
Heterogeneous chemistry in the 3-D state: an original approach to generate bioactive, mechanically-competent bone scaffolds
Anna
Tampieri
 *a,
Andrea
Ruffini
a,
Alberto
Ballardini
a,
Monica
Montesi
a,
Silvia
Panseri
a,
Francesca
Salamanna
b,
Milena
Fini
b and
Simone
Sprio
*a
*a,
Andrea
Ruffini
a,
Alberto
Ballardini
a,
Monica
Montesi
a,
Silvia
Panseri
a,
Francesca
Salamanna
b,
Milena
Fini
b and
Simone
Sprio
*a
aInstitute of Science and Technology for Ceramics, National Research Council, Via Granarolo 64, 48018 Faenza, Italy. E-mail: simone.sprio@istec.cnr.it; anna.tampieri@istec.cnr.it
bLaboratory of Preclinical and Surgical Studies, Rizzoli-RIT Department, IRCCS-Rizzoli Orthopedic Institute Bologna, Via di Barbiano 1/10, 40136, Italy
First published on 13th November 2018
Abstract
The present work investigates heterogeneous gas–solid reactions involved in the biomorphic transformation of natural wood into large 3-D hydroxyapatite (HA) scaffolds recapitulating physico-chemical, morphological and mechanical features typical of natural bone. In particular, we found that the use of a reactive CO2/H2O gas mixture, under supercritical conditions at high pressure, permits to control heterogeneous CaO-CO2 reactions throughout the whole bulk and to direct the nucleation-growth of CaCO3 at a relatively low temperature, thus obtaining a highly reactive 3-D precursor enabling the formation of a large biomorphic HA scaffold preserving fine nanostructure by a hydrothermal process. To the best of our knowledge, the application of heterogeneous chemical reactions in the 3-D state is an original way to generate large HA scaffolds maintaining bio-relevant ionic substitutions, with specific regard to Mg2+, Sr2+ and CO32− ions, conferring a superior ability to guide cell fate. We hypothesize that the original nanostructure of the final 3-D HA scaffold, not achievable by the classic sintering procedure, and the multi-scale hierarchical organization inherited by the original template, account for its high compression strength with damage-tolerant mechanical behaviour. The ability of the new scaffold to induce bone regeneration is attested by the overexpression of genes, early and late markers of the osteogenic differentiation pathway, and by the in vivo osteoinductivity. We hypothesize that the unique association of bioactive chemical composition, nanostructure and multi-scale hierarchy can synergistically act as instructing signals for cells to generate new bone tissue with organized 3-D architecture. These results point to its great applicative potential for the regeneration of large bone defects, which is a still unmet clinical need.
1. Introduction
Nanotechnology is a pioneering discipline aimed at fabricating new devices capable of superior smart performance, thanks to the unusual properties of nanomaterials even when assembled into 3-D solid bodies. The great majority of previous studies report the fabrication of low-dimensionality nanometric systems (e.g. 0-D to 2-D materials such as nano-dots, nano-wires and nano-wells) by chemical synthesis or supramolecular organization of atomic building blocks.1 Conversely, the fabrication of functional 3-D inorganic materials retaining nanostructure poses significant challenges. In fact, the current processing to consolidate inorganic materials with relevant mechanical performance requires the use of sintering treatments, which normally degrade the metastable phases, the crystal ordering and nano-size. Lattice distortion, atom vacancies or site-specific ion doping are all features which raise the internal crystal energy, thus triggering higher reactivity and responsiveness to external stimuli, in respect to phases characterized by more ordered crystal domains and stoichiometric composition.2–6A very relevant case study is given by nanocrystalline calcium phosphates (CaPs) exhibiting multiple ionic substitutions (e.g. Mg2+, CO32−, and Sr2+), which are well known as the elective phases to develop scaffolds for bone regeneration.7–11 Although, when in the form of nano-powders, the synthesis of such bioactive ceramics is not a critical task,12,13 the production of ion-doped hydroxyapatite (HA) as 3-D scaffolds endowed with adequate mechanical performance is a challenge that the scientific community is intensively facing. In fact, current fabrication methods do not allow accurate control of crystal and compositional changes, particularly during the consolidation process, where irreversible crystal growth and chemical stabilization occurs, reducing the scaffold bioactivity.5
In attempting to preserve such an “inorganic metabolic phase” in the form of 3-D scaffolds, previous studies reported a biomineralization process, i.e. the nucleation of apatite nano-phase onto self-assembling collagen fibrils. This process activates multi-scale control mechanisms: the mineral phase is confined to the nano-scale by interaction with a collagen matrix, thus maintaining phase instability and dynamic ion exchange.5 The final product of such a technology is fibrous, porous scaffolds able to deform elastically in liquid media, thus showing remarkable adaptability to tissue defects but, at the same time, inability to retain its original volume under compressive forces. On the other hand, self-hardening bioactive phases such as CaP cements could also represent an option to obtain bioactive 3-D scaffolds.14 Unfortunately, the chemical reaction and setting of metastable CaP yielding bioactive HA cannot be controlled to give pore size and interconnectivity favouring osteoconduction and extensive vascularization, coupled with adequate mechanical performance.15
To obtain scaffolds with mechanical strength enabling their application in critical size bone defects, bioactive ceramic powders have been used in association with biocompatible and bio-erodible polymers. The as-obtained printable pastes have been used for the fabrication of 3-D porous scaffolds by additive manufacturing techniques.16 However, besides the scarce compositional and mechanical similarity with bone tissue, polymer-based scaffolds often exhibit non-enzymatic dissolution profiles for which acidic and harmful by-products can be released, altering cell metabolism and vitality, thus jeopardizing tissue healing, particularly in the case of large bone defects.17
Striving to generate 3-D bodies with unique features, new technological approaches are needed, relying on the control of chemical reactions and mass transfer under non-equilibrium conditions, rather than under stable thermodynamic regimes.18,19 Emerging technologies to build 3-D bioceramics might be conceived as a process governed by concomitant nucleation/growth/assembling phenomena directly occurring in the 3-D state, preventing crystal growth and ordering.
In a previous study20 we investigated the biomorphic transformation of natural wood into 3-D HA scaffolds by a multi-step process; however, some inherent limitations came up, since only small pieces could be transformed, and the biological and mechanical features of the final product were still poor. In the present work we explore in depth one stage of a multi-step process consisting in the transformation of CaO into CaCO3 in the 3-D state, with the purpose of obtaining a highly reactive precursor enabling subsequent transformation into 3-D apatitic scaffolds with superior biological and mechanical properties.
We established heterogeneous reactions between CO2 gas in the supercritical state and solid biomorphic CaO templates, and we found that the kinetic control of the temperature- and time-dependent grain growth makes it possible to obtain final 3-D scaffolds with bioactive composition and nanostructure, at the basis of desired functional performances. Previous studies described the use of heterogeneous solid–gas reactions, using supercritical carbon dioxide under wet conditions, to transform micro-nanoparticles into calcium carbonate.21–23 However, to the best of our knowledge, no studies have been reported so far on the nucleation/transformation of ceramics, occurring in the 3-D state, where interface reactions and diffusive processes play a crucial role, particularly in activating internal tensions and deformations hindering the maintenance of 3-D structural integrity.
The obtained results confirm the potential of 3-D heterogeneous chemistry as an original approach to generate bone scaffolds suitable for the regeneration of large portions of bone, hitherto prevented by critical limitations in the existing bone graft materials.
2. Materials and methods
2.1 Materials synthesis
Cylindrical Rattan wood pieces (Calamus manna) were used as 3-D templates guiding the transformation process. The template was preliminarily heated at 1000 °C (heating/cooling rate: 1 °C min−1) under nitrogen gas flow, to eliminate the organic components and achieve a porous carbon body. The obtained body was then placed in a vessel containing metallic Ca granules (Sigma Aldrich, USA), and subjected to heating up to 1250 °C (heating/cooling rate: 2 °C min−1) to allow sublimation of calcium in an oxygen-free environment and under partial vacuum (P = 100 mbar), thus activating the gas–solid Ca–C reaction. The resulting CaC2 was converted into calcium oxide by heating at 900 °C under air atmosphere. Then, further conversion of the resulting CaO into CaCO3 was carried out by heating under a flux of carbon dioxide under non-isothermal and isobaric conditions (P = 100 atm) maintained by a back-pressure regulator. This process was carried out under dry or wet conditions from room temperature to above 800 °C. Wet conditions were established by using hydrated carbon dioxide with a molar ratio of CO2/H2O of ∼10. Under the above applied pressure, CO2 is in the supercritical state in the whole range of temperatures investigated.Then, the obtained CaCO3 body was placed in a closed reactor containing a 2.0 M solution of (NH4)2HPO4 (Sigma Aldrich, USA) buffered at pH 8.5, and heated at 220 °C under water vapour pressure (∼20 bar) to activate CO3 ↔ PO4 ion exchange in the whole solid. Finally, a conclusive treatment by soaking in an aqueous solution containing 0.5 M Sr(NO3)2 and 0.5 M Mg(NO3)2 ions for 24 h at 50 °C, buffered at pH 7.0, was carried out to obtain a biomorphic apatite body presenting multiple doping with Mg2+ and Sr2+ ions (henceforth termed B-HA).
2.2 Materials characterization
Physico-chemical characterization was carried out on samples crushed into fine powders by mortar and pestle.The evaluation of the carbonate content was made by thermogravimetric analysis (TGA) of dried samples using a Stanton STA 1500 (Stanton, London, UK). About 10 mg of the sample was weighed in an alumina crucible and heated from room temperature to 1100 °C under nitrogen flow. The heating rate was 10 °C min−1. The carbonate content was evaluated on the basis of the weight loss detected between 600 °C and 1100 °C, following the method described in a previous study.24
Fourier transform infrared spectroscopy (FTIR) was performed by using a Nicolet 380 spectrometer (Thermo-Fisher Scientific Inc., Waltham, MA, USA). The FTIR spectra were recorded in the wavelength range of 4000 to 400 cm−1 on small pellets obtained by mixing 1 mg of the sample powder with 150 mg of anhydrous potassium bromide (KBr). A pure KBr disk was used as a blank.
The ion release profile was obtained by two methods: (i) by immersing tablets obtained by uniaxial pressing of pulverized samples (1.5 g of powder, 10 mm in diameter, 2 mm in height) in 1 litre of pH = 7.3 buffer solution (TRIZMA buffer, Sigma Aldrich); (ii) by immersing scaffolds in cell culture medium (DMEM Glutamax medium (Gibco) containing 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin (100 U ml−1–100 μg mL−1)) to simulate the conditions of in vitro cell culture (see below, Section 2.2.6). The scaffold weight/medium volume ratio was about 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15. In both methods, the solution containing the released ions was maintained at 37 °C. 1 ml of solution was taken out at scheduled times (i.e. after 0.25, 1, 2, 3, 7 and 14 days) and analysed by ICP-OES for the quantitative determination of ions released by the sample. All the experiments were performed in triplicate.
:15. In both methods, the solution containing the released ions was maintained at 37 °C. 1 ml of solution was taken out at scheduled times (i.e. after 0.25, 1, 2, 3, 7 and 14 days) and analysed by ICP-OES for the quantitative determination of ions released by the sample. All the experiments were performed in triplicate.
The B-HA scaffolds were scanned with the high-resolution microtomography system Skyscan 1176 (Bruker Micro-CT, Belgium) at 50 kV and 500 μA using an aluminium filter of 0.50 mm while the ex vivo samples were scanned at 80 kV and 300 μA using a copper and aluminium filter. The nominal resolution (pixel size) was set for all samples at 17.50 μm. The images obtained from acquisition were then reconstructed by using the software NRecon (Bruker MicroCT, Belgium) with corrections for alignment, depending on acquisition, beam hardening correction and ring artefact reduction. The resulting images were saved in jpg 8-bit format.
Mercury porosimetry was used to evaluate pore size distribution (<50 μm) by two different apparatus (Carlo-Erba Porosimeter 2000 and Macropores Unit 120) working on separate pore size ranges. The open and total porosity of the studied ceramics was measured by Archimedes’ method and geometrical weight-volume evaluation, respectively. The specific surface area (SSA) of the scaffold was measured by the nitrogen adsorption method, following the Brunauer–Emmett–Teller (BET) model (Sorpty 1750, Carlo Erba, Milan, Italy).
Each sample (diameter 8 mm, height 4 mm), sterilized by 25 kGy γ-ray radiation prior to use, was placed one per well in a 24-well plate and pre-soaked in culture medium for 72 h at 37 °C. A sintered commercial porous HA body (S-HA) was used as the control.
For the cell viability and cell morphology study, the samples were seeded by carefully dropping 20 μl of cell suspension (5.0 × 104 cells) onto the scaffold upper surface, and allowing cell attachment for 20 min in the incubator, before the addition of 1 ml of cell culture medium (DMEM Glutamax Gibco, 10% FBS and 1% penicillin–streptomycin (100 U ml−1–100 μg mL−1)).
For the osteogenic gene expression profile, the U-CUP perfusion bioreactor system (Cellec Biotek AG) was used. Briefly, mMSCs were seeded at 2.0 × 106 cells per scaffold with a bidirectional flow rate of 3 ml min−1 for 18 h, and then all the media were collected and the seeding efficiency was evaluated by counting the number of cells left in the culture media after trypan blue staining.
The cell-seeded constructs were then cultured with a bidirectional perfusion flow rate of 0.3 ml min−1 for an additional 14 days.25 The medium was changed twice a week. All the cell-handling procedures were performed in a sterile laminar flow hood. All cell-culture incubation steps were performed at 37 °C with 5% CO2.
2.2.6.1 Cell viability assay. A Live/Dead assay kit (Invitrogen) was used according to the manufacturer's instructions. Briefly, the samples were washed with 1× PBS for 5 min and incubated with calceinacetoxymethyl (Calcein AM) (2 μM) plus ethidium homodimer-1 (EthD-1) (4 μM) for 15 min at 37 °C in the dark; then the samples were rinsed in 1× PBS.26 Images were acquired using a Nikon inverted Ti-E fluorescence microscope (Nikon). One sample per group was analysed at day 1.
2.2.6.2 Cell morphology evaluation. One sample per group was used for fluorescence and SEM analysis at day 1 and day 3 of cell culture. In order to visualize actin filaments, samples were washed with PBS 1× for 5 min, fixed with 4% (w/v) paraformaldehyde for 15 min and washed with PBS 1× for 5 min. Permeabilization was performed with PBS 1× with 0.1% (v/v) Triton X-100 for 5 min. FITC-conjugated Phalloidin (Invitrogen) (38 nM) in PBS 1× was added for 20 min at room temperature in the dark.27
Cells were washed with PBS 1× for 5 min and incubated with the nuclear stain DAPI (Invitrogen) (300 nM) in 1× PBS for 5 min. The nuclear morphological changes were also evaluated. Images were acquired by using an Inverted Ti-E fluorescence microscope (Nikon).
For SEM analysis, after 1 day one sample per group was washed with 0.1 M sodium cacodylate buffer, pH 7.4, and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, for 2 h at 4 °C, washed with 0.1 M sodium cacodylate buffer, pH 7.4, and dehydrated in a graded series of ethanol for 10 min each. Dehydrated samples were sputter-coated with gold and observed using a Quanta Scanning Electron Microscope (ESEM Quanta 200, FEI).
2.2.6.3 Quantitative real-time polymerase chain reaction (q-PCR). At day 14, cells grown on B-HA and S-HA samples, used as a calibrator, were homogenized and total RNA extraction was performed by the use of the Tri Reagent, followed by a Direct-zol™ RNA MiniPrep kit (Euroclone) according to the manufacturer's instructions. RNA integrity was analysed by native agarose gel electrophoresis and quantification performed by using a Qubit® 2.0 fluorometer together with a Qubit® RNA BR assay kit following the manufacturer's instructions. Total RNA (500 ng) was reverse transcribed to cDNA using a High-Capacity cDNA Reverse Transcription kit according to the manufacturer's instructions. Quantification of gene expression, using Taqman assays (Applied Biosystems), for Runt-related transcription factor 2 (Runx2, Mm01340178), alkaline phosphatase (ALP, Mm00475834), bone morphogenetic protein 2 (BMP2, Mm01340178), Osteopontin (OPN, spp1 Mm00436767), Collagen 15 (Col15a1, Mm00456584) and glyceraldehyde 3-phosphate dehydrogenase, used as a housekeeping gene (GAPDH, Mm99999915), was performed by the use of a StepOne™ Real-Time PCR System (Applied Biosystems). 4 scaffolds for each sample were analysed using three technical replicates for each experiment. Data were collected using the OneStep Software (v.2.2.2) and relative quantification was performed using the comparative threshold (Ct) method (ΔΔCt), where relative gene expression level equals 2−ΔΔCt.28
2.2.6.4 Statistical analysis. Results were expressed as the mean ± SEM plotted on graphs. Statistical analysis was made by two-way ANOVA using ΔCt values of gene expression analysis by using GraphPad Prism software (version 6.0), with statistical significance set at p ≤ 0.05.
During the course of a safety study on the B-HA scaffold, in addition to the evaluation of the lack of adverse effects after the subcutaneous implantation at 12 weeks, we also evaluated the osteoinductive potential of the scaffold. In vivo osteoconductivity of the B-HA scaffold was assessed in 3 male rabbits (Oryctolagus cuniculus, Charles River, Lecco, Italy), 2.4 ± 0.2 kg body weight. After skin shaving and disinfection, animals were anaesthetized, and then placed in a prone position for the subcutaneous surgical implantation. 5 disks of B-HA (diameter 8 mm, height 4 mm) were implanted into the subcutaneous tissue of each rabbit at 2 to 3 cm from the midline and parallel to the spinal column, and about 2 cm apart from each other; then the surgical wound was closed with a surgical thread. The animals were euthanized at 12 weeks.
For histological examination, the retrieved implants were fixed in 4% formaldehyde, subsequently washed with distilled water, superficially decalcified, dehydrated in a graded series of alcohols and, finally, embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) and with Goldner's Trichrome. Histological analyses of osteoinduction were carried out on 6 histological sections (2 for each animal) with a digital scanner with a resolution of 0.5 μm (CS System, Aperio Technologies, Vista, CA, USA) and with an optical microscope (Olympus BX51). All sections were observed for evidence of ectopic bone formation and vascularization and a histological score was adopted according to the qualitative scoring of osteoinduction by Ranly et al. (Ranly et al., J. Bone Joint Surg. Am., 2005, 87, 2052–2064). With this system, sections with no evidence of materials or new tissue were valued as 0. The qualitative score was 1 when the material was observed but no new bone was present, 2 when an ossicle was observed, 3 when two or more ossicles were present, and 4 when at least 70% of the slide (at 10× magnification) was covered by an ossicle.
3. Results and discussion
3.1 The relevance of reactive precursors in 3-D heterogeneous chemistry
In a previous study20 we applied a multi-step process to transform natural wood into 3-D hydroxyapatite bioceramics with hierarchically organized porosity. These previous experiments gave 3-D hydroxyapatite bodies reproducing the original wood structure, but limited in size (i.e. <1 cm) and with poor mechanical strength (i.e. σC ∼2–4 MPa). After a thorough evaluation of the various steps we realized that the reaction playing a critical role in determining the features of the final product is the transformation of calcium oxide into CaCO3, which was previously performed in a CO2 atmosphere without any applied pressure. Under such conditions the formation of the calcite scale at the CaO surface was observed, limiting further penetration of reactive CO2 in the bulk template and hampering complete transformation into calcium carbonate. Moreover, the uncontrolled grain growth of CaCO3 and the lowering of specific surface area led to a poorly reactive phase affecting the subsequent transformation of CaCO3 into hydroxyapatite by the hydrothermal method.Hence, these preliminary results suggested that the achievement of 3-D CaCO3 precursors with high reactivity plays a key role in the formation of the final biomorphic apatite with large dimensions and improved biologic and mechanical performance. To achieve this aim, specific attention is paid to the ensemble of physico-chemical phenomena governing the carbonation reaction: (i) gas–solid interaction at the interface between CO2 gas and the CaO template, (ii) CO2 diffusion from the surface to the bulk, (iii) nucleation/growth of the calcium carbonate phase in the 3-D state.
The completion of the carbonation reaction is achieved by using wet CO2 gas under supercritical conditions (sc-CO2), and at a pressure of 100 atm. Under these conditions, the process can be well described by the Avrami–Erofeyev and 3-D diffusion–Jander kinetic models29 and follows a nearly linear profile up to completion at ∼650 °C (Fig. 1a, solid line). The carbonation reaction is supported by the catalytic function of thin CO2-rich aqueous film formed at the surface of the CaO template which activates the formation of Ca(OH)2 phase at the interface.30 Then, the dissolution of Ca(OH)2 in the acidic CO2/H2O layer generates Ca2+ ions at the interface, acting as nucleation sites for the CaCO3 phase, following the reaction Ca2+ + CO32− → CaCO3. By analysing the temperature-dependent equilibrium curves of the reactions involved in the carbonation process (Fig. 1b), the limiting step in the wet carbonation process is the reaction: CO2 + H2O → HCO3− + H+, which generates HCO3− ions pushing the crystallization of the CaCO3 phase. This reaction is strongly enhanced under supercritical conditions (106 times higher than in wet CO2 flux, see Fig. 1b): the use of wet sc-CO2 at a high CO2/H2O molar ratio (10:1) enhances the reaction rate, induces the formation of reactive Ca2+ ions by increased Ca(OH)2 phase dissolution into the CO2/H2O layer, and sustains the progress of the reaction Ca2+ + HCO3− → CaCO3 + H+ into the bulk until complete transformation into 3-D CaCO3. It was previously reported that the formation of Ca(OH)2 as an intermediate phase is a key aspect to achieve the complete transformation of CaO particles into CaCO3;22,23 however, when the transformation process is directly applied to 3-D bodies with multi-scale hierarchic structure, the uncontrolled formation of Ca(OH)2 is a serious drawback leading to complete disruption of the 3-D body.20 Here, the use of wet sc-CO2 under pressure and the control of the gas mixture composition allow enhanced kinetic control of the reaction and the formation of Ca(OH)2 only as a transient phase at the gas–solid interface. In fact, it is observed that the final CaCO3 body presents no damage or structural alteration; therefore, it can be hypothesized that Ca(OH)2 phase is rapidly dissociated into reacting ions to give the final CaCO3 (calcite) product already at 650 °C (Fig. 1a).
 | ||
| Fig. 1 (a) Calcium carbonate formation under dry and wet supercritical conditions; (b) equilibria of relevant reactions involved in the carbonation process. | ||
The use of high pressure pushing the CO2 gas penetration into the bulk template assure the complete carbonation. In fact, by using wet pressure-less CO2 the yield of the carbonation process is limited to ∼30% (Fig. 1a, dotted line). It was previously reported that the use of wet CO2 can prevent the formation of a passivating CaCO3 scale and renders the carbonation reaction self-sustaining when transforming CaO particles.22 However, here the process is applied to 3-D CaO bodies, that poses additional problems related to the diffusion of CO2 into the bulk structure, so that in absence of high CO2 pressure the dissolution/re-precipitation phenomena yielding the formation of CaCO3 phase could not be sustained up to the inner core of the 3-D template.
In our experiments complete carbonation could occur also using dry CO2 under similar supercritical conditions;31 however, with dry sc-CO2 at P = 100 atm complete transformation of CaO into CaCO3 could not be obtained below ∼800 °C (Fig. 1a, dashed line). In spite of the fact that both dry and wet sc-CO2 conditions can yield complete carbonation of 3-D CaO bodies, the different thermal energies involved in the two processes remarkably affect the nucleation/growth of the CaCO3 phase and, consequently, its final microstructure. In fact, Fig. 2 shows that the CaO-CO2 reaction under dry supercritical conditions generates CaCO3 phase presenting coarse grains, up to ∼10–20 μm in size (Fig. 2a). The occurrence of extensive grain growth phenomena during dry carbonation is due to the much higher temperature (i.e. ∼800 °C) required for completing the process. Conversely, when CaCO3 is obtained under wet supercritical conditions, the mechanism of dissolution/precipitation promotes nucleation phenomena and the final grain growth is limited to ∼1–2 μm (Fig. 2b). The high CO2 pressure and the presence of a thin aqueous surface film induce precipitation of CaCO3 at the particles’ contact interface and enhance the inter-particle forces. The much lower temperature needed for carbonation under wet conditions (see Fig. 1a) permits the maintenance of a higher specific surface, at the basis of the high reactivity of the as-formed CaCO3. Such a precursor maintains a perfect physical integrity and a remarkable strength thanks to the small particle size with a high specific surface, yielding enhanced intergranular cohesion. Conversely, the 3-D calcite bodies obtained under dry conditions cannot be further treated since they undergo complete disintegration in a short time. Residual mechanical stresses related to enhanced grain growth can be hypothesized.
 | ||
| Fig. 2 Microstructure of the CaCO3 precursor obtained under dry (a), and wet (b) supercritical conditions. | ||
3.2 Phosphatization of the highly reactive CaCO3 precursor and ion doping
The phosphatization process was applied to the reactive CaCO3 precursor obtained by reaction in wet sc-CO2 at 100 atm. An alkaline and buffered hydrothermal solution within the pH range of hydroxyapatite stability (∼5–11) was employed according to the solubility isotherms typical of Ca–P systems.32 SEM analysis in Fig. 3 shows the microstructure of the CaCO3 precursor at an intermediate stage during the hydrothermal process, showing the coverage of the original CaCO3 particles with lamellar apatite nanoparticles. This suggests that our experimental conditions induce dissolution of calcite grains and subsequent re-precipitation of hydroxyapatite nanocrystals as highly textured hexagonal nanosized platelets (Fig. 3a). Apatite nano-platelets are ∼20 ± 10 nm in thickness and ∼150 ± 50 nm in width (Fig. 3b), and are oriented along specific crystal directions, thus suggesting that their development occurs under a crystallographic control exerted by the CaCO3 precursor during the transformation process. This assumption confirms previous studies reporting that the heterogeneous nucleation of apatite phase preferentially occurs by epitaxial development on the surface of dissolving calcite crystals, as driven by the structural affinity between the lattice planes of calcite and of apatite crystals.33 After the hydrothermal process, a subsequent treatment was carried out in an aqueous solution containing Mg2+ and Sr2+ ions to allow their diffusion and exchange with Ca2+ ions in the 3-D structure of the as-prepared apatite scaffold. The ion exchange process firstly occurs at the surface of the apatite nanocrystals and subsequently diffuses into the crystal lattice. The doping strategy, designed to introduce the desired amount of Mg2+ and Sr2+ ions, was performed by varying the parameters influencing the diffusion rate of doping ions, i.e. T (temperature) and ion concentration, according to the Gibbs equation for ion exchange phenomena in a closed system (i.e. ΔG = RTln(Q/Kq), where Q is the reaction quotient and Kq is the standard equilibrium constant). After preliminary experiments, a temperature of 50 °C and concentrations of Mg- and Sr-nitrate salts of 0.5 M were found to be adequate to obtain ion doping to extents close to those found in natural bone tissue.34 The as-obtained 3-D biomorphic apatite scaffold presenting multiple ion doping is henceforth termed B-HA.
 | ||
| Fig. 3 SEM images of the CaCO3 microstructure collected during the phosphatization process. (a) Coverage of the original CaCO3 particles with lamellar apatite; (b) high resolution image of the newly formed apatite crystals. | ||
3.3 Physico-chemical analysis of the biomorphic apatite
Semi-quantitative analysis by XRD (Fig. 4a) reveals hydroxyapatite as the main crystalline phase, with minor amounts of β-tricalcium phosphate (βTCP, ∼5 wt%)35,36 and no other crystalline phases detected. The FTIR spectrum in Fig. 4b shows the presence of absorption bands belonging to vibration modes of CO3 groups when replacing PO43− ions in the apatite structure11 (i.e. ∼870, 1430 and 1450 cm−1). Such an ionic substitution (B-type carbonation) typically occurs in newly formed bone tissue and is a major source of structural disorder in hydroxyapatite, increasing the apatite solubility without altering the surface polar properties and the affinity of the osteoblast cells, thus favouring the bone regeneration process.36 B-type carbonation cannot be obtained in sintered hydroxyapatite bodies, since high temperature treatments provoke phase decomposition and elimination of CO32− ions. Conversely, by our process, the transformation of CaCO3 into 3-D biomorphic hydroxyapatite occurs at a very low temperature (i.e. 220 °C), thus permitting to retain nanocrystallinity and CO32− substitution in the final material.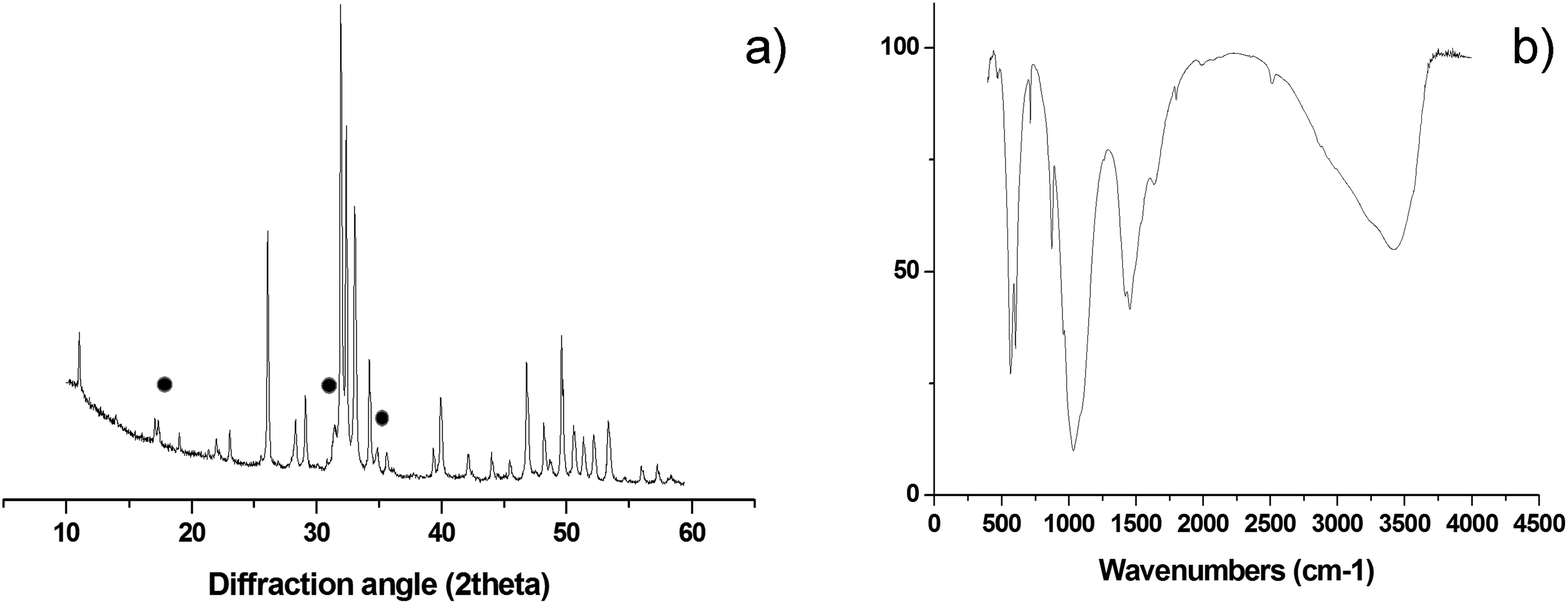 | ||
| Fig. 4 (a) XRD pattern of B-HA scaffold; the symbols point to XRD peaks belonging to the bTCP phase. All the remaining, non-marked, peaks belong to hydroxyapatite phase. (b) FTIR spectrum of B-HA scaffold. | ||
ICP-OES and TG analyses in Table 1 confirm the presence of magnesium, strontium and carbonate ions in amounts close to those found in natural bone tissue.36 The replacement of Ca2+ with Mg2+ and Sr2+ ions in the apatite structure is confirmed by the much lower Ca/P ratio, in comparison with the typical value of stoichiometric hydroxyapatite (i.e. 1.67), and also by the lower extent of calcium detected by ICP-OES. In particular, for B-HA the Ca content is found to be 33.4 wt%, whereas in stoichiometric hydroxyapatite, calcium content is 39.9 wt%. Therefore, physicochemical analyses confirm that the doping process generates a Mg, Sr-doped carbonated apatite with general formula Ca5−x−yMgxSry(PO4)3−z(CO3)zOH.
| Ca/P (molar ratio) | Mg/Ca (mol%) | Sr/Ca (mol%) | Cations/PO4 (molar ratio) | CO3/PO4 (mol%) | |
|---|---|---|---|---|---|
| B-HA | 1.55 | 3.89 | 0.97 | 1.62 | 12 |
The multiple ionic doping is further confirmed by full profile analysis of the XRD patterns, revealing a slight increase of both a and c cell parameters in the apatite phase present in B-HA, with a lattice volume expansion of ∼1% in respect to the nominal value of stoichiometric apatite (Table 2). These results agree with previous studies carried out on ion-doped apatite nanopowders synthesized by wet methods.23,37 Slight lattice distortion was detected also in the βTCP phase formed in B-HA, particularly a decrease of a parameter and, to a lesser extent, of c parameter, thus departing from the crystal structure typical of βTCP towards the values reported for the whitlockite crystal model (i.e. βTCP phase partially substituted with magnesium).38 This finding suggests that partial replacement of Ca2+ with Mg2+ occurs in both the HA and βTCP phases.
| a (Å) | c (Å) | Cell volume (Å3) | c/a | D av (nm) | |
|---|---|---|---|---|---|
| Apatite phase into B-HA | 9.45556 (56) | 6.90039 (42) | 534.29 | 0.730 | 66 ± 1 |
| βTCP phase into B-HA | 10.38307 (105) | 37.29588 (549) | 3482.12 | 3.592 | 42 ± 1 |
| Whitlockite crystal model | 10.337 | 37.068 | 3430.2 | 3.586 | — |
B-HA is capable of sustained ion release along the whole period of investigation (Fig. 5). B-HA releases Ca2+, PO43− and the doping Mg2+ and Sr2+ ions at a high release rate during the first 24 hours, followed by a slower release rate culminating in a plateau, particularly for P and Sr (Fig. 5a). Fig. 5b shows that B-HA releases similar relative amounts of Ca and P ions and – to a much higher extent – Sr2+ and, particularly, Mg2+ ions. A commercial sintered hydroxyapatite showing the same porosity extent (S-HA) is used as a control material. The release of calcium ions from S-HA is negligible; this result is reasonable, since the sintering process provokes stabilization of hydroxyapatite into a stoichiometric phase since any doping ions cannot be retained in the apatite structure. Conversely, our new synthesis approach gives a nanocrystalline apatite phase, directly as a 3-D nanostructured scaffold, presenting bone-like levels of bioactive doping ions.23,39,40 The ion release from B-HA is an index of higher solubility in respect to stoichiometric HA, which is useful to achieve bio-resorbability in vivo. This effect can be further supported by the presence of an ion-doped βTCP phase in B-HA, able to regulate cell-mediated degradation and resorption in vivo.41
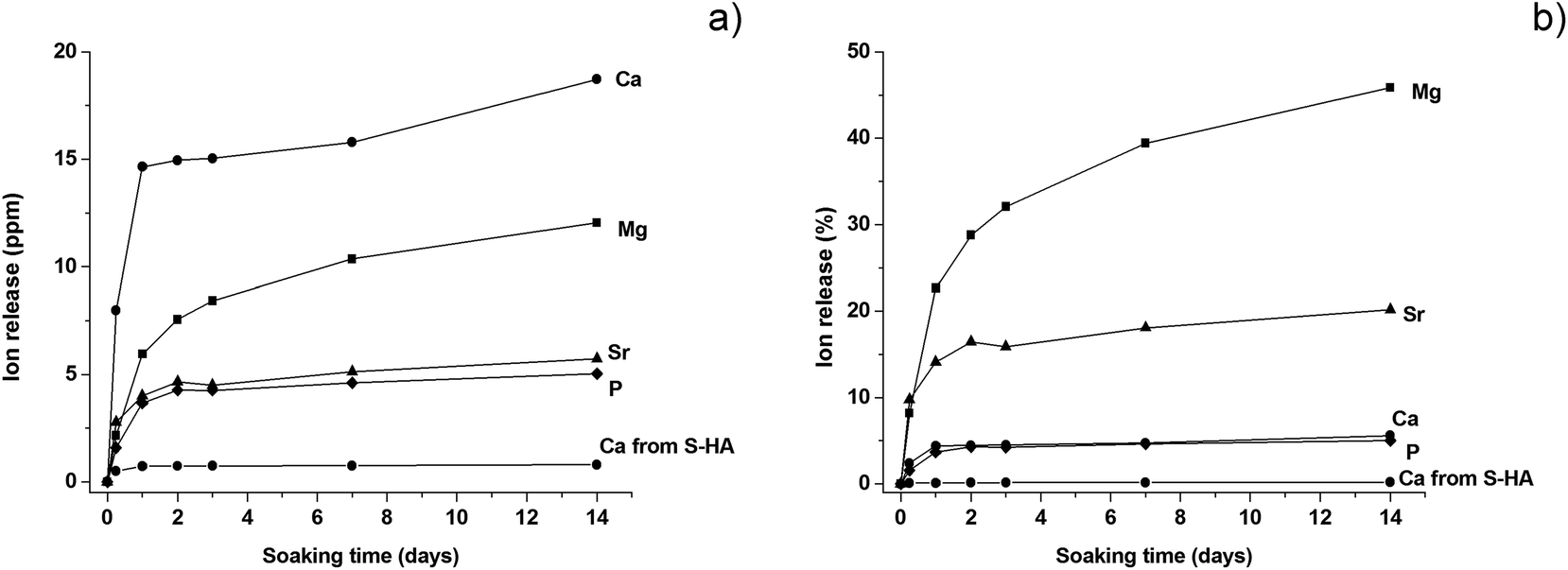 | ||
| Fig. 5 Ions release from B-HA and S-HA along 14 days of soaking in physiologic body fluid. (a) Absolute values; (b) relative release in respect to the initial ion amount. | ||
Fig. 6 shows the results of an ion release test carried out in DMEM medium, reported as ion concentrations (Fig. 6a) and as wt% in respect to the total amount of ions initially contained in B-HA (Fig. 6b). A substantial release of Sr2+ ions is observed, particularly in the first 3 days, corresponding to the pre-conditioning time (i.e. the process applied to the scaffold prior to carry out the in vitro cell tests), where most of the release (∼45 wt%) is observed, while only ∼5 wt% is released in the following 10 days (Fig. 6b).
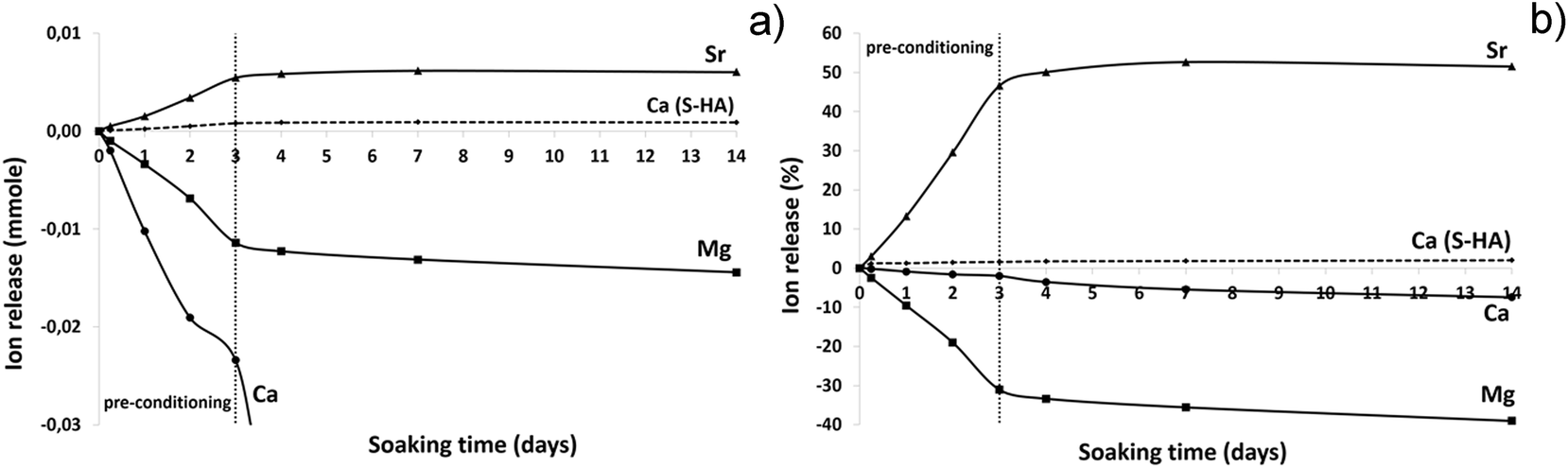 | ||
| Fig. 6 Ions release from B-HA and S-HA along 14 days of soaking in DMEM. (a) Ion concentration; (b) relative release in respect to the initial ion amount. | ||
As concerns the detection of Mg2+ and Ca2+, such ions are present in the DMEM medium at a concentration of 42.29 and 17.22 ppm, respectively and, accordingly, an ion uptake is recorded, rather than their release (Fig. 6a and b). During the step corresponding to the preconditioning, most Mg2+ ions are taken up (∼30 wt%) and then ∼10 wt% is absorbed in the following 10 days. Similar behaviour is observed for Ca2+ ions which are absorbed from DMEM to the extent of ∼10 wt%. We can hypothesize that Sr2+ ions are released since they are not present in DMEM while in the case of Mg2+and Ca2+ ions, present in large amounts in DMEM, the equilibrium is shifted towards the ion adsorption into the B-HA scaffold rather than their release from the scaffold to the medium. The high specific surface area and the exposure of charged functional groups at the surface of B-HA are responsible for the ability of ion exchange in aqueous media, much larger than in sintered apatite (S-HA), where ion release/uptake is negligible (Fig. 6a and b). The adsorption of Ca2+ and Mg2+ ions, favoured by the high surface activity, can be justified by an ion enrichment of the so-called maturation layer,42 which is a thin disordered layer in the apatite structure, playing a key role in the dynamic equilibrium of ion exchange with the physiological environment.
3.4 Morphological and mechanical analysis
The overall porosity of the B-HA scaffold is ∼60 vol%, and the extent of open porosity is ∼50 vol%, with a specific surface area of 12.30 m2 g−1. In particular, Fig. 7 shows that B-HA has anisotropic structure, with oriented channels (average diameter ∼300 μm) and diffuse smaller pores (Fig. 7a). This structure closely mimics the structural hierarchy typical of the osteonic system of compact bone,20 and the major channels are characterized by an intricate tubular morphology (Fig. 7b). Analysis at larger magnification reveals prismatic nanocrystals (i.e. ∼200 × 20 nm) with a hexagonal-like shape (Fig. 7c), forming a textured structure featuring strong inter-particle interaction, and resembling the organization of mineral crystals in natural bones.43 | ||
| Fig. 7 SEM analysis of B-HA. (a) View from the top of the scaffold, showing macroscopic longitudinal channels; (b) view of the transversal section showing interconnected smaller channels; (c) high magnification image showing the lamellar nanostructure composing the whole B-HA scaffold. | ||
B-HA shows open pores, quite uniformly distributed from the macro to the nanoscale (Fig. 8a). Micro-CT imaging (Fig. 8b) confirms that the open channels in B-HA run along the entire length of the scaffold without evident interruptions, which, thanks to extensive interconnection at the multi-scale, is extremely beneficial to ensure new bone penetration and vascularization in the whole scaffold.
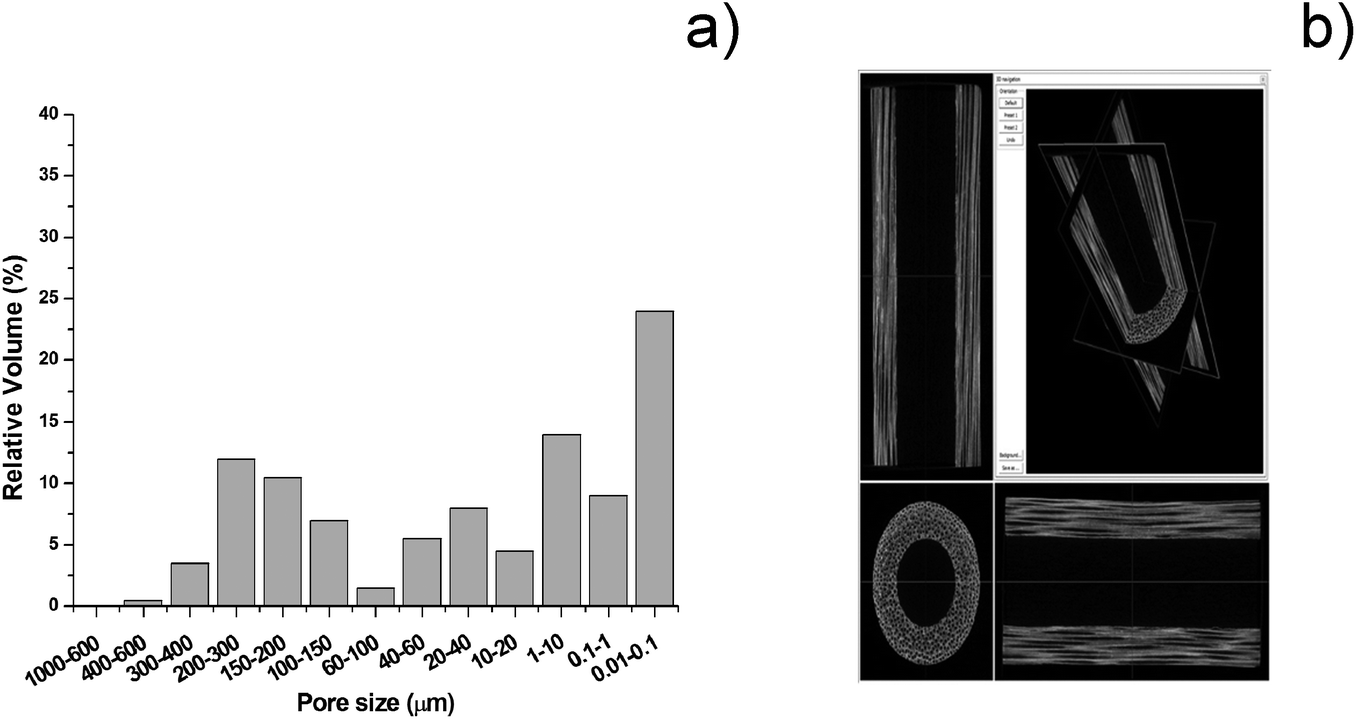 | ||
| Fig. 8 (a) Pore size distribution in B-HA. (b) Micro-CT scan of B-HA scaffolds. | ||
Mechanical tests show that B-HA exhibits high compression strength (Fig. 9, left) and a damage-tolerant mechanical behaviour. While sintered ceramics typically experience a brittle fracture mode, according to Ashby's model,44 the fracture profile of B-HA under compression is comparable to that exhibited by natural bone.45 Two main regimes can here be evidenced: (i) a linear elastic behaviour followed by (ii) a stress plateau where B-HA starts to fracture but without reaching complete failure and maintaining good structural integrity, at least up to a deformation of ∼14%. The high strength of B-HA can be related to the formation of twin boundaries between the lamellar apatite grains during their epitaxial growth from the reactive CaCO3 precursor, during the hydrothermal process.46 Damage-tolerant properties shown by B-HA can be explained on the basis of multi-scale hierarchical porosity able to absorb energy and arrest crack propagation. The overall mechanical behaviour is the result of a fracturing process that initially involves only the tiniest struts, without affecting the strength and integrity of the whole scaffold, similarly as occurs in natural bone. Damage resistance of B-HA is also shown by the easy insertion of surgical screws without undergoing fragmentation or even catastrophic rupture (Fig. 9, right). This behaviour is absolutely original for ceramic bodies that are among the best examples of brittle materials and cannot be screwed without damaging. Scaffolds with such damage-tolerant behaviour are promising candidates for clinical applications, even in load-bearing sites, since they permit the use of bone plates or screws for the scaffold fixation, aiding in sustaining early mechanical loads, as well as in preventing dislocations and micro-movement. The channel-like interconnected porosity shown by B-HA is able to induce extensive bone penetration and vascularization throughout the whole scaffold, which are key aspects to achieve an ever increasing load-bearing ability. Further investigations in relevant pre-clinical models are required to confirm this hypothesis.
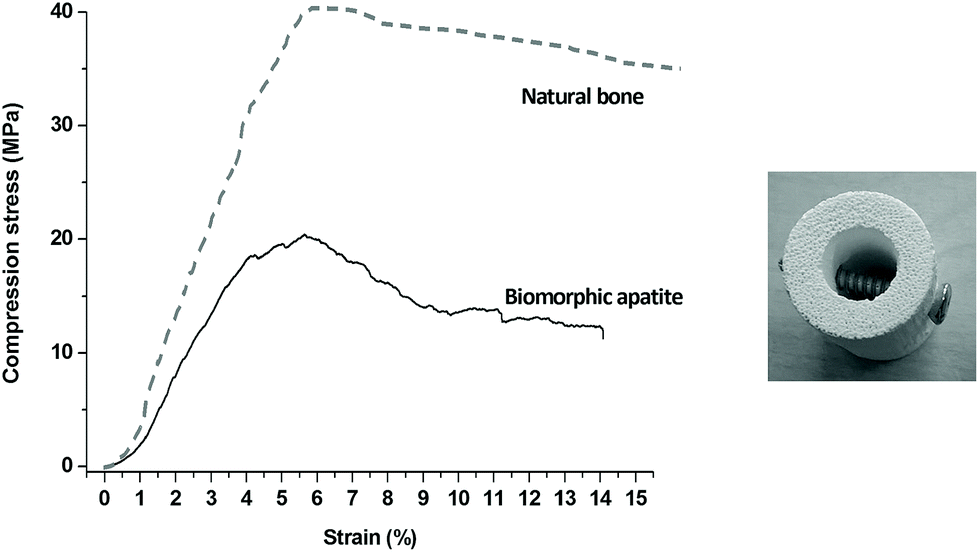 | ||
| Fig. 9 Mechanical behaviour of biomorphic B-HA. Left: Compressive test in comparison with natural bone.47 Right: Picture showing the easy insertion of a screw in B-HA. | ||
3.5 In vitro biological evaluations
The regenerative potential of the B-HA scaffold, in comparison with a porous sintered apatite (S-HA), was evaluated through an in vitro 3-D culture model under both standard and bioreactor conditions.The cell culture was firstly analysed for cell viability with the Live/Dead assay based on the simultaneous determination of live and dead cells with two probes, Calcein and EthD-1, that measure recognized parameters of cell viability: intracellular esterase activity and plasma membrane integrity, respectively. A very high ratio of viable cells is seen with no significant differences among biomorphic B-HA and the control S-HA groups (Fig. 10a and b). Morphological analysis by phalloidin staining and SEM showed that mMSCs well adhered to the B-HA samples without any difference compared to the S-HA control group (Fig. 10c and d). After 3 days of cell culture, cells grow into the porous scaffold structure and infiltrate the scaffolds as shown in Fig. 10c and d. Moreover, the nuclear morphological qualitative analysis confirms that B-HA is not cytotoxic. In fact, the cell nuclei show their native morphology and no abnormal alterations (e.g. nuclear fragmentation and chromatin condensation) are detected (Fig. 10c and d insets). In addition, a detailed cell morphology analysis by SEM shows, after 24 hours, cells firmly attached to the sample surface, with their typical spindle/fibroblast-like morphology (Fig. 10e and f).
