Conflicts of interest
The authors declare that they have no conflicts of interest to disclose.References
- D. W. Deamer, R. C. Prince and A. R. Crofts, Biochim. Biophys. Acta, 1972, 274, 323–335 CrossRef CAS.
- E. Mayhew, D. Papahadjopoulos, Y. M. Rustum and C. Dave, Ann. N. Y. Acad. Sci., 1978, 308, 371–386 CrossRef CAS PubMed.
- Y. Barenholz, J. Liposome Res., 2003, 13, 1–8 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- G. Batist, J. Barton, P. Chaikin, C. Swenson and L. Welles, Expert Opin. Pharmacother., 2002, 3, 1739–1751 CrossRef CAS PubMed.
- D. D. Lasic, B. Ceh, M. C. Stuart, L. Guo, P. M. Frederik and Y. Barenholz, Biochim. Biophys. Acta, 1995, 1239, 145–156 CrossRef.
- V. Wasserman, P. Kizelsztein, O. Garbuzenko, R. Kohen, H. Ovadia, R. Tabakman and Y. Barenholz, Langmuir, 2007, 23, 1937–1947 CrossRef CAS PubMed.
- S. Clerc and Y. Barenholz, Biochim. Biophys. Acta, 1995, 1240, 257–265 CrossRef.
- Y. Avnir, R. Ulmansky, V. Wasserman, S. Even-Chen, M. Broyer, Y. Barenholz and Y. Naparstek, Arthritis Rheum., 2008, 58, 119–129 CrossRef CAS PubMed.
- D. C. Drummond, C. O. Noble, Z. X. Guo, K. Hong, J. W. Park and D. B. Kirpotin, Cancer Res., 2006, 66, 3271–3277 CrossRef CAS PubMed.
- C. Li, J. Cui, C. Wang, Y. Li, L. Zhang, X. Xiu, Y. Li, N. Wei, L. Zhang and P. Wang, J. Pharm. Pharmacol., 2011, 63, 765–773 CrossRef CAS PubMed.
- K. M. Laginha, S. Verwoert, G. J. R. Charrois and T. M. Allen, Clin. Cancer Res., 2005, 11, 6944–6949 CrossRef CAS PubMed.
- M. J. W. Johnston, S. C. Semple, S. K. Klimuk, K. Edwards, M. L. Eisenhardt, E. C. Leng, G. Karlsson, D. Yanko and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 55–64 CrossRef CAS PubMed.
- G. Haran, R. Cohen, L. K. Bar and Y. Barenholz, Biochim. Biophys. Acta, 1993, 1151, 201–215 CrossRef CAS.
- T. M. Allen, T. Mehra, C. Hansen and Y. C. Chin, Cancer Res., 1992, 52, 2431–2439 CAS.
- M. B. Bally, R. Nayar, D. Masin, P. R. Cullis and L. D. Mayer, Cancer Chemother. Pharmacol., 1990, 27, 13–19 CrossRef CAS PubMed.
- M. J. Johnston, S. C. Semple, S. K. Klimuk, K. Edwards, M. L. Eisenhardt, E. C. Leng, G. Karlsson, D. Yanko and P. R. Cullis, Biochim. Biophys. Acta, 2006, 1758, 55–64 CrossRef CAS PubMed.
- C. Li, J. Cui, Y. Li, C. Wang, Y. Li, L. Zhang, L. Zhang, W. Guo, J. Wang, H. Zhang, Y. Hao and Y. Wang, Eur. J. Pharm. Sci., 2008, 34, 333–344 CrossRef CAS PubMed.
- C. Li, J. Cui, C. Wang, J. Cao, L. Zhang, Y. Li, M. Liang, X. Xiu, Y. Li, N. Wei and C. Deng, J. Liposome Res., 2012, 22, 42–54 CrossRef CAS PubMed.
- C. Li, J. Cui, Y. Li, C. Wang, Y. Li, L. Zhang, L. Zhang, W. Guo, J. Wang, H. Zhang, Y. Hao and Y. Wang, Eur. J. Pharm. Sci., 2008, 34, 333–344 CrossRef CAS PubMed.
- J. Cui, C. Li, W. Guo, Y. Li, C. Wang, L. Zhang, L. Zhang, Y. Hao and Y. Wang, J. Controlled Release, 2007, 118, 204–215 CrossRef CAS PubMed.
- A. L. Seynhaeve, S. Hoving, D. Schipper, C. E. Vermeulen, G. de Wiel-Ambagtsheer, S. T. van Tiel, A. M. Eggermont and T. L. Ten Hagen, Cancer Res., 2007, 67, 9455–9462 CrossRef CAS PubMed.
- M. Ramesh, P. Ahlawat and N. R. Srinivas, Biomed. Chromatogr., 2010, 24, 104–123 CrossRef PubMed.
- J. Fassberg and V. J. Stella, J. Pharm. Sci., 1992, 81, 676–684 CrossRef CAS PubMed.
- F. C. Passero Jr., D. Grapsa, K. N. Syrigos and M. W. Saif, Expert Rev. Anticancer Ther., 2016, 16, 697–703 CrossRef PubMed.
- W. Yang, Z. Yang, J. Liu, D. Liu and Y. Wang, Asian J. Pharm. Sci., 2018 DOI:10.1016/j.ajps.2018.08.003.
- D. B. Fenske and P. R. Cullis, in Liposomes, Pt E, ed. N. Duzgunes, Elsevier Academic Press Inc., San Diego, 2005, vol. 391, pp. 7–40 Search PubMed.
- D. B. Fenske, K. F. Wong, E. Maurer, N. Maurer, J. M. Leenhouts, N. Boman, L. Amankwa and P. R. Cullis, Biochim. Biophys. Acta, 1998, 1414, 188–204 CrossRef CAS.
- X. Li, D. J. Hirsh, D. Cabral-Lilly, A. Zirkel, S. M. Gruner, A. S. Janoff and W. R. Perkins, Biochim. Biophys. Acta, 1998, 1415, 23–40 CrossRef CAS.
- J. Cui, C. Li, W. Guo, Y. Li, C. Wang, L. Zhang, L. Zhang, Y. Hao and Y. Wang, J. Controlled Release, 2007, 118, 204–215 CrossRef CAS PubMed.
- J. X. Cui, C. L. Li, W. M. Guo, Y. H. Li, C. X. Wang, L. Zhang, L. Zhang, Y. L. Hao and Y. L. Wang, J. Controlled Release, 2007, 118, 204–215 CrossRef CAS PubMed.
- P. R. Harrigan, K. F. Wong, T. E. Redelmeier, J. J. Wheeler and P. R. Cullis, Biochim. Biophys. Acta, 1993, 1149, 329–338 CrossRef CAS.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419–436 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |