
A hepatitis B virus-derived human hepatic cell-specific heparin-binding peptide: identification and application to a drug delivery system†
Qiushi
Liu
ab,
Masaharu
Somiya
 a,
Masumi
Iijima
ac,
Kenji
Tatematsu
a and
Shun'ichi
Kuroda
*a
a,
Masumi
Iijima
ac,
Kenji
Tatematsu
a and
Shun'ichi
Kuroda
*a
aThe Institute of Scientific and Industrial Research, Osaka University, Osaka 567-0047, Japan. E-mail: skuroda@sanken.osaka-u.ac.jp
bThe Graduate School of Frontier Biosciences, Osaka University, Osaka 565-0871, Japan
cFaculty of Applied Bioscience, Tokyo University of Agriculture, Tokyo 156-8502, Japan
First published on 12th November 2018
Abstract
Viruses are naturally evolved nanocarriers that can evade host immune systems, attach specifically to the surfaces of target cells, enter the cells through endocytosis, escape from endosomes efficiently, and then transfer their genomes to host cells. Hepatitis B virus (HBV) is a ∼42 nm enveloped DNA virus that can specifically infect human hepatic cells. To utilize the HBV-derived early infection machinery in synthetic nanocarriers, the human hepatic cell-binding site (i.e., the sodium taurocholate co-transporting polypeptide (NTCP)-binding site, with myristoylated pre-S1(2–47)) and the low pH-dependent fusogenic domain (pre-S1(9–24)) are indispensable for targeting and endosomal escape, respectively. However, cell-surface NTCP has recently been shown not to be involved in the initial attachment of HBV. In this study, we identified a novel heparin-binding site (pre-S1(30–42)) in the N-terminal half of the pre-S1 region, which presumably interacts with cell-surface heparan sulfate proteoglycan (HSPG) and plays a pivotal role in the initial attachment of HBV to human hepatic cells. The evolutionarily conserved amino acid residues Asp-31, Trp-32, and Asp-33 are indispensable for the heparin-binding activity. Liposomes (LPs) displaying the peptide were endocytosed by human hepatic cells in a cell-surface heparin-dependent manner and delivered doxorubicin to human hepatic cells more efficiently than myristoylated pre-S1(2–47)-displaying LPs. These results demonstrated that the pre-S1(30–42) peptide is the most promising HBV-derived targeting peptide for synthetic nanocarriers, and that this peptide exhibits high specificity for human hepatic cells and efficiently induces endocytosis.
Introduction
Drug delivery system (DDS) nanocarriers that are systemically administered should escape from the mononuclear phagocyte system (MPS), which includes monocytes, macrophages, and Kupffer cells,1,2 and then deliver their payloads to target tissues specifically.3 Furthermore, they should trigger endocytosis and then release their payloads into the cytoplasm by perturbing the endosomal membrane.4,5 Thus far, conventional synthetic nanocarriers have not fully demonstrated these functions that are found in the early infection machineries of naturally occurring viruses. In the last decade, viruses have been engineered as DDS nanocarriers for the delivery of drugs and/or genes. The innate properties of viruses, such as broad tissue tropism, diverse cellular receptors, high immunogenicity, carcinogenicity, and limited payload capacity, have hampered their clinical application.6 Furthermore, it is sometimes difficult to obtain a large amount of these viruses in mass-scale production. These situations have led us to reconsider using intact viruses and to instead develop synthetic nanoparticles equipped with the viral early infection machinery with stringent specificity for target cells and tissues.Hepatitis B virus (HBV) is a ∼42 nm enveloped DNA virus that can specifically infect human hepatic cells. There are three types of envelope proteins on the surface of HBV: small (S), middle (M, pre-S2 + S region), and large (L, pre-S1 + pre-S2 + S region) proteins.7 HBV is believed to interact first with heparan sulfate proteoglycan (HSPG) on human hepatocytes,8,9 promptly move to interacting with sodium taurocholate-cotransporting polypeptide (NTCP; an essential receptor for HBV infection),10 and then enter these cells by receptor-mediated endocytosis.11 We have established an HBV-mimicking nanoparticle, bio-nanocapsule (BNC), which is composed of HBV envelope L proteins embedded in a lipid bilayer and demonstrates human liver targeting, cell entry, and endosomal escape.12,13 BNCs can deliver drugs and genes to human hepatic tissues specifically and efficiently in vitro and in vivo.14 Furthermore, BNCs can fuse with liposomes (LPs) and form BNC-virosomes under acidic conditions to deliver doxorubicin (DOX) to human hepatic tumors in xenograft mice and efficiently inhibit tumor growth.15–17 Although BNCs and BNC-virosomes are promising HBV-mimicking nanoparticles, biologics-related issues, such as immunogenicity, quality control, and production costs, have obstructed the conduct of clinical research on these drugs. Thus, as next-generation HBV-mimicking nanoparticles, conventional synthetic nanocarriers should deploy the early infection machinery of HBV by displaying functional domains, such as a cell targeting peptide, a cell entry peptide, and an endosomal escaping peptide.
To date, several functional domains responsible for early infection have been identified in the HBV L protein. First, polymerized-albumin receptor (PAR) in the pre-S2 region18 has been shown to contribute to the escape from MPS by recruiting polymerized human serum albumin.13 Second, both the human hepatic cell recognition site at the N-terminal peptide of the pre-S1 region (from Pro-10 to Pro-36)19 and the NTCP-binding site at the myristoylated N-terminal peptide of the pre-S1 region (from Gly-2 to Val-47; myr-pre-S1(2–47))10 contribute to cellular targeting and entry. Third, the low pH-dependent fusogenic domain of the N-terminal peptide in the pre-S1 region (from Asn-9 to Ala-24) contributes to endosomal escape.20,21 Recently, LPs modified with the NTCP-targeting peptide have been used as a human hepatic tissue-specific nanocarrier;22 however, we found that HSPG, rather than cell-surface NTCP, is responsible for the initial attachment of HBV to cells and the induction of endocytosis.23 HBV cell entry can be blocked by heparin completely, indicating that the interaction of HBV with human hepatic tissues is mediated by heparin.8 Recently, several groups have reported that the pre-S region contains a heparin-binding domain indispensable for the initial cellular attachment of HBV.24,25 Thus, even if conventional synthetic nanocarriers harbor the HBV-derived functional domains mentioned above, it is still necessary to introduce the heparin-dependent cellular attachment domain to maximize the efficiency of the initial attachment. In this study, we identified a novel heparin-binding site in the pre-S1 region using peptide-displaying LPs. The heparin-binding peptide (from Pro-30 to Pro-42) showed higher specificity to human hepatic cells than other heparin-binding peptides and induced endocytosis efficiently. Furthermore, we showed that the peptide-displaying LPs are promising HBV-mimicking nanoparticles for human hepatic cell-specific drug delivery.
Experimental
Peptides
The HBV pre-S1-derived peptide (encompassing myristoylated Gly-2 to Val-77, with C-terminal Cys), its truncated mutant peptides (with C-terminal Cys), and conventional heparin-binding peptides (human immunodeficiency virus (HIV, gp120, RPNNNTRKRIRC), vitronectin (GKKQRFRHRNRKGC)) were synthesized by Synpeptide Co. Ltd (Shanghai, China) and SCRUM Inc. (Tokyo, Japan).Preparation of peptide-displaying liposomes
To prepare maleimide-displaying LPs, ethanol containing dipalmitoylphosphatidylcholine (DPPC; NOF, Tokyo, Japan), cholesterol (Nacalai Tesque, Kyoto, Japan), and N-(3-maleimide-1-oxopropyl)-L-α-phosphatidylethanolamine (DOPE-MAL; NOF) were injected at a molar ratio of 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:5 into 10 mM Tris-HCl buffer (pH 8.0) with gentle stirring at room temperature and then subjected to gel filtration chromatography using a Sephadex G-25 column (GE Healthcare, Buckinghamshire, UK) equilibrated with phosphate-buffered saline (PBS). To prepare PEGylated maleimide-displaying LPs, a mixture of DPPC, cholesterol, and N-[(3-maleimide-1-oxopropyl) aminopropyl polyethyleneglycol-carbamyl] distearoylphosphatidyl-ethanolamine (DSPE-PEG-MAL) at a molar ratio of 55:40:5 was processed by the microfluidics technique using a NanoAssemblr Benchtop system (Precision Nanosystems, Vancouver, Canada). Peptides with C-terminal Cys were displayed on the LP surface using a maleimide–thiol coupling reaction.21 Peptide-displaying LPs were labeled with the lipophilic dye CellVue Claret (Sigma-Aldrich, St Louis, MO, USA).
:40:5 into 10 mM Tris-HCl buffer (pH 8.0) with gentle stirring at room temperature and then subjected to gel filtration chromatography using a Sephadex G-25 column (GE Healthcare, Buckinghamshire, UK) equilibrated with phosphate-buffered saline (PBS). To prepare PEGylated maleimide-displaying LPs, a mixture of DPPC, cholesterol, and N-[(3-maleimide-1-oxopropyl) aminopropyl polyethyleneglycol-carbamyl] distearoylphosphatidyl-ethanolamine (DSPE-PEG-MAL) at a molar ratio of 55:40:5 was processed by the microfluidics technique using a NanoAssemblr Benchtop system (Precision Nanosystems, Vancouver, Canada). Peptides with C-terminal Cys were displayed on the LP surface using a maleimide–thiol coupling reaction.21 Peptide-displaying LPs were labeled with the lipophilic dye CellVue Claret (Sigma-Aldrich, St Louis, MO, USA).
Physicochemical analyses
The size, polydispersity index (PDI), and ζ-potential of the LPs were measured with a Zetasizer Nano ZS (Malvern, Worcestershire, UK). The concentration of lipids in the LPs was calculated with the cholesterol content determined using a Cholesterol E-Test Wako kit (Wako, Osaka, Japan).Cell culture
Human hepatocellular carcinoma-derived HepG2 cells expressing NTCP (HepG2/NTCP) were kindly provided by Dr Ryosuke Suzuki (National Institute of Infectious Diseases, Tokyo, Japan). Human hepatocellular carcinoma-derived NuE cells were a gift from Prof. Takushi Tadakuma (National Defense Medical College, Tokorozawa, Japan). Primary human hepatocytes (PHH, originating from a 54-year-old Caucasian female) were purchased from Lonza (Basel, Switzerland). Human embryonic kidney-derived HEK293 T cells, rat hepatocellular carcinoma-derived MH1C1 cells, human colon carcinoma-derived WiDr cells, human lung carcinoma-derived A549 cells, and human cervical carcinoma-derived HeLa cells were purchased from RIKEN Cell Bank (Tsukuba, Japan). The cells were maintained in Ham's F-12/Dulbecco's modified Eagle's medium (DMEM) (50%/50%, v/v, Nacalai Tesque, Kyoto, Japan) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma-Aldrich) at 37 °C under a humidified atmosphere containing 5% (v/v) CO2.Flow cytometry analysis
Approximately 1.0 × 105 HepG2 cells were seeded in 6-well plates (Corning Inc., New York, NY, USA) and incubated in a culture medium containing 3% (v/v) DMSO at 37 °C for at least 24 h. The cells were incubated with CellVue-labeled peptide-displaying LPs (10 μg mL−1 as lipid) at 37 °C for 6 h, washed twice with Dulbecco's PBS (D-PBS, Nacalai Tesque), and then analyzed using a FACS Canto II flow cytometer (BD Biosciences, San Diego, CA, USA). To evaluate the effect of heparin on the cellular attachment of peptide-displaying LPs, heparin sodium salt (10 μg mL−1 final, Nacalai Tesque) was added to the culture medium 5 min before the addition of CellVue-labeled peptide-displaying LPs (10 μg mL−1 as lipid). To analyze the endocytosis pathway of peptide-displaying LPs, the cells were treated with the endocytosis inhibitor chlorpromazine (12.5 μg ml−1, 30 min) or amiloride (1 mM, 15 min), and then incubated with CellVue-labeled peptide-displaying LPs (10 μg mL−1 as lipid) for 6 h.Intracellular trafficking analysis
Approximately 5.0 × 104 HepG2 or HepG2/NTCP cells per well were seeded in 8-well glass bottomed chamber slides (Corning Inc.), and incubated in a culture medium containing 3% DMSO for 24 h. After fixation and staining with Hoechst 33342 (Nacalai Tesque), cell-surface NTCP was visualized with a rabbit polyclonal anti-NTCP antibody (HPA042727, Sigma-Aldrich) and an Alexa Fluor-488 labeled goat anti-rabbit IgG (H + L) secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA).Approximately 5.0 × 104 HepG2 or HepG2-NTCP cells per well grown on 8-well glass-bottomed chamber slides were incubated in a culture medium containing 3% DMSO for 24 h, and then incubated in a culture medium containing 5 μg mL−1 (as a lipid) of CellVue-labeled peptide-LPs at 37 °C for 5.5 h, followed by a 30 min incubation with 50 nM LysoTracker Red (Thermo Fisher Scientific, Waltham, MA, USA). After fixation with 4% (w/v) paraformaldehyde, the cells were stained with Hoechst 33342 (Nacalai Tesque). The fluorescence images obtained with an FV-1000 confocal laser scanning microscope (LSM) (Olympus, Tokyo, Japan) were analyzed using the ImageJ version 1.47 g software (National Institutes of Health, Bethesda, MA, USA).
For visualizing the intracellular localization of DOX, approximately 5.0 × 104 HepG2 cells per well were grown on 8-well glass-bottomed chamber slides at 37 °C for 24 h, then incubated with DOX, LPs containing DOX, pre-S1(30–42)-displaying LPs containing DOX, myr-pre-S1(2–47)-displaying LPs containing DOX, and pre-S1(47–77)-displaying LPs containing DOX (25 μg mL−1 as DOX) for 8 h. The fluorescence derived from DOX and Hoechst 33342 was observed under an FV-1000D confocal LSM.
To label early endosomes and late endosomes/lysosomes, HepG2 cells were fixed with 4% (w/v) paraformaldehyde, stained with Hoechst 33342, permeabilized with 0.1% (w/v) Triton X-100 (Wako), and then treated with a mouse monoclonal anti-EEA1 (early endosome antigen-1) antibody (Abcam, Cambridge, UK) or mouse monoclonal anti-LAMP2 (lysosome-associated membrane protein-2) antibody (Abcam). Both EEA1 and LAMP2 were visualized with an Alexa Fluor 555-labeled goat anti-mouse IgG secondary antibody (Sigma-Aldrich).
Evaluation of cell-surface proteoglycans
Approximately 5.0 × 104 HepG2 cells per well were grown on 8-well glass bottomed chamber slides for 24 h. To evaluate the effect of heparin on the cellular attachment of peptide-displaying LPs, the cells pre-treated with heparin sodium salt (10 μg mL−1; see above) were incubated with CellVue-labeled peptide-LPs at 37 °C for 6 h, observed under a LSM, and then analyzed using the ImageJ software. For the removal of sulfate groups on cell-surface HSPG, the cells were incubated with sodium chlorate (100 mM, Nacalai Tesque) for 16 h and with CellVue-labeled peptide-LPs at 37 °C for the next 6 h, and then observed under a LSM. For the ablation of heparin sulfate groups on cell-surface HSPG, the cells were incubated with 8 units per mL heparinase I (Sigma-Aldrich) for 60 min at 37 °C without serum, incubated with CellVue-labeled peptide-LPs at 37 °C for 6 h, and then subjected to LSM observation.Heparin-binding assay
Peptide-displaying LPs (20 μg mL−1, as a lipid) labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD; Thermo Fisher Scientific) were incubated with 50 μL (50% slurry) of heparin Sepharose 6 beads (GE Healthcare) or Sepharose 6B beads (GE Healthcare) in PBS for 1 h, and then washed twice with PBS. The fluorophores were eluted from bound LPs with PBS containing 0.1% (v/v) Triton X-100, and their fluorescence intensity was measured using a Varioskan fluorescence microplate reader (Thermo Electron, Vantaa, Finland).Encapsulation of doxorubicin into LPs
Doxorubicin (DOX; Sigma-Aldrich) was encapsulated into LPs using a remote loading method.17,26 Free DOX was removed by using a Sephadex G-25 gel filtration column equilibrated with 10 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 3.4% (w/v) sucrose. Following disruption with 0.1% (w/v) sodium dodecyl sulfate (SDS) and 0.1 N HCl, the total DOX concentration was calculated from the fluorescence intensity derived from DOX (excitation at 475 nm; emission at 515 nm), which was measured using a Varioskan fluorescence microplate reader.Cytotoxicity assay
HepG2 cells (approximately 5 × 103 cells per well) seeded in 96-well cell culture plates (Nalge Nunc, Naperville, IL) were cultured for 24 h, and then treated with DOX, DOX-containing PEGylated LPs, and DOX-containing peptide-displaying PEGylated LPs at DOX concentrations of 0.195–25 μg mL−1 at 37 °C for 8 h. As a control, the cells were treated with 40 μg mL−1 of LPs without DOX encapsulation. Following a fresh medium exchange, the cells were further incubated at 37 °C for 24 h. Cell viability was determined using a WST-8 assay reagent (Nacalai Tesque) according to the manufacturer's protocol. The formation of tetrazolium salts from WST-8 was measured using a Varioskan fluorescence microplate reader.Results
Identification of the cellular attachment domain in the pre-S1 region
Because the pre-S region is responsible for the heparin-dependent binding of HBV,24,25 we delineated the HepG2 cell-attachment domain using truncated pre-S1 peptide-displaying CellVue-labeled LPs (Fig. 1A). The size, PDI, and ζ-potential of each sample are shown in Fig. S1.† All samples exhibited a comparable size (from ∼100 nm to ∼200 nm; PDI from ∼0.2 to ∼0.3) and negative charge (from ∼−30 mV to ∼−50 mV). After incubation at 37 °C for 6 h, the cells were subjected to flow cytometry analysis. Compared with the myr-pre-S1(2–47) peptide, truncated peptides encompassing Asn-9 to Val-47 exhibited higher cellular attachment activity with HepG2 cells, whereas the pre-S1(47–77) peptide exhibited no activity (Fig. 1B). The pre-S1(30–42) peptide showed the highest affinity for HepG2 cells. | ||
| Fig. 1 Delineation of the cellular attachment domain in the pre-S1 region. Pre-S1-derived synthetic peptides (A) were displayed on LPs, and then subjected to a cellular attachment analysis (B) using HepG2 cells. Blue, myr-(2–47) peptide; gray, (9–24) peptide; yellow, (17–30) peptide; red, (30–42) peptide; green, (24–42) peptide; purple, (24–47) peptide; and black, (47–77) peptide. N = 3, error bars represent SDs. | ||
Cellular attachment of pre-S1-derived peptide-displaying LPs
Pre-S1-derived peptide-displaying LPs were applied to HepG2 cells with or without NTCP overexpression (Fig. S2†). After incubation at 37 °C for 6 h, the cells were observed under a LSM (Fig. 2A). Pre-S1(30–42)-displaying LPs exhibited significantly higher cellular attachment activity than myr-pre-S1(2–47)-displaying LPs, which was not affected by NTCP overexpression (Fig. 2B). Meanwhile, pre-S1(2–47)-displaying LPs showed a comparable level of cellular attachment activity to that of myr-pre-S1(2–47)-displaying LPs in HepG2 cells, but slightly lower activity in HepG2/NTCP cells (Fig. 2B). These results indicated that cell-surface NTCP does not substantially contribute to the cellular attachment of pre-S1(30–42)-displaying LPs. These data indicated that the pre-S1(30–42) domain plays a pivotal role in the initial cellular attachment of HBV in a NTCP-independent manner and that the flanking regions of the pre-S1(30–42) domain may suppress cellular attachment activity.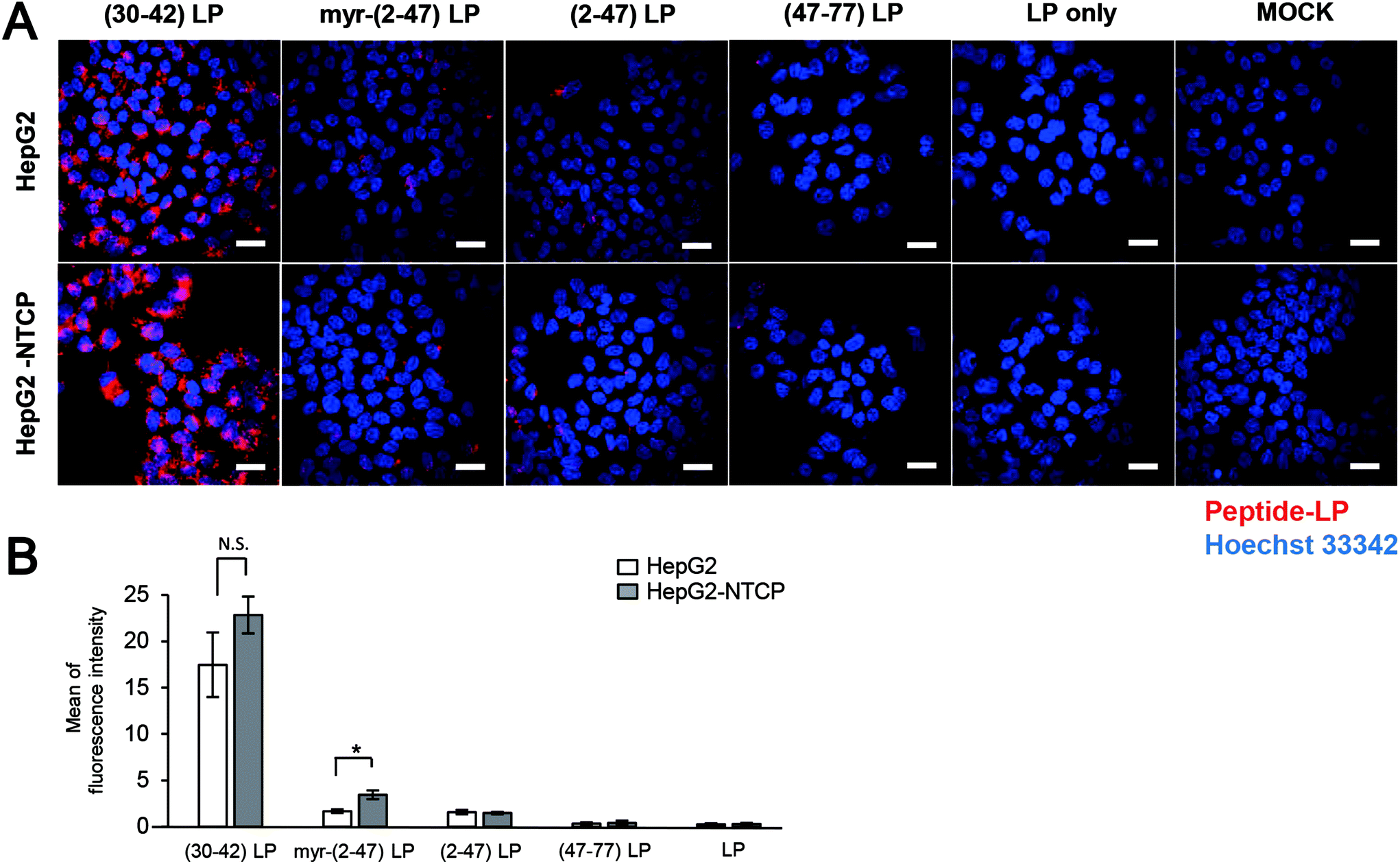 | ||
| Fig. 2 Cellular attachment of pre-S1-derived peptide-displaying LPs. Pre-S1-derived peptide-displaying CellVue-labeled LPs were incubated with HepG2 cells with or without NTCP expression. After staining the nuclei with Hoechst 33342 (blue), CellVue-derived fluorescence (red) was observed using a confocal laser scanning microscope (A). The peptides used were the pre-S1(30–42) peptide, myr-pre-S1(2–47) peptide, pre-S1(2–47) peptide, and pre-S1(47–77) peptide. Scale bars represent 20 μm. The amounts of fluorophore in each cell were determined using the ImageJ program (B). N = 3, error bars represent SDs. Student's t-test; *, p < 0.005. N.S., not significant. | ||
Cell entry and intracellular trafficking of pre-S1(30–42)-displaying LPs
The conjugation of the pre-S1(30–42) peptide on the LPs was optimized. The sizes, PDI and ζ-potential of pre-S1(30–42)-displaying LPs with different ratios of DOPE-MAL were determined (Fig. S3†). The results indicated that pre-S1(30–42)-displaying LPs with less than 5 mol% of DOPE-MAL exhibited an optimal particle size and PDI. HepG2 cells were incubated with CellVue-labeled pre-S1(30–42)-displaying LPs at 37 °C for 6 h, and then subjected to flow cytometry. The pre-S1(30–42)-displaying LPs with 5 mol% of DOPE-MAL exhibited higher cellular attachment activity with HepG2 cells than that with 1 to 4 mol% DOPE-MAL (Fig. S4†). No significant difference was found in pre-S1(30–42)-displaying LPs with 5 to 10 mol% of DOPE-MAL, indicating that pre-S1(30–42)-displaying LPs with 5 mol% DOPE-MAL is the optimal condition for cellular delivery. Consequently, pre-S1(30–42)-displaying LPs with 5 mol% of DOPE-MAL were used for further experiments.HepG2 cells were incubated with CellVue-labeled pre-S1(30–42)-displaying LPs at 37 °C for 6 h, and then trypsinized at 37 °C for 20 min to remove extracellular proteins and LPs.23 The cells were next subjected to the flow cytometry assay. As shown in Fig. 3A, no significant difference was found between trypsinized and intact HepG2 cells, indicating that nearly all pre-S1(30–42)-displaying LPs were incorporated into HepG2 cells. When HepG2 cells were incubated with CellVue-labeled pre-S1(30–42)-displaying LPs at 4 °C for 1 h, weak fluorescence was observed in the cells (Fig. 3B), suggesting that the cell entry of pre-S1(30–42)-displaying LPs is temperature dependent. Next, HepG2 cells were treated with endocytosis inhibitors (chlorpromazine or amiloride), and then with CellVue-labeled pre-S1(30–42)-displaying LPs at 37 °C for 6 h. As shown in Fig. 3C, both inhibitors (amiloride and chlorpromazine (CPZ)) significantly repressed the incorporation of pre-S1(30–42)-displaying LPs, indicating that cell entry is mainly driven by clathrin-mediated endocytosis and micropinocytosis, similar to that of BNCs and HBV virions.27,28
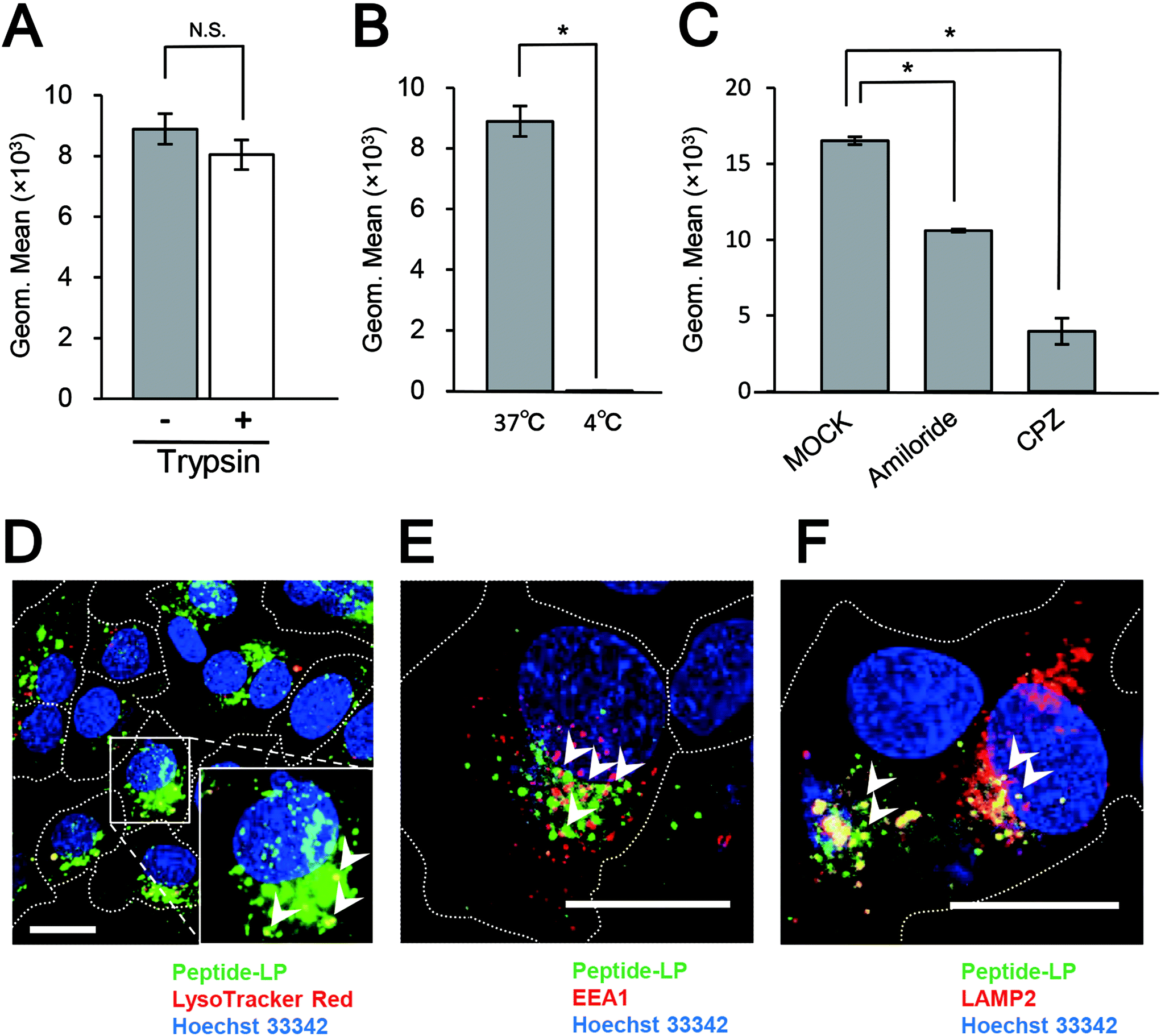 | ||
| Fig. 3 Intracellular trafficking analysis of pre-S1(30–42)-displaying LPs. HepG2 cells were incubated with pre-S1(30–42)-displaying CellVue-labeled LPs under various conditions (with trypsinization (A), incubation at 4 °C (B), and endocytosis inhibitors (C)), and then analyzed by flow cytometry. N = 3, error bars represent SDs. Student's t-test; *, p < 0.005. Subcellular localizations of pre-S1(30–42)-displaying CellVue-labeled LPs in HepG2 cells. LysoTracker (D), EEA1 (E), and LAMP2 (F) were analyzed using a confocal laser scanning microscope. Scale bars represent 20 μm. | ||
The materials incorporated by receptor-mediated endocytosis are mainly sorted to late endosomes/lysosomes via early endosomes.29 HepG2 cells were incubated with CellVue-labeled pre-S1(30–42)-displaying LPs at 37 °C for 6 h, and then subjected to LSM observation. The pre-S1(30–42)-displaying LPs were found to partially co-localize with LysoTracker Red (Fig. 3D), early endosome marker EEA-1 (Fig. 3E), and late endosome/lysosome marker LAMP-2 (Fig. 3F). The time course study of the uptake of pre-S1(30–42)-displaying LPs, pre-S1(47–77)-displaying LPs and unmodified LPs in the HepG2 cells is shown in Fig. S5.† We found that the cellular attachment and entry of pre-S1(30–42)-displaying LPs were increased in a time-dependent manner. The intracellular trafficking of unmodified LPs was evaluated as a control (Fig. S6A†). These data indicated that pre-S1(30–42)-displaying LPs could enter HepG2 cells with the help of the endocytosis machinery.
Effect of heparin on the cellular attachment of pre-S1(30–42)-displaying LPs
Because heparin can inhibit the HBV infection efficiently in vitro,8 probably by interaction with the pre-S1 region,24,25 we evaluated the effect of heparin on the cellular attachment of pre-S1(30–42)-displaying LPs. HepG2 cells were pre-treated with heparin sodium salt (from 0.001 to 10 μg mL−1) at 37 °C for 5 min, and then incubated with CellVue-labeled pre-S1(30–42)-displaying LPs for 6 h. Upon flow cytometry analysis and LSM observation, heparin was found to inhibit the cellular attachment of pre-S1(30–42)-displaying LPs in a dose-dependent manner, and block the cellular attachment of pre-S1(30–42)- and myr-pre-S1(2–47)-displaying LPs completely at a heparin concentration of 10 μg mL−1 (Fig. 4A, B and C). When HepG2 cells were pre-treated with sodium chlorate (from 1 to 100 mM) for 16 h or heparinase I for 1 h, the cellular attachment of CellVue-labeled pre-S1(30–42)-displaying LPs was significantly decreased (Fig. 4D, E, and F). The effect of heparin on the cellular uptake of unmodified LPs was evaluated as a control (Fig. S6B†). These results strongly suggested that the pre-S1(30–42) region could interact with sulfated glycosaminoglycan chains of cell-surface proteoglycans, which may be involved in the initial cellular attachment of various viruses (e.g., HBV,25 HIV,30 dengue virus,31 adeno-associated virus,32 and herpes simplex virus33).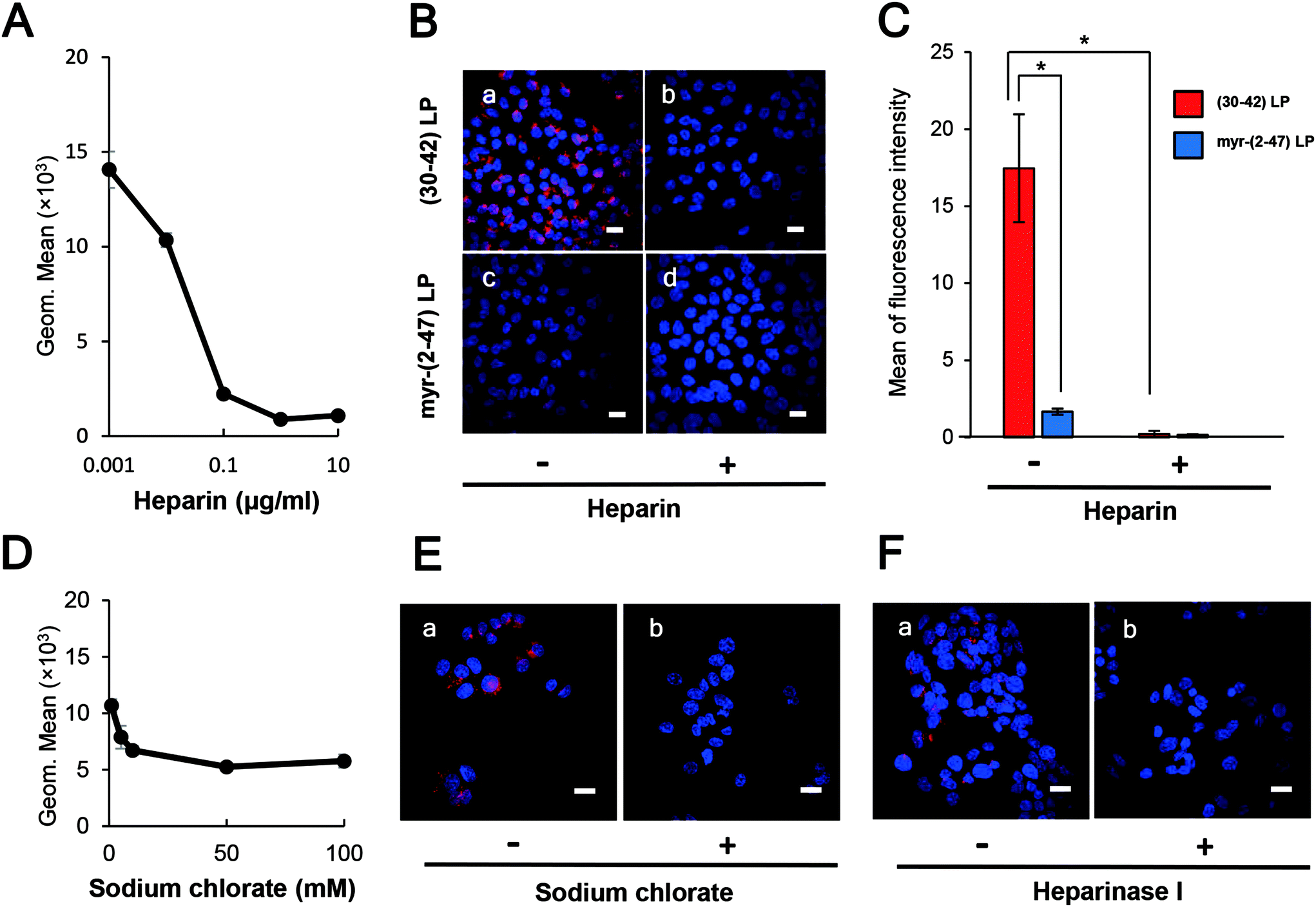 | ||
| Fig. 4 Effect of cell-surface heparin on the cellular attachment of pre-S1(30–42)-displaying LPs. The cellular attachment of pre-S1(30–42)- and myr-pre-S1(2–47)-displaying CellVue-labeled LPs on HepG2 cells was analyzed by using flow cytometry analysis and confocal laser scanning microscopy. Treatment with heparin (A, B, and C), sodium chlorate (D and E), and heparinase I (F). Scale bars represent 20 μm. The amounts of fluorophore in each cell were determined using the ImageJ program (B). N = 3, error bars represent SDs. | ||
Delineation of the heparin-binding site in the pre-S1 region
Pre-S1-derived peptides were displayed on DiD-labeled LPs and mixed with heparin Sepharose 6 beads. After extensive washing, the fluorophores were eluted with Triton X-100 and quantitated using a fluorescence microplate reader. As shown in Fig. 5A, both pre-S1(30–42)- and myr-pre-S1(2–47)-displaying LPs exhibited a significantly higher affinity to the beads. Furthermore, pre-S1(9–24)-displaying LPs exhibited medium affinity, with a comparable level of LPs displaying the conventional heparin-binding peptide from HIV gp12034 and vitronectin,35 whereas pre-S1(47–77)-displaying LPs did not exhibit any affinity. Because the affinity of all peptides was nearly lost with a large excess of heparin, these data indicated that the N-terminal half of the pre-S1 region (i.e., from Gly2 to Val47) displays heparin-binding activity and that the pre-S1(30–42) region plays a pivotal role in direct binding with heparin. Next, the LPs displaying three mutated pre-S1(30–42) peptides were subjected to the heparin-binding assay using heparin Sepharose 6 beads (Fig. 5B). When Trp-32, Phe-34, and Trp-41 were substituted with Ala (pre-S1(30–42, WFW/AAA) peptide), the heparin-binding activity of these LPs decreased significantly (Fig. 5C). The evolutionarily conserved amino acids of the peptide (Asp-31, Trp-32, and Asp-33; pre-S1(30–42, DWD/AAA) peptide) were substituted with Ala, and the heparin-binding activity of the LPs was completely lost, whereas the peptide harboring mutations at Pro-30, Pro-36, and Pro-42 (pre-S1(30–42, PPP/AAA) peptide) resulted in no significant change in activity. These results strongly suggested that the essential residues for heparin-binding activity are Asp-31, Trp-32, and Asp-33. It is interesting that these amino acids have been well conserved during HBV evolution (Fig. 6).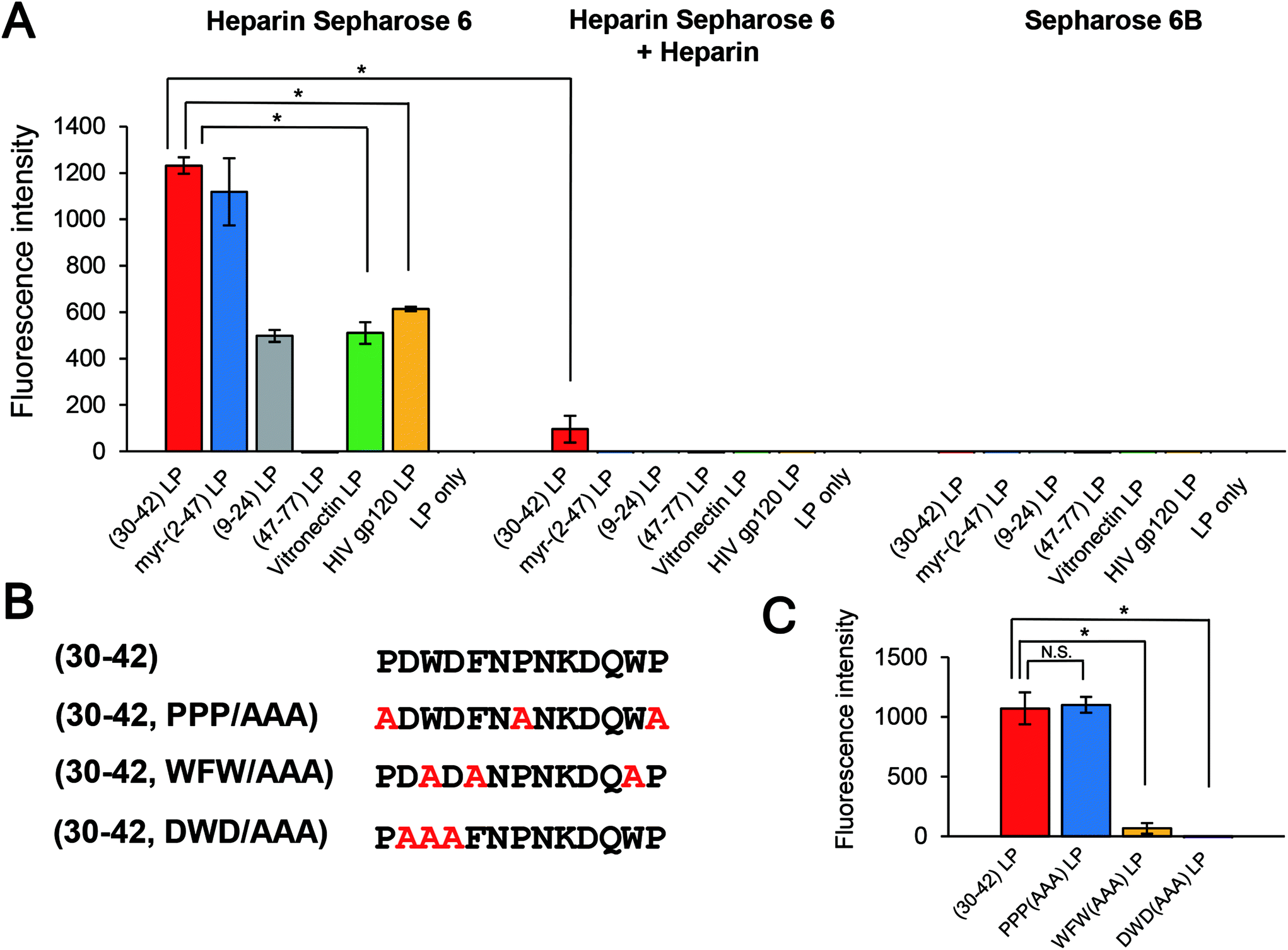 | ||
| Fig. 5 Direct binding of pre-S1(30–42)-displaying LPs to heparin. Pre-S1-derived peptide-displaying DiD-labeled LPs (A) and mutated pre-S1-derived peptide-displaying DiD-labeled LPs (B, C) were incubated with heparin Sepharose 6. The fluorophores derived from bound LPs were quantitated using a fluorescence microplate reader. Conventional heparin-binding peptides (vitronectin and HIV gp120) were used as controls. N = 3, error bars represent SDs. | ||
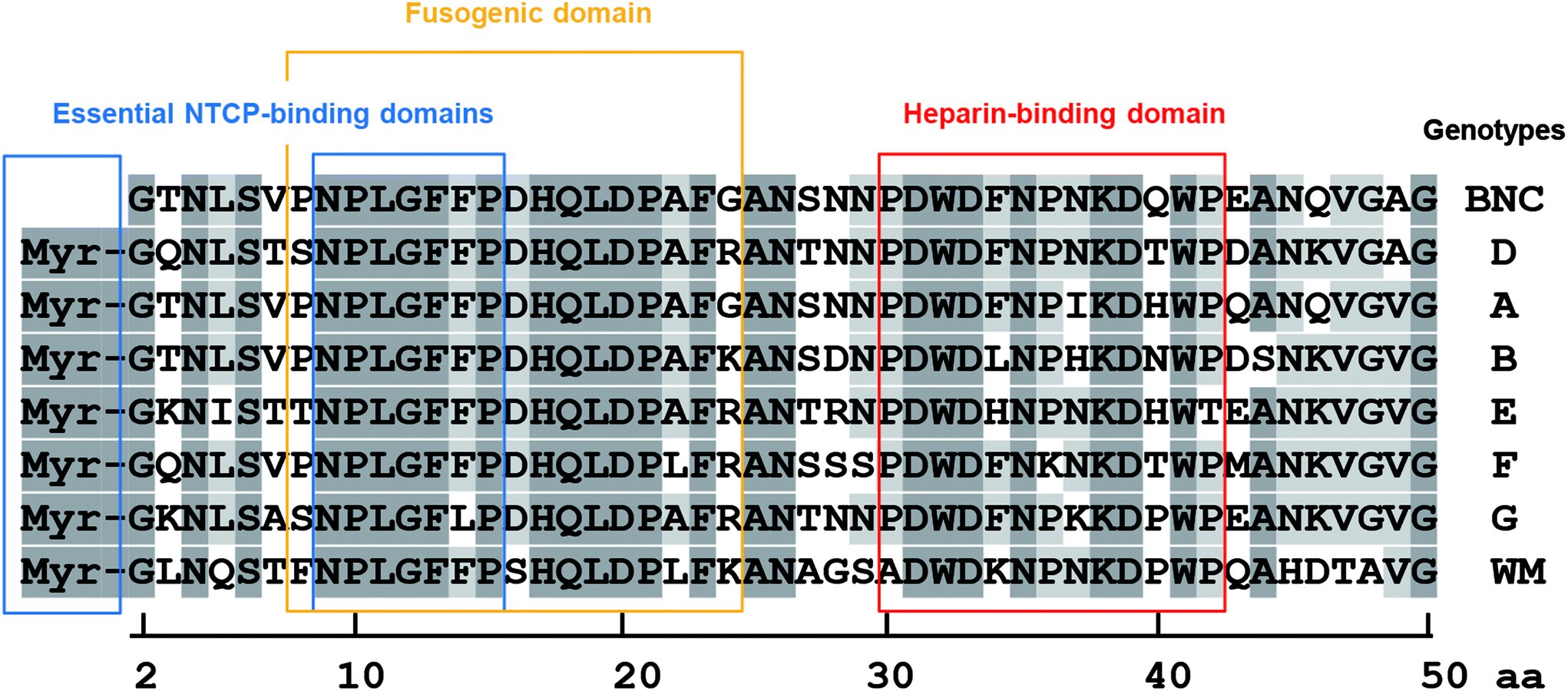 | ||
| Fig. 6 Comparison of the N-terminal pre-S1 regions of various HBV genotypes. Essential NTCP-binding domains (blue boxes), fusogenic domain (yellow box), and heparin-binding domain (red box). Dark gray highlights indicate well-conserved amino acids, and light gray highlights indicate slightly conserved amino acids. | ||
Cell specificity of the pre-S1(30–42)-displaying LPs
The pre-S1(30–42)-displaying LPs were shown to be promising HBV-mimicking nanocarriers because they could efficiently attach to HepG2 cells in a heparin-dependent manner and then promptly enter the cells via the endocytic pathway (Fig. 2). We therefore incubated CellVue-labeled pre-S1(30–42)-displaying LPs with various cells at 37 °C for 6 h and analyzed cell specificity by flow cytometry. As shown in Fig. 7A, the pre-S1(30–42)-displaying LPs exhibited higher cellular attachment activity to human hepatic cells (HepG2, primary hepatocyte, Huh7, and NuE cells) and human liver endothelial cells (Sk-Hep-1), and low activity to rat hepatic MH1C1 cells. No significant activity was observed with human non-hepatic cells (HeLa, A549, HEK293, and WiDr cells). The myr-pre-S1(2–47)-displaying LPs exhibited lower activity to human hepatic cells than pre-S1(30–42)-displaying LPs (Fig. 7B). Furthermore, the pre-S1(47–77)-displaying LPs could not attach to any of the cells examined (Fig. 7C). Notably, under the same conditions, the LPs displaying conventional heparin-binding peptides (vitronectin and HIV gp120) exhibited broad cellular attachment activity to various cells, including human non-hepatic cells (HeLa, A549, and HEK293, Fig. 7D and E). These data indicated that the pre-S1(30–42) peptide preferentially interacts with human hepatic cells. Thus, the pre-S1(30–42)-displaying LPs can be used as human hepatic cell-specific HBV-mimicking nanocarriers.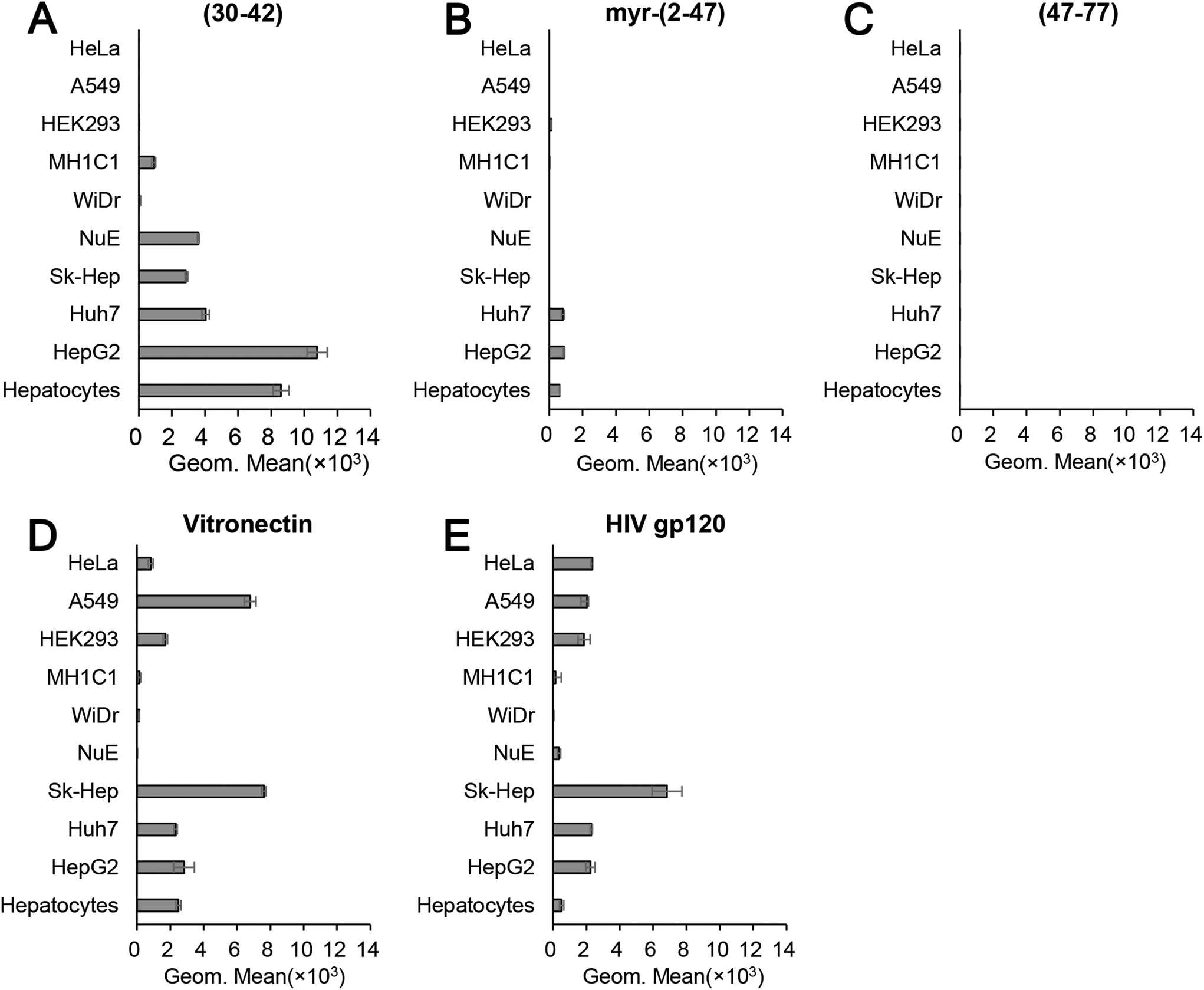 | ||
| Fig. 7 Cell specificity of pre-S1(30–42)-displaying LPs. Pre-S1-derived peptide-displaying CellVue-labeled LPs (A, pre-S1(30–42); B, myr-pre-S1(2–47); C, pre-S1(47–77)) were incubated with various cells, and then analyzed by flow cytometry. Conventional heparin-binding peptides (D, vitronectin; E, HIV gp120) were used as controls. N = 3, error bars represent SDs. | ||
Effect of the pre-S1(30–42) peptide on the cytotoxicity of DOX-containing LPs
DOX-containing LPs were modified with the pre-S1(30–42) peptide, the myr-pre-S1(2–47) peptide, and the pre-S1(47–77) peptide, and incubated with HepG2 cells for the assessment of cytotoxicity. The sizes and ζ-potentials of peptide-displaying LPs containing DOX are shown in Fig. 8A and B, respectively. The efficiency of the DOX incorporation of pre-S1(30–42)-displaying LPs, myr-pre-S1(2–47)-displaying LPs, pre-S1(47–77)-displaying LPs, and LPs was estimated to be 93.9% ± 0.28%, 92.8% ± 0.52%, 88.4% ± 0.51%, and 94.0% ± 1.10% (mean ± SD, n = 3), respectively. As shown in Fig. 8C, pre-S1(30–42)-displaying LPs exhibited the highest cytotoxicity among all samples. The IC50 value (50% inhibitory concentration for cell viability) of each sample was ∼0.8 μg mL−1 (pre-S1(30–42)-displaying LPs), ∼1.5 μg mL−1 (myr-pre-S1(2–47)-displaying LPs), ∼6.0 μg mL−1 (pre-S1(47–77)-displaying LPs), ∼6.2 μg mL−1 (LPs alone), and ∼1.5 μg mL−1 (free DOX). Furthermore, the intracellular distribution of DOX after the treatment is shown in Fig. S7.† The intracellular DOX was mainly localized in the nuclei. No cytotoxicity was found in pre-S1(30–42)-displaying LPs without DOX (Fig. S8†). These results indicated that the pre-S1(30–42) peptide, rather than other peptides, can enhance the efficient cellular uptake of DOX-containing LPs by HepG2 cells. | ||
| Fig. 8 Cytotoxicity of pre-S1(30–42)-displaying DOX-containing LPs. The size (A) and zeta potential (B) of pre-S1-derived peptide-displaying LPs are shown. Pre-S1(30–42) peptide, red; myr-pre-S1(2–47) peptide, blue; LP only, green; pre-S1(47–77) peptide, black. Cytotoxicity of pre-S1-derived peptide-displaying DOX-containing LPs to HepG2 cells (C). Pre-S1(30–42) peptide, red; myr-pre-S1(2–47) peptide, blue; pre-S1(47–77) peptide, green; LP only, purple; and free DOX, black. N = 3, error bars represent SDs. Student's t-test; *, p < 0.005. | ||
Discussion
HBV was recently proposed to interact with HSPG as a primary receptor8,25 and subsequently be transferred to NTCP as a secondary receptor for the establishment of infection.10,36 The key viral determinant of HBV infection is the N-terminal myristoylated pre-S1(2–47) domain, because the associated peptide can block HBV infection by inhibiting NTCP-dependent cell entry.10 However, cell-surface NTCP is not involved in the initial cellular attachment of HBV;23 rather, the HSPG-interacting domain is important in the early infection machinery of HBV. For a long time, there has been a controversy about whether the HSPG-interacting domain is located in the pre-S region25 or the S region.37–39 Recently, HBV virions have been revealed to change their morphology in the bloodstream from an N-type virion to a B-type virion, and then to acquire infectivity to human hepatic cells.24 The N-type virion, which hides the pre-S region in its interior cavity, cannot bind to HSPG and primary human hepatocytes, whereas the B-type virion, which exposes the pre-S region to its exterior space, can bind to HSPG and primary human hepatocytes to establish infection. Because both N and B virion types expose the S region to its exterior space, the pre-S region might be a key determinant for the interaction with HSPG. Our results indicated that the N-terminal half of the pre-S1 region (from Asn-9 to Val-47) can interact with HSPG (Fig. 1 and 5). The pre-S1(30–42) domain, particularly, plays a pivotal role in interactions with HSPG, and evolutionarily conserved residues (Asp-31, Trp-32, and Asp-33) are indispensable for this interaction (Fig. 6). Another research group reported that neutralizing antibody KR127 directly interacted with the pre-S1(26–34) region,40 suggesting that the KR127 antibody inhibits HBV infection by restricting the function of the evolutionarily conserved amino acids (Asp-31, Trp-32, and Asp-33). N-Terminal pre-S1-derived peptides, especially the myr-pre-S1(2–48) peptide41–43 and the myr-pre-S1(2–47) peptide,10 have been revealed to inhibit HBV infection efficiently. In an in vitro HBV infection assay, the myr-pre-S1(2–18) peptide inhibited the HBV infection of Tupaia hepatocytes, whereas neither the myr-pre-S1(2–8) nor myr-pre-S1(19–48) peptides block the HBV infection in these cells.41 Notably, the myr-pre-S1(2–48) peptide showed the highest inhibition activity. Furthermore, point mutations between Asn-9 and Phe-14 in the myr-pre-S1(2–48) peptide abrogated its inhibition of HBV infection.10,43 These results indicated that the myr-pre-S1(9–18) region is essential for HBV infection (presumably by acting as an essential NTCP-binding domain; see Fig. 6) and that the pre-S1(19–48) region supports the function of the myr-pre-S1(9–18) region. Therefore, the pre-S1(30–42) domain may be a key determinant in the enhancement of HBV infection.Generally, cell entry by viruses is executed through complicated steps, including receptor attachment, endocytosis, membrane fusion, and endosomal escape. Interestingly, the evolutionarily conserved residues in the pre-S1(9–47) region harbor many functional domains (Fig. 6), including the essential NTCP-binding domain (N-terminal myristoylation, from Asn-9 to Pro-15), low pH-dependent fusogenic domain (from Asn-9 to Gly-24), and heparin-binding domain (from Pro-30 to Pro-42). Recently, by using a targeted RNA interference screening, glypican 5, a family of glycosylphosphatidylinositol-anchored HSPG, has been found to be one of the initial attachment receptors for HBV.44 These data have led us to propose a new model of HBV early infection: initial attachment of the pre-S1(30–42) region to cell-surface HSPG, cell entry by endocytosis, interactions of myristoyl residues and the pre-S1(9–15) region with endosomal NTCP, exposure of the pre-S1(9–24) region through a conformational change in the L protein, induction of full membrane fusion between the HBV membrane and the endosomal membrane under acidic conditions, and then the escape of the virus core from late endosomes.
HSPG is widely utilized as an initial cell attachment receptor for various viruses and nanocarriers, including hepatitis C virus,45 porcine circovirus 2,46 herpes simplex virus,33 exosomes,47 apolipoprotein E-derived peptide-modified LPs,48 and cell-penetrating peptide-modified LPs,49 because HSPG is ubiquitously expressed in various tissues.35 Many heparin-binding peptides have been identified in viral and heparin-binding proteins that can interact with various types of cells. It is noteworthy that hepatitis C virus could infect hepatic cells specifically with the help of highly sulfated glycosaminoglycans (GAGs) found in the liver.45,50 The liver-specific infection of HBV could be blocked by heparin or other highly sulfated GAGs, but not by lower sulfated GAGs.8 These studies suggested that highly sulfated GAGs specifically expressed in the liver may be responsible for the liver-specific uptake of HBV.
In this study, we identified a novel heparin-binding peptide, pre-S1(30–42), that shows a strong affinity for human hepatic cells (Fig. 7). The surface charges of conventional heparin-binding peptides are often positive. The theoretical isoelectric points of heparin-binding peptides of HIV gp120, vitronectin, adeno-associated virus 2 VP3, equine arteritis virus E protein, and eastern equine encephalitis virus E2 glycoprotein are 12.48, 12.48, 12.00, 11.83, and 10.30, respectively,51–56 whereas that of the pre-S1(30–42) peptide is 3.93. We postulated that the pre-S1(30–42) peptide, especially the hydrophobic segment consisting of Asp-31, Trp-32, and Asp-33, interacts with heparin through hydrophobic interactions rather than electrostatic interactions. Because highly sulfated heparin is commonly found in human hepatic cells50 and treatment with sodium chlorate abrogated the interactions between pre-S1(30–42)-displaying LPs and sulfated glycosaminoglycan chains (Fig. 4C), as described above, the strong affinity of the pre-S1(30–42) peptide to human hepatic cells may be sustained by the highly sulfated residues in HSPG. Although the cellular uptake of pre-S1(30–42)-displaying LPs was significantly higher than that of myr-pre-S1(2–47)-displaying LPs (Fig. 1B and B), the binding affinity to heparin Sepharose 6 of these LPs was similar (Fig. 5A). Unlike the pre-S1(30–42) peptide, the myr-pre-S1(2–47) peptide contains two membrane-interacting domains, the N-terminal myristoyl group and the fusogenic domain pre-S1(9–24).20 It was suggested that these domains may hamper the cellular entry of myr-pre-S1(2–47)-displaying LPs by interacting with the plasma membrane.
The pre-S1(30–42) peptide shows a negative charge under physiological conditions, while conventional heparin-binding peptides exhibit a positive charge. Owing to this unique property, pre-S1(30–42)-displaying nanocarriers are expected to show a reduced non-specific interaction with biomolecules and cells during blood circulation. In particular, macrophages prefer to engulf positively charged materials more efficiently than negatively charged ones.57 Collectively, the stringent hepatotrophic property of HBV is mediated by two domains residing in the myr-pre-S1(2–47) region. The pre-S1(30–42) region first interacts with highly sulfated heparin in human hepatic cells. After HSPG-mediated endocytosis, the myr-pre-S1(9–15) region interacts with NTCP in endosomes. This working hypothesis agrees well with the present model for the mechanism underlying HBV early infection.23,58
Conclusions
We identified a novel heparin-binding domain in the N-terminal half of the pre-S1 region (from Pro-30 to Pro-42), localized close to the low-pH fusogenic domain (from Asn-9 to Gly-24) and NTCP-binding site (N-terminal myristoyl residue and from Asn-9 to Pro-15). The pre-S1(30–42) peptide showed higher specificity for human hepatic cell-derived heparin, and the evolutionarily conserved residues Asp-31, Trp-32, and Asp-33 were found to be indispensable for this activity. Our results shed light on the early infection machinery of HBV, elucidating how functional domains in the myr-pre-S1(2–42) region are coordinated on a molecular basis. Finally, since the pre-S1(30–42) domain is involved in the initial cellular attachment of HBV, pre-S1(30–42)-displaying LPs can be used as efficient human hepatic cell-specific HBV-mimicking DDS nanocarriers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the JSPS KAKENHI (Grant-in-Aid for Scientific Research (S) 16H06314 to SK; Grant-in-Aid for Scientific Research (C) 15K07840 to KT; Grant-in-Aid for Scientific Research (C) 16K04888 to MI; Grant-in-Aid for Early-Career Scientists 18K18386 to MS; Grant-in-Aid for JSPS Fellows 17J06926 to QL), the Japan Agency for Medical Research and Development (AMED) (17cm0106214h0002 and 17fk0310105h0001 to SK), and the “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” (MEXT).References
- M. J. Ernsting, M. Murakami, A. Roy and S. D. Li, Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- L. C. Davies, S. J. Jenkins, J. E. Allen and P. R. Taylor, Tissue-resident macrophages, Nat. Immunol., 2013, 14, 986–995 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard Jr., Nanoparticle-based targeted drug delivery, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, Insight into nanoparticle cellular uptake and intracellular targeting, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Drug Delivery Systems: Entering the Mainstream, Science, 2014, 303, 1818–1822 CrossRef PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Non-viral vectors for gene-based therapy, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- P. Tiollais, C. Pourcel and A. Dejean, The hepatitis B virus, Nature, 1985, 317, 489–495 CrossRef CAS PubMed.
- C. M. Leistner, S. Gruen-Bernhard and D. Glebe, Role of glycosaminoglycans for binding and infection of hepatitis B virus, Cell. Microbiol., 2008, 10, 122–133 CAS.
- O. Lamas Longarela, T. T. Schmidt, K. Schoneweis, R. Romeo, H. Wedemeyer, S. Urban and A. Schulze, Proteoglycans act as cellular hepatitis delta virus attachment receptors, PLoS One, 2013, 3, e58340 CrossRef PubMed.
- H. Yan, G. Zhong, G. Xu, W. He, Z. Jing and Z. Gao, et al., Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus, eLife, 2012, 1, e00049 CrossRef PubMed.
- H. C. Huang, C. C. Chen, W. C. Chang, M. H. Tao and C. Huang, Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis, J. Virol., 2012, 86, 9443–9453 CrossRef CAS PubMed.
- S. Kuroda, S. Otaka, T. Miyazaki, M. Nakao and Y. Fujisawa, Hepatitis B virus envelope L protein particles: synthesis and assembly in Saccharomyces cerevisiae, purification and characterization, J. Biol. Chem., 1992, 267, 1953–1961 CAS.
- M. Somiya, Q. Liu and S. Kuroda, Current Progress of Virus-mimicking Nanocarriers for Drug Delivery, Nanotheranostics, 2017, 1, 415–429 CrossRef PubMed.
- T. Yamada, Y. Iwasaki, H. Tada, H. Iwabuki, M. K. L. Chuah and T. VandenDriessche, et al., Nanoparticles for the delivery of genes and drugs to human hepatocytes, Nat. Biotechnol., 2003, 21, 885–890 CrossRef CAS PubMed.
- J. Jung, T. Matsuzaki, K. Tatematsu, T. Okajima, K. Tanizawa and S. Kuroda, Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various materials, J. Controlled Release, 2008, 126, 255–264 CrossRef CAS PubMed.
- T. Kasuya, J. Jung, R. Kinoshita, Y. Goh, T. Matsuzaki, M. Iijima, N. Yoshimoto, K. Tanizawa and S. i. Kuroda, Bio-Nanocapsule–Liposome Conjugates for In Vivo Pinpoint Drug and Gene Delivery, Methods Enzymol., 2009, 464, 147–166 CAS.
- Q. Liu, J. Jung, M. Somiya, M. Iijima, N. Yoshimoto, T. Niimi, A. D. Maturana, S. H. Shin, S. Y. Jeong, E. K. Choi and S. Kuroda, Virosomes of hepatitis B virus envelope L proteins containing doxorubicin: synergistic enhancement of human liver-specific antitumor growth activity by radiotherapy, Int. J. Nanomed., 2015, 10, 4159–4172 CAS.
- Y. Itoh, S. Kuroda, T. Miyazaki, S. Otaka and Y. Fujisawa, Identification of polymerized-albumin receptor domain in the pre-S2 region of hepatitis B virus surface antigen M protein, J. Biotechnol., 1992, 23, 71–82 CrossRef CAS PubMed.
- A. R. Neurath, S. B. Kent, N. Strick and K. Parker, Identification and chemical synthesis of a host cell receptor binding site on hepatitis B virus, Cell, 1986, 46, 429–436 CrossRef CAS PubMed.
- M. Somiya, Y. Sasaki, T. Matsuzaki, Q. Liu, M. Iijima, N. Yoshimoto, T. Niimi, A. D. Maturana and S. Kuroda, Intracellular trafficking of bio-nanocapsule-liposome complex: Identification of fusogenic activity in the pre-S1 region of hepatitis B virus surface antigen L protein, J. Controlled Release, 2015, 212, 10–18 CrossRef CAS PubMed.
- Q. Liu, M. Somiya, N. Shimada, W. Sakamoto, N. Yoshimoto, M. Iijima, K. Tatematsu, T. Nakai, T. Okajima, A. Maruyama and S. Kuroda, Mutational analysis of hepatitis B virus pre-S1 (9–24) fusogenic peptide, Biochem. Biophys. Res. Commun., 2016, 474, 406–412 CrossRef CAS PubMed.