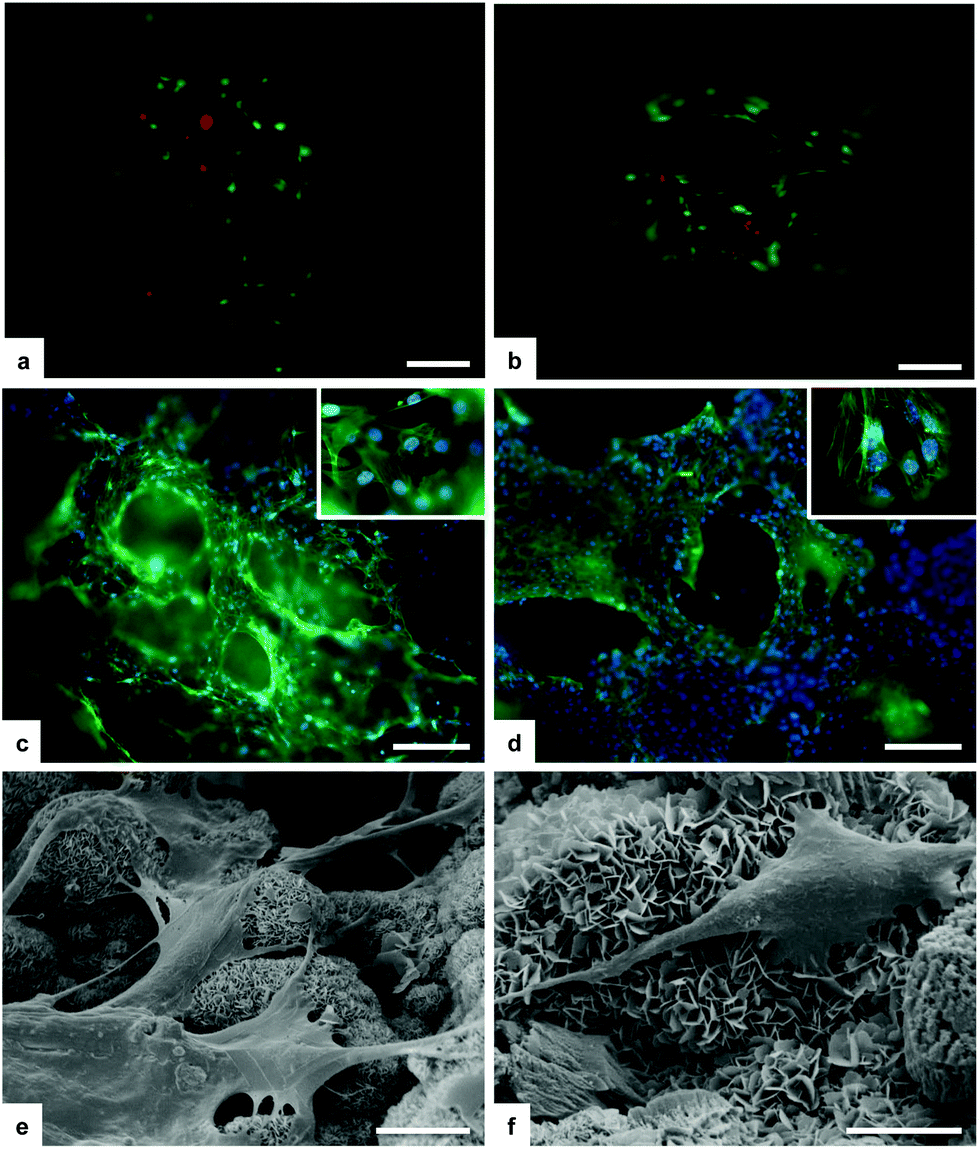 | ||
| Fig. 10 Cell viability analysed by the Live/Dead assay. Calcein AM stains for live cells are shown in green, and EthD-1 stains for dead cells in red. (a) and (b) show mMSCs seeded on B-HA and S-HA samples, respectively, after 1 day. Analysis of cell morphology by phalloidin staining at day 3. Phalloidin in green stains for actin filaments and DAPI in blue stains for cell nuclei. (c) B-HA and (d) S-HA. Analysis of cell morphology assessed by SEM. (e) and (f) show mMSCs seeded on a B-HA sample at day 1. Scale bars: (a–d) 200 μm; (e and f) 10 μm. | ||
The bioreactor was used as a tool for predictive studies, since it replicates physiological processes and can be used as a clinically-reflective approach for initial in vitro screening, reducing in that way the number of animals required for preclinical assessment, thus complying with the 3R principles.
18 hours after seeding in the bioreactor, the seeding efficiency was evaluated by counting the number of cells left in the culture media. No statistical differences were observed between the two materials: for B-HA, 57475 ± 12720 and for S-HA, 32466 ± 7973 (mean ± SEM) cells. The very high ratio of cells seeded on both the scaffolds, 97% and 98%, respectively, confirms the excellent biocompatibility of B-HA.
The mRNA level of genes involved in osteogenic differentiation was analysed by q-PCR after 14 days of dynamic cell culture conditions (Fig. 11). The gene expression profile observed is a strong index of the inductive effect exerted by B-HA. In fact, the results highlight a significant effect of B-HA on the up-regulation of genes involved in both early (BMP2, Runx2 and ALP) and late (OPN and Col15a1) stages of osteogenic commitment,47 when compared to the sintered apatite.
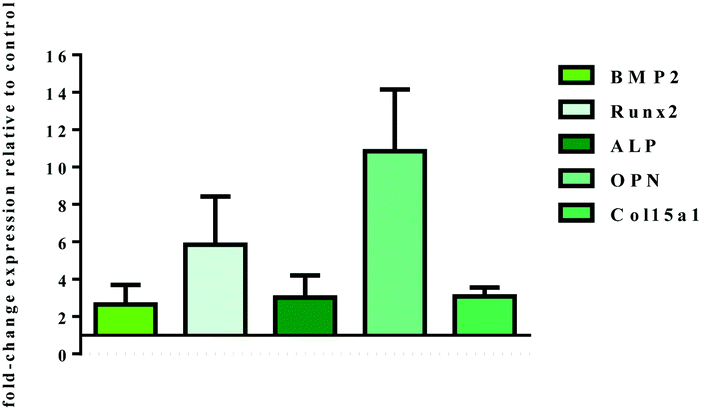 | ||
| Fig. 11 Relative quantification (2−ΔΔCt) of gene expression after 14 days of dynamic cell culture. Fold-change expression of the target genes induced by B-HA samples in respect to S-HA samples, used as a calibrator, are shown in the graph. Two-way ANOVA analysis indicated statistical difference between the samples with p ≤ 0.0001. | ||
In vivo bone formation is a gradual and well-orchestrated process associated with characteristic temporal modifications in gene expression and composed of different stages: (i) cell proliferation, (ii) extracellular matrix production and maturation, and (iii) matrix mineralization.48,49 It is well known that BMP2 is an essential regulator of the bone formation process, and acts as an upstream signal of RUNX2, the most important transcription factor necessary to tune the cascade of molecular events inducing osteoblast differentiation.47 Once triggered, this molecular machinery leads to the expression of osteoblast differentiation markers genes, including ALP, COL, OPN. In this study all these genes, both early and late markers, are overexpressed in respect to the sintered apatite S-HA, indicating the inductive effect exerted by B-HA and the well consolidated osteogenic differentiation pathway. Of particular relevance is the high level of OPN expression, a late marker of differentiation, because of its role in the triggering of bone matrix mineralization.50,51
The obtained results become even more relevant considering that sintered hydroxyapatite, used in this study as a calibrator, had already demonstrated inductive effects on MSC differentiation in a previous bioreactor study.52 In this respect, the enhanced activation of the osteoinductive signalling pathway exerted by B-HA is ascribable to its ability to dynamically activate the uptake/release of Mg2+ and Sr2+ ions which are well known triggers of bone regeneration,53,54 but are not achievable in sintered hydroxyapatite as doping ions.
3.6 In vivo osteoinductivity test
In vivo tests demonstrate the excellent osteoinductive ability of the B-HA scaffold, showing well developed bone tissue, formed in the ectopic site 12 weeks after subcutaneous implantation in rabbits (Fig. 12). These results were further confirmed by the qualitative scoring system by Ranly et al. that showed six or more ossicles in all the examined sections (mean score = 3.0 ± 0.0). In detail, histological analyses show both osteogenesis and angiogenesis, starting from the fibrous capsule, in the ectopic site (Fig. 12a and b). In particular, abundant woven bone structures composed of collagen fibrils randomly orientated and with a felted texture are seen, with the presence of a neovascular network penetrating through the scaffold (Fig. 12a). Abundant osteoid tissue covered by numerous osteoblasts is also observed in the neo-mineralized tissue and osteocytes, with lenticular form, are embedded in the dense matrix (Fig. 12b). In addition, some osteoclasts can also be seen at the new woven bone surface; they lay at Howship's lacunae level, surface depressions caused by the resorption of bone, typical of normal bone remodelling (Fig. 12a). | ||
| Fig. 12 Histological analysis of B-HA implanted subcutaneously in a rabbit model after 12 weeks. (a) Hematoxylin–eosin stain and (b) Goldner's Trichrome stain. Magnification 20×. | ||
These results are obtained in the ambit of a study performed following ISO 10993 rules that was aimed at investigating B-HA safety. Therefore, even if they may be considered as a first in vivo evaluation step, they are of outstanding interest, considering that we subcutaneously implanted the B-HA scaffold without adding cells, growth factors or other biological stimuli able to elicit bone formation. Additionally, it is important to underline that osteogenesis depends on many factors55–57 and one of them is the animal species used. Rabbit is reported as one of the in vivo models with the lowest osteogenic capacity. Some authors implanting Ca–P ceramics in several animals at different experimental times and until 120 days reported ectopic osteogenesis in dogs and pigs, but no or minimal ectopic osteogenesis in rabbits.55,58 However, in this preliminary study B-HA scaffolds have high osteogenic activity, although we have not yet tested in other animals except rabbits and in comparison with other materials with known osteoinductivity or a benchmark ceramic.
Even though the underlying mechanisms and potential signalling pathways at the basis of B-HA osteoinductivity still need to be further elucidated, the obtained results are a clear indication of the relevance of biomimetic physicochemical, morphological and mechanical features as bio-cues able to direct the cell fate to bone tissue regeneration. The control of gas–solid reactions under supercritical conditions at high pressure was effective to generate, for the first time, a bone scaffold recapitulating all these properties, particularly associating bioactive composition, cell-conducive macro/micro-porosity and hierarchical structure at the basis of superior mechanical performance. Considering also the large scaffold size achievable with our method, the results obtained in the present work are very promising for further preclinical tests aimed at obtaining bone regeneration in critical size load-bearing defects. In this respect, the bone-mimicking hierarchical structure and mechanical properties of the B-HA scaffold are both factors useful to activate cell mechanotransduction mechanisms and promote the regeneration of mechanically functional bone tissue.59,60
With this potential, B-HA could also be used in osteoporotic patients, where the sustained release of Mg2+ and Sr2+ ions can help to rebalance the natural bone turnover and boost the osteointegration process.61,62 In addition, apatite scaffolds with the ability of ion release are promising to contrast bacterial proliferation and improve the clinical outcome, as suggested by previous in vitro studies showing that co-doping with Mg2+ and CO32− ions enhances the antibacterial ability of hydroxyapatite and at the same time improves the osteogenic differentiation ability of human mesenchymal stem cells.63
All these assumptions require substantial demonstration by means of relevant preclinical studies. However, the experimental evidence herein provided supports the conclusion that bone-mimicking composition, morphology and mechanics are features very relevant to be obtained altogether in a bone scaffold to promise effective regenerative ability, particularly when large, load bearing bone parts have to be treated.
5. Conclusions
The present work highlights that heterogeneous chemistry in the 3-D state is an original approach for the development of 3-D bulk nanostructured materials with superior performance, bypassing the common synthesis/consolidation processing which easily degrades functionally relevant chemical, structural and morphological features.We showed that the pivotal aspect in processing labile phases when in the 3-D state is the management of complex chemical interplay under strictly designed environmental conditions. Gas–solid reactions carried out under pressure under supercritical conditions could be kinetically controlled by the mediation of a thin aqueous surface layer, generating 3-D reactive precursors permitting to finely tailor the crystallinity and the structure of the final product at the multi-scale. This paves the way towards a new concept of function-by-design in materials science.
Until now, bioactive, bioresorbable materials able to dynamically exchange information with physiological systems and to act as instructors for cells could not be obtained as 3-D mechanically-competent scaffolds. The outstanding bioactivity and osteoinductivity demonstrated by the biomorphic apatite scaffold in vitro and in vivo is extremely promising to solve clinical needs for which no regenerative solutions still exist. The biomorphic apatite demonstrates the validity of the “biomimetic concept” and – most importantly – suggests that biomimetic scaffolds alone can favour metabolic processes yielding tissue regeneration without the aid of additional growth factors, bioactive molecules and cells. This has a great impact on translational processes where it will be possible to avoid complex regulatory and storage issues, thus drastically shortening the time required for translation “from the bench to patient” and reducing the healthcare costs.
Disclosure
The authors declare no conflicts of interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Project NIPROGEN (POR FESR 2014-2020; CUP B42I16000020005) for providing financial support. The authors kindly acknowledge Mr C. Melandri and Mrs A. Piancastelli for the execution of mechanical and porosimetry tests and Ms C. Piccinini for technical help in performing part of the in vitro study.References
- J. W. Steed, D. R. Turner and K. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry, John Wiley and Sons, 2007 Search PubMed.
- R. Wongmaneerung, R. Yimnirun and S. Ananta, Fabrication and characterization of perovskite ferroelectric PMN/PT ceramic nanocomposites, J. Mater. Sci., 2009, 44, 5428–5440 CrossRef CAS.
- F. Roulland, G. Allainmat, M. Pollet and S. Marinel, Low temperature sintering of the binary complex perovskite oxides xBa(Zn1/3Ta2/3)O3 + (1−x)Ba(Mg1/3Ta2/3)O3, J. Eur. Ceram. Soc., 2005, 25, 2763–2768 CrossRef CAS.
- S. Qian, K. Zhu, X. Pang, J. Wang, J. Liu and J. Qiu, Influence of sintering temperature on electrical properties of (K0.4425Na0.52Li0.0375)(Nb0.8825Sb0.07Ta0.0475)O3 ceramics without phase transition induced by sintering temperature, J. Adv. Ceram., 2013, 2(4), 353–359 CrossRef CAS.
- A. Tampieri, S. Sprio, M. Sandri and F. Valentini, Mimicking natural bio-mineralization processes: a new tool for osteo-chondral scaffold development, Trends Biotechnol., 2011, 29(10), 526–535 CrossRef CAS PubMed.
- M. F. Limonov and R. M. De La Rue, Optical Properties of Photonic Structures: Interplay of Order and Disorder, CRC Press, 2012 Search PubMed.
- S. Cazalbou, D. Eichert, X. Ranz, C. Drouet, C. Combes, M. F. Harmand and C. Rey, Ion exchanges in apatites for biomedical application, J. Mater. Sci.: Mater. Med., 2005, 16, 405–409 CrossRef CAS PubMed.
- S. Cazalbou, C. Combes, D. Eichert, C. Rey and M. J. Glimcher, Poorly crystalline apatites: evolution and maturation in vitro and in vivo, J. Bone Miner. Metab., 2004, 22, 310–317 CrossRef PubMed.
- L. Bertinetti, C. Drouet, C. Combes, C. Rey, A. Tampieri, S. Coluccia and G. Martra, Surface characteristics of nanocrystalline apatites: effect of Mg surface enrichment on morphology, surface hydration species, and cationic environments, Langmuir, 2009, 25, 5647–5654 CrossRef CAS PubMed.
- S. G. Dahl, P. Allain, P. J. Marie, Y. Mauras, G. Boivin, P. Ammann, Y. Tsouderos, P. D. Delmas and C. Christiansen, Incorporation and distribution of strontium in bone, Bone, 2001, 28, 446–453 CrossRef CAS PubMed.
- C. Rey, B. Collins, T. Goehl, I. R. Dickson and M. J. Glimcher, The Carbonate Environment in Bone-Mineral - a Resolution-Enhanced Fourier-Transform Infrared-Spectroscopy Study, Calcif. Tissue Int., 1989, 45, 157–164 CrossRef CAS PubMed.
- M. Iafisco, A. Ruffini, A. Adamiano, S. Sprio and A. Tampieri, Biomimetic magnesium-carbonate-apatite nanocrystals endowed with strontium ions as anti-osteoporotic trigger, Mater. Sci. Eng., C, 2013, 35(1), 212–219 Search PubMed.
- S. Sprio, A. Tampieri, E. Landi, M. Sandri, S. Martorana, G. Celotti and G. Logroscino, Physico-chemical properties and solubility behaviour of multi-substituted hydroxyapatite powders containing silicon, Mater. Sci. Eng., C, 2008, 28, 179–187 CrossRef CAS.
- M. Schumacher and M. Gelinsky, Strontium modified calcium phosphate cements—Approaches towards targeted stimulation of bone turnover, J. Mater. Chem. B, 2015, 3, 4626–4640 RSC.
- J. T. Zhang, W. Z. Liu, V. Schnitzler, F. Tancret and J. M. Bouler, Calcium phosphate cements for bone substitution: Chemistry, handling and mechanical properties, Acta Biomater., 2014, 10, 1035–1049 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- L. Huinan, B. S. Elliott and T. J. Webster, Less harmful acidic degradation of poly(lactic-co-glycolic acid) bone tissue engineering scaffolds through titania nanoparticle addition, Int. J. Nanomed., 2006, 1(4), 541–545 CrossRef.
- C. B. Carter and M. G. Norton, Ceramic Materials: Science and Engineering, Springer, Berlin, 2013 Search PubMed.
- C. Suryanarayana, Non-equilibrium Processing of Materials, Pergamon Materials Series, Elsevier, The Netherlands, 1999, vol. 2 Search PubMed.
- A. Tampieri, S. Sprio, A. Ruffini, G. Celotti, I. G. Lesci and N. Roveri, From Wood to Bone: multi-step process to convert wood hierarchical structures into biomimetic hydroxyapatite scaffolds for bone tissue engineering, J. Mater. Chem., 2009, 19(28), 4973–4980 RSC.
- K. Vance, G. Falzone, I. Pignatelli, M. Bauchy, M. Balonis and G. Sant, Direct Carbonation of Ca(OH)2 Using Liquid and Supercritical CO2: Implications for Carbon-Neutral Cementation, Ind. Eng. Chem. Res., 2015, 54, 8908–8918 CrossRef CAS.
- D. T. Beruto and R. Botter, Liquid-like H2O Adsorption layers to catalyze the Ca(OH)2/CO2 solid−gas reaction and to form a non-protective solid product layer at 20 °C, J. Eur. Ceram. Soc., 2000, 20(4), 497 CrossRef CAS.
- W. Gu, D. W. Bousfield and C. P. Tripp, Formation of calcium carbonate particles by direct contact of Ca(OH)2 powders with supercritical CO2, J. Mater. Chem., 2006, 16, 3312–3317 RSC.