- X. Zhang, Q. Zhang, Q. Peng, J. Zhou, L. Liao, X. Sun, L. Zhang and T. Gong, Hepatitis B virus preS1-derived lipopeptide functionalized liposomes for targeting of hepatic cells, Biomaterials, 2014, 35, 6130–6141 CrossRef CAS PubMed.
- M. Somiya, Q. Liu, N. Yoshimoto, M. Iijima, K. Tatematsu, T. Nakai, T. Okajima, K. Kuroki, K. Ueda and S. Kuroda, Cellular uptake of hepatitis B virus envelope L particles is independent of sodium taurocholate cotransporting polypeptide, but dependent on heparan sulfate proteoglycan, Virology, 2016, 497, 23–32 CrossRef CAS PubMed.
- S. Seitz, C. Iancu, T. Volz, W. Mier, M. Dandri, S. Urban and R. Bartenschlager, A Slow Maturation Process Renders Hepatitis B Virus Infectious, Cell Host Microbe, 2016, 20, 25–35 CrossRef CAS PubMed.
- A. Schulze, P. Gripon and S. Urban, Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans, Hepatology, 2007, 46, 1759–1768 CrossRef CAS PubMed.
- A. Fritze, F. Hens, A. Kimpfler, R. Schubert and R. Peschka-Suss, Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient, Biochim. Biophys. Acta, 2006, 1758, 1633–1640 CrossRef CAS PubMed.
- M. Yamada, A. Oeda, J. Jung, M. Iijima, N. Yoshimoto, T. Niimi, S. Y. Jeong, E. K. Choi, K. Tanizawa and S. Kuroda, Hepatitis B virus envelope L protein-derived bio-nanocapsules: mechanisms of cellular attachment and entry into human hepatic cells, J. Controlled Release, 2012, 160, 322–329 CrossRef CAS PubMed.
- H. C. Huang, C. C. Chen, W. C. Chang, M. H. Tao and C. Huang, Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis, J. Virol., 2012, 86, 9443–9453 CrossRef CAS PubMed.
- E. Smythe and G. Warren, The mechanism of receptor-mediated endocytosis, Eur. J. Biochem., 1991, 202, 689–699 CrossRef CAS PubMed.
- L. de Witte, M. Bobardt, U. Chatterji, G. Degeest, G. David, T. B. Geijtenbeek and P. Gallay, Syndecan-3 is a dendritic cell-specific attachment receptor for HIV-1, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19464–19469 CrossRef CAS PubMed.
- Y. Chen, T. Maguire, R. E. Hileman, J. R. Fromm, J. D. Esko, R. J. Linhardt and R. M. Marks, Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate, Nat. Med., 1997, 3, 866–871 CrossRef CAS PubMed.
- J. O'Donnell, K. A. Taylor and M. S. Chapman, Adeno-associated virus-2 and its primary cellular receptor–Cryo-EM structure of a heparin complex, Virology, 2009, 385, 434–443 CrossRef PubMed.
- M. T. Shieh, D. WuDunn, R. I. Montgomery, J. D. Esko and P. G. Spear, Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans, J. Cell Biol., 1992, 166, 1273–1281 CrossRef.
- E. Crublet, J. P. Andrieu, R. R. Vives and H. Lortat-Jacob, The HIV-1 envelope glycoprotein gp120 features four heparan sulfate binding domains, including the co-receptor binding site, J. Biol. Chem., 2008, 283, 15193–15200 CrossRef CAS PubMed.
- M. Bernfield, M. Götte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum and M. Zako, Functions of cell surface heparan sulfate proteoglycans, Annu. Rev. Biochem., 1999, 68, 729–777 CrossRef CAS PubMed.
- Y. Ni, F. A. Lempp, S. Mehrle, S. Nkongolo, C. Kaufman, M. Falth, J. Stindt, C. Koniger, M. Nassal, R. Kubitz, H. Sultmann and S. Urban, Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes, Gastroenterology, 2014, 146, 1070–1083 CrossRef CAS PubMed.
- G. Abou-Jaoude and C. Sureau, Entry of hepatitis delta virus requires the conserved cysteine residues of the hepatitis B virus envelope protein antigenic loop and is blocked by inhibitors of thiol-disulfide exchange, J. Virol., 2007, 81, 13057–13066 CrossRef CAS PubMed.
- J. Salisse and C. Sureau, A function essential to viral entry underlies the hepatitis B virus “a” determinant, J. Virol., 2009, 83, 9321–9328 CrossRef CAS PubMed.
- C. Sureau and J. Salisse, A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant, Hepatology, 2013, 57, 985–994 CrossRef CAS PubMed.
- H. J. Hong, C. J. Ryu, H. Hur, S. Kim, H. K. Oh, M. S. Oh and S. Y. Park, In vivo neutralization of hepatitis B virus infection by an anti-preS1 humanized antibody in chimpanzees, Virology, 2004, 318, 134–141 CrossRef CAS PubMed.
- D. Glebe, S. Urban, E. V. Knoop, N. Çaǧ, P. Krass, S. Grün, A. Bulavaite, K. Sasnauskas and W. H. Gerlich, Mapping of the Hepatitis B Virus Attachment Site by Use of Infection-Inhibiting preS1 Lipopeptides and Tupaia Hepatocytes, Gastroenterology, 2005, 129, 234–245 CrossRef CAS.
- P. Gripon, I. Cannie and S. Urban, Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein, J. Virol., 2005, 79, 1613–1622 CrossRef CAS PubMed.
- M. Engelke, K. Mills, S. Seitz, P. Simon, P. Gripon, M. Schnolzer and S. Urban, Characterization of a hepatitis B and hepatitis delta virus receptor binding site, Hepatology, 2006, 43, 750–760 CrossRef CAS PubMed.
- E. R. Verrier, C. C. Colpitts, C. Bach, L. Heydmann, A. Weiss, M. Renaud, S. C. Durand, F. Habersetzer, D. Durantel, G. Abou-Jaoudé, M. M. López Ledesma, D. J. Felmlee, M. Soumillon, T. Croonenborghs, N. Pochet, M. Nassal, C. Schuster, L. Brino, C. Sureau, M. B. Zeisel and T. F. Baumert, A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses, Hepatology, 2016, 63, 35–48 CrossRef CAS PubMed.
- H. Barth, C. Schafer, M. I. Adah, F. Zhang, R. J. Linhardt, H. Toyoda, A. Kinoshita-Toyoda, T. Toida, T. H. Van Kuppevelt, E. Depla, F. Von Weizsacker, H. E. Blum and T. F. Baumert, Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate, J. Biol. Chem., 2003, 278, 41003–41012 CrossRef CAS PubMed.
- G. Misinzo, P. L. Delputte, P. Meerts, D. J. Lefebvre and H. J. Nauwynck, Porcine circovirus 2 uses heparan sulfate and chondroitin sulfate B glycosaminoglycans as receptors for its attachment to host cells, J. Virol., 2006, 80, 3487–3494 CrossRef CAS PubMed.
- H. C. Christianson, K. J. Svensson, T. H. van Kuppevelt, J.-P. Li and M. Belting, Cancer cell exosomes depend on cell-surface heparin sulfate proteoglycans for their internalization and functional activity, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17380–17385 CrossRef CAS PubMed.
- I. Sauer, I. R. Dunay, K. Weisgraber, M. Bienert and M. Dathe, An Apolipoprotein E-Derived Peptide Mediates Uptake of Sterically StabilizedLiposomes into Brain Capillary Endothelial Cells, Biochemistry, 2015, 44, 2021–2029 CrossRef PubMed.
- C. Marty, C. Meylan, H. Schott, K. Ballmer-Hofer and R. A. Schwendener, Enhanced heparan sulfate proteoglycan-mediated uptake of cell-penetrating peptide-modified liposomes, Cell. Mol. Life Sci., 2004, 61, 1785–1794 CAS.
- P. Vongchan, M. Warda, H. Toyoda, T. Toida, R. M. Marks and R. J. Linhardt, Structural characterization of human liver heparan sulfate, Biochim. Biophys. Acta, 2005, 1721, 1–8 CrossRef CAS PubMed.
- B. Bjellqvist, G. J. Hughes, C. Pasquali, N. Paquet, F. Ravier, J. C. Sanchez, S. Frutiger and D. F. Hochstrasser, The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences, Electrophoresis, 1993, 14, 1023–1031 CrossRef CAS PubMed.
- B. Bjellqvist, B. Basse, E. Olsen and J. E. Celis, Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions, Electrophoresis, 1994, 15, 529–539 CrossRef CAS PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein Identification and Analysis Tools on the ExPASy Server, Methods Mol. Biol., 1999, 112, 531–552 Search PubMed.
- A. Kern, K. Schmidt, C. Leder, O. J. Muller, C. E. Wobus, K. Bettinger, C. W. Von der Lieth, J. A. King and J. A. Kleinschmidt, Identification of a Heparin-Binding Motif on Adeno-Associated Virus Type 2 Capsids, J. Virol., 2003, 77, 11072–11081 CrossRef CAS PubMed.
- Z. Lu, S. Sarkar, J. Zhang and U. B. Balasuriya, Conserved arginine residues in the carboxyl terminus of the equine arteritis virus E protein may play a role in heparin binding but may not affect viral infectivity in equine endothelial cells, Arch. Virol., 2016, 161, 873–886 CrossRef CAS PubMed.
- C. L. Gardner, J. Choi-Nurvitadhi, C. Sun, A. Bayer, J. Hritz, K. D. Ryman and W. B. Klimstra, Natural variation in the heparan sulfate binding domain of the eastern equine encephalitis virus E2 glycoprotein alters interactions with cell surfaces and virulence in mice, J. Virol., 2013, 87, 8582–8590 CrossRef CAS PubMed.
- Y. Tabata and Y. Ikada, Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage, Biomaterials, 1988, 9, 256–362 CrossRef.
- S. Urban, R. Bartenschlager, R. Kubitz and F. Zoulim, Strategies to inhibit entry of HBV and HDV into hepatocytes, Gastroenterology, 2014, 147, 48–64 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01134f |
| This journal is © The Royal Society of Chemistry 2019 |