- E. Landi, S. Sprio, M. Sandri, G. Celotti and A. Tampieri, Development of Sr and CO3 co-substituted hydroxyapatites for biomedical applications, Acta Biomater., 2008, 4(3), 656–663 CrossRef CAS PubMed.
- G. Nagy, T. Lorand, Z. Patonai, G. Montsko, I. Bajnoczky, A. Marcsik and L. Mark, Analysis of pathological and non-pathological human skeletal remains by FT-IR spectroscopy, Forensic Sci. Int., 2008, 175(1), 55–60 CrossRef CAS PubMed.
- N. G. Papadopoulos, G. V. Dedoussis, G. Spanakos, A. D. Gritzapis, C.N. Baxevanis and M. Papamichail, An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry, J. Immunol. Methods, 1994, 177, 101–111 CrossRef CAS PubMed.
- H. Faulstich, S. Zobeley, G. Rinnerthaler and J. V. Small, Fluorescent phallotoxins as probes for filamentous actin, J. Muscle Res. Cell Motil., 1988, 5, 370–383 CrossRef.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- A. Khawam and D. R. Flanagan, Solid-State Kinetic Models: Basics and Mathematical Fundamentals, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- P. Baglioni, D. Chelazzi and R. Giorgi, Nanotechnologies in the Conservation of Cultural Heritage: A compendium of materials and techniques, Springer, Dordrecht, 2014, pp. 38–41 Search PubMed.
- M. Ramezani, P. Tremain, E. Doroodchi and B. Moghtaderi, Determination of carbonation/calcination reaction kinetics of a limestone sorbent in low CO2 partial pressures using TGA experiments, Energy Procedia, 2017, 114, 259–270 CrossRef CAS.
- I. J. Macha, Comparative study of Coral Conversion, Part 2: Microstructural evolution of calcium phosphate, J. Aust. Ceram. Soc., 2015, 51(2), 149–159 CAS.
- P. Álvarez-Lloret, A. B. Rodriguez-Navarro, G. Falini, S. Fermani and M. Ortega-Huertas, Crystallographic control of the hydrothermal conversion of calcitic sea urchin spine (Paracentrotus lividus) into apatite, Cryst. Growth Des., 2010, 10, 5227–5232 CrossRef.
- C. Rey, Calcium phosphate biomaterials and bone mineral. Differences in composition, structures and properties, Biomaterials, 1990, 11, 13–15 CrossRef CAS PubMed.
- L. M. Rodriguez-Lorenzo, J. N. Hart and K. A. Gross, Structural and chemical analysis of well-crystallized hydroxyfluorapatites, J. Phys. Chem. B, 2003, 107(33), 8316–8320 CrossRef CAS.
- M. Yashima, A. Sakai, T. Kamiyama and A. Hoshikawa, Crystal structure analysis of beta-tricalcium phosphate Ca3(PO4)(2) by neutron powder diffraction, J. Solid State Chem., 2003, 175(2), 272–277 CrossRef CAS.
- A. Tampieri, G. Celotti, E. Landi and M. Sandri, Magnesium Doped Hydroxyapatite: Synthesis and Characterization, Key Eng. Mater., 2004, 264–268, 2051–2054 CAS.
- L. W. Schroeder, B. Dickens and W. E. Brown, Crystallographic studies of the role of Mg as a stabilizing impurity in beta-Ca3(PO4)2. II Refinement of Mg-containing beta-Ca3(PO4)2, J. Solid State Chem., 1977, 22, 253–262 CrossRef CAS.
- E. Landi, G. Logroscino, L. Proietti, A. Tampieri, M. Sandri and S. Sprio, Biomimetic Mg-substituted Hydroxyapatite: from synthesis to in vivo behaviour, J. Mater. Sci.: Mater. Med., 2008, 19, 239–247 CrossRef CAS PubMed.
- E. Landi, A. Tampieri, G. Celotti, S. Sprio, M. Sandri and G. Logroscino, Sr-subtituted hydroxyapatites for osteoporotic bone replacement, Acta Biomater., 2007, 3(6), 961–969 CrossRef CAS PubMed.
- M. Roy and S. Bose, Osteoclastogenesis and osteoclastic resorption of tricalcium phosphate: Effect of strontium and magnesium doping, J. Biomed. Mater. Res., Part A, 2012, 100, 2450–2461 Search PubMed.
- C. Rey, C. Combes, C. Drouet, S. Cazalbou, D. Grossin, F. Brouillet and S. Sarda, Surface properties of biomimetic nanocrystalline apatites: applications in biomaterials, Prog. Cryst. Growth Charact. Mater., 2014, 60(3–4), 63–73 CrossRef CAS.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Biomimetic Systems for Hydroxyapatite Mineralization Inspired By Bone and Enamel, Chem. Rev., 2008, 108(11), 4754–4783 CrossRef CAS PubMed.
- T. M. Keaveny and W. C. Hayes, Mechanical properties of cortical and trabecular bone, in Bone, ed. B. K. Hall, CRC Press, Boca Raton, FL, 1993, pp. 285–344 Search PubMed.
- L. J. Gibson and M. F. Ashby, Cellular Ceramics – Structure and Properties, ed. D. R. Clarke, S. Suresh and I. M. Ward, Cambridge University Press, UK, 2nd edn, 1999 Search PubMed.
- Q. An, W. A. Goddard III, K. Y. Xie, G. Sim, K. J. Hemker, T. Munhollon, M. F. Toksoy and R. A. Haber, Superstrength through nanotwinning, Nano Lett., 2016, 16, 7573–7579 CrossRef CAS PubMed.
- B. Orlando, L. Giacomelli, M. Ricci, A. Barone and U. Covani, Leader genes in osteogenesis: a theoretical study, Arch. Oral Biol., 2013, 58(1), 42–49 CrossRef CAS PubMed.
- J. Isaac, J. Nohra, J. Lao, E. Jallot, J. M. Nedelec, A. Berdal and J. M. Sautier, Effects of strontium-doped bioactive glass on the differentiation of cultured osteogenic cells, Eur. Cells Mater., 2011, 21, 130–143 CrossRef CAS PubMed.
- H. Siggelkow, K. Rebenstorff, W. Kurre, C. Niedhart, I. Engel, H. Schulz, M. J. Atkinson and M. J. Hufner, Development of the osteoblast phenotype in primary human osteoblasts in culture: comparison with rat calvarial cells in osteoblast differentiation, Cell. Biochem., 1999, 75, 22–35 CrossRef CAS.
- P. J. Thurner, C. G. Chen, S. Ionova-Martin, L. Sun, A. Harman, A. Porter, J. W. Ager 3rd, R. O. Ritchie and T. Alliston, Osteopontin deficiency increases bone fragility but preserves bone mass, Bone, 2010, 46, 1564–1573 CrossRef CAS PubMed.
- B. A. Allo, S. Lin, K. Mequanint and A. S. Rizkalla, Role of bioactive 3D hybrid fibrous scaffolds on mechanical behavior and spatiotemporal osteoblast gene expression, ACS Appl. Mater. Interfaces, 2013, 5(15), 7574–7583 CrossRef CAS PubMed.
- A. Papadimitropoulos, E. Piccinini, S. Brachat, A. Braccini, D. Wendt, A. Barbero, C. Jacobi and I. Martin, Expansion of human mesenchymal stromal cells from fresh bone marrow in a 3-D scaffold-based system under direct perfusion, PLoS One, 2014, 9(7), e102359 CrossRef PubMed.
- M. Li, P. He, Y. Wu, Y. Zhang, H. Xia, Y. Zheng and Y. Han, Stimulatory effects of the degradation products from Mg-Ca-Sr alloy on the osteogenesis through regulating ERK signaling pathway, Sci. Rep., 2016, 6, 32323 CrossRef CAS PubMed.
- M. Montesi, S. Panseri, M. Dapporto, A. Tampieri and S. Sprio, Sr-substituted bone cements direct mesenchymal stem cells, osteoblasts and osteoclasts fate, PLoS One, 2017, 12(2), e0172100 CrossRef PubMed.
- Z. Yang, H. Yuan, W. Tong, P. Zou, W. Chen and X. Zhang, Osteogenesis in extraskeletally implanted porous calcium phosphate ceramics: variability among different kinds of animals, Biomaterials, 1996, 17(22), 2131–2137 CrossRef CAS PubMed.
- R. Z. LeGeros, Calcium phosphate-based osteoinductive materials, Chem. Rev., 2008, 108(11), 4742–4753 CrossRef PubMed.
- R. Yang, F. Ye, L. Cheng, J. Wang, X. Lu, Y. Shi, H. Fan, X. Zhang and H. Bu, Osteoinduction by Ca-P biomaterials implanted into the muscles of mice, J. Zhejiang Univ., Sci., B, 2011, 12(7), 582–590 CrossRef CAS PubMed.
- U. Ripamonti, Osteoinduction in porous hydroxyapatite implanted in heterotopic sites of different animal models, Biomaterials, 1996, 17(1), 31–35 CrossRef CAS PubMed.
- D. E. Ingber, Cellular tensegrity: defining new rules of biological design that govern the cytoskeleton, J. Cell Sci., 1993, 104, 613–627 Search PubMed.
- F. M. Pavalko, S. M. Norvell, D. B. Burr, C. H. Turner, R. L. Duncan and J. P. Bidwell, A model for mechanotransduction in bone cells: The load-bearing mechanosomes, J. Cell. Biochem., 2003, 88, 104–112 CrossRef CAS PubMed.
- S. Galli, M. Stocchero, M. Andersson, J. Karlsson, W. He, T. Lilin, A. Wennerberg and R. Jimbo, The effect of magnesium on early osseointegration in osteoporotic bone: a histological and gene expression investigation, Osteoporosis Int., 2017, 28(7), 2195–2205 CrossRef CAS PubMed.
- Z. Saidak and P. J. Marie, Strontium signaling: Molecular mechanisms and therapeutic implications in osteoporosis, Pharmacol. Ther., 2012, 136(2), 216–226 CrossRef CAS PubMed.
- A. Ballardini, M. Montesi, S. Panseri, A. Vandini, P. G. Balboni, A. Tampieri and S. Sprio, New hydroxyapatite nanophases with double antibacterial and osteogenic ability, J. Biomed. Mater. Res., 2017, 106(2), 521–530 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |