DOI:
10.1039/C8BM00788H
(Paper)
Biomater. Sci., 2019,
7, 336-346
Anti-biofouling therapeutic nanoparticles with removable shell and highly efficient internalization by cancer cells†
Received
11th July 2018
, Accepted 30th October 2018
First published on 13th November 2018
Abstract
Cationic gelatin nanoparticles ((+)nGNPs) were prepared by in situ polymerization upon the surfaces of monodispersed gelatin nanoparticles (GNPs) using N-(3-Aminopropyl)methacrylamide (APm) as monomer, which were then decorated with doxorubicin terminated poly(2-methylacryloyloxyethyl phosphorylcholine) (DOX-pMPC) via EDC/NHS conjugation to obtain core–shell nanoparticles ((+)nGNPs@DOX-pMPC) for cancer therapy. The non-fouling pMPC shell could effectively shield the positively charged surface of inner nanoparticle and prevent non-specific protein adsorption, thus endowing the materials with potential for long-acting cancer treatment. Furthermore, the acyl hydrazone bond connecting DOX and pMPC chain could be easily hydrolyzed in the weakly acidic tumor microenvironment. After decladding of the pMPC shell, electropositive (+)nGNPs carrying the drugs can be effectively internalized by cancer cells to induce apoptosis, avoiding undesirable hindrance caused by the superhydrophilic outer layer. On combining the above properties, this drug delivery system can be a promising candidate for long-acting, low-toxicity and high-efficiency cancer therapy.
Introduction
High relapse and mortality rates make cancer one of the greatest medical challenges over the past few decades.1,2 Differing from small molecular anti-cancer drugs, nanomedicine has recently emerged as a promising tool for cancer therapy due to its remarkable characteristics, such as high drug loading capacity, relative low toxicity to normal tissues and passive targeting capability based on enhanced permeability and retention (EPR) effects.3–6 Unfortunately, due to their inherent exogenetic natures, almost all nanomedicines suffer from biological fouling on their surfaces (e.g. plasma protein binding, platelet adhesion and thrombus formation) in vivo, resulting in immune recognition or ‘opsonization’.7,8 The undesirable effects lead to a rapid clearance of nanomedicine from the blood by immunocytes and reticuloendothelial system (RES), thus severely restricting its therapeutic efficacy.9,10
Opsonization is reportedly largely affected by the surface properties of nanomedicines.11,12 Thus, methods involving masking of these surface properties using polymeric stabilizers to create a more neutral and biologically inert surface for nanomedicines have been considerably studied. The most common tactic, ‘PEGylation’, uses poly(ethylene glycol) (PEG) to decorate the surfaces of nanomedicines by chemical attachments to increase the solution viscosity, form hydrated sheath and create an anti-biofouling surface.13,14 However, according to some studies, PEG immunogenicity has been strongly correlated with its amphiphilic characteristics.15,16 Consequently, recent works have focused on developing anti-biofouling drug or protein delivery systems (DDSs/PDSs) decorated by a truly hydrophilic zwitterionic polymer, such as poly(2-methacryloyloxyethyl phosphoryl-choline) (pMPC), which is a polyzwitterion containing both positively and negatively charged groups that possess high resistance to biofouling.17–21 Hence, many efforts have been made on designing anti-biofouling nanomedicines using pMPC as coatings and pMPC-based materials have been considerable developed.22–28 Conversely, the dense hydration layer formed by pMPC coatings also severely hinders the interactions between nanomedicine and cancer cells, resulting in extremely low cellular internalization of the drugs and limited therapeutic efficacy.29–31 On the other side, cationic nanoparticles are often used for facilitating uptake by cells due to their strong cellular interactions with negatively charged biological membranes. To take advantage of the specific physiology of tumor microenvironment,32–35 it is therefore desirable to fabricate nanomedicines with stimuli-responsive properties for the enhancement of their predesigned functions, by combining selected features into the same system.36–41
To endow the nanomedicine with features of both biofouling resistance and higher cellular internalization, herein, novel core–shell nanoparticles named as (+)nGNPs@DOX-pMPC are successfully fabricated. As shown in Scheme 1, gelatin nanoparticles (GNPs) were selected as the drug carriers due to their biocompatible & modifiable features.42,43 Next, they were transformed into cationic derivatives ((+)nGNPs) by surface in situ polymerization using the monomer N-(3-Aminopropyl) methacrylamide (APm) for facilitating internalization by cancer cells. Afterwards, (+)nGNPs were encapsulated by covalent attachment with anticancer drug doxorubicin (DOX) terminated pMPC via EDC/NHS conjugation to obtain the final product. Benefiting from the peripheral pMPC layer, inner cationic carriers can be prevented from the undesirable accumulation of biological species. After arrival of (+)nGNPs@DOX-pMPC at tumor sites, pMPC layer can be gradually removed via hydrolysis of acyl hydrazone bond, linking DOX and pMPC chain, under the weakly acidic conditions (pH value between 6.5–7.0), while the drug still remains on the cationic carriers. The re-exposed positively charged surface allows the nanoparticles to easily penetrate the cell membrane. Although the amide linkage between DOX and GNPs is relatively stable toward pH, intracellular localized nanoparticles can still be slowly decomposed by the multiple hydrolytic enzymes in the cytoplasm to induce cell apoptosis. Compared with conventional nano-carriers DOX-GNPs, which are prepared by conjugating DOX onto the GNPs with direct covalent bonding, the core–shell nanoparticles presented here exhibit both enhanced anti-biofouling properties and higher cellular internalization efficiency, thus making them a promising candidate for long-acting and effective cancer therapy.
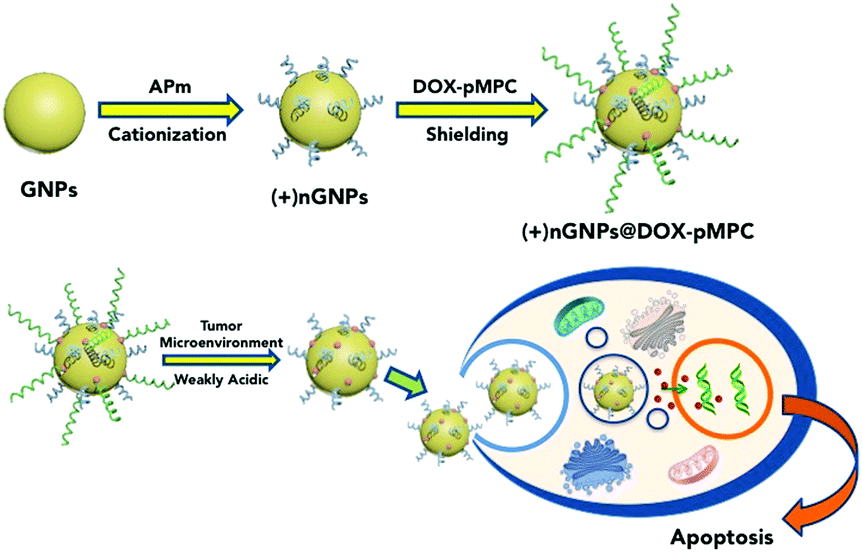
 |
| | Scheme 1 Schematic illustration of the fabrication of (+)nGNPs@DOX-pMPC and its stimuli-responsive decladding in tumor microenvironment for high cellular internalization and cancer therapy. | |
Experimental section
Materials and characterization
Gelatin type A from porcine skin (GelA, 240 Bloom), and glutaraldehyde (AR, 50% in H2O) were purchased from Aladdin Chemical Reagent Co. Ltd (Shanghai, China). N-Acryloxysuccinimide (NAS, 99%), fluorescein isothiocyanate isomer I (FITC, 95%), 2-methacryloyloxyethyl phosphoryl-choline (MPC, 96%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydro-chloride (EDC, 99%), and N-hydroxysuccinimide (NHS, 98%) were purchased from J&K Scientific Co. Ltd (Beijing, China). N-(3-Aminopropyl)methacrylamide hydrochloride (APm·HCl, 98%) was purchased from Macklin Reagent Co. Ltd (Shanghai, China). Ammonium persulfate (APS, ≥98%), N,N,N′,N′-tetra-methylethylenediamine (TEMED, 99%), 4-cyano-4-(phenyl-carbonothioylthio) pentanoic acid (CTP), and hydrazine mono-hydrate (N2H4·H2O, 80%, v/v) were purchased from Sigma-Aldrich (St Louis, USA). Biochemical reagents, including methyl tetrazolium (MTT), tris acetate-EDTA buffer (TAE, 50×) and agarose, were provided by Beyotime Biotechnology (Shanghai, China). FITC-Annexin V apoptosis detection kit, and normal mouse serum were obtained from Thermo Fisher Scientific (USA). Polystyrene tissue culture treated 6-well, 12-well and 96-well plates were purchased from Corning Costar. All other reagents and solvents were at least analytical grade and used without further purification.
1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCEIII 400 spectrometer with D2O as solvent at 298 K and tetramethylsilane (TMS) as the internal reference. Molecular weights of synthetic polymers were determined by gel permeation chromatography (GPC). PerkinElmer series 200 system (Viscotek A6000M, GeneralMixed, 300 mm × 8.0 mm aqueous column, polyethylene glycol calibration) was equipped with a refractive index (RI) detector. 0.1 mol L−1 sodium nitrate aqueous solution was used as the mobile phase at a flow rate of 1 mL min−1 at 30 °C. Dynamic light scattering (DLS) measurements were performed with a Malvern Zetasizer Nano S apparatus equipped with a 4.0 mW laser operating at λ = 633 nm. All samples with gelatin concentration of 0.5 mg mL−1 were measured in phosphate buffer (PB) solution at a scattering angle of 173°. Transmission electron microscopy (TEM) studies were performed with a JEOL JEM-100CX-II instrument at a voltage of 200 kV. Samples were prepared by drop-casting their solutions onto carbon-coated copper grids and then air-drying at room temperature before measurement.
Synthesis of poly(2-methacryloyloxyethyl phosphorylcholine) (pMPC)
RAFT polymerization method was utilized for the synthesis of pMPC. Typically, MPC (0.3 g, 1.02 mmol), CTP (7 mg, 0.025 mmol), and ACVA (2.9 mg, 0.01 mmol) were dissolved in 4 mL ultra-dry methanol, added into a glass reaction bulb, and then degassed via several freeze–pump–thaw cycles. The bulb was then immersed into an oil bath heated to 70 °C to initiate the polymerization. The mixture was kept at 70 °C under continuous stirring for another 10 h. Next, the bulb was dipped into liquid nitrogen to terminate the reaction. The solution was transferred into a dialysis bag (MWCO 3.5 kDa) and thoroughly dialyzed against ultrapure water to remove the unreacted monomer, chain transfer reagent and initiator. Finally, the product was frozen and lyophilized to afford product pMPC as light pink powder (yield: 86%). 1H NMR (400 MHz, D2O) δ (ppm): 7.86 (d, 2H), 7.58 (t, 1H), 7.42 (t, 2H), 4.19 (br, 2H), 4.12 (br, 2H), 3.98 (br, 2H), 3.57 (br, 2H), 3.13 (s, 9H), 2.10–1.70 (br, 2H), 0.98–0.79 (br, 3H).
Synthesis of hydrazide terminated poly(2-methacryloyloxyethyl phosphorylcholine) (NH2NH-pMPC)
NH2NH-pMPC was synthesized by a reaction between the reserved carboxyl group at the terminus of pMPC chain and hydrazine, as reported in a previous publication.44 Briefly, 200 mg of pMPC was refluxed for 4 h in 4 mL of ultra-dry methanol with an addition of 0.05 mL of sulfuric acid. The reaction mixture was then cooled to room temperature and hydrazine hydrate (80%, v/v) (0.05 mL) was added. The system was refluxed with vigorously stirring for another 1 h. Afterwards, the mixture was maintained at low temperature to yield NH2NH-pMPC. The final product was dialyzed against ultrapure water to remove impurities and lyophilized to a white powder NH2NH-pMPC (yield: 90%). 1H NMR (400 MHz, D2O) δ (ppm): 4.18 (br, 2H), 4.10 (br, 2H), 3.96 (br, 2H), 3.86 (br, 2H), 3.56 (br, 2H), 3.14 (s, 9H), 2.10–1.70 (br, 2H), 0.96–0.77 (br, 3H).
Synthesis of doxorubicin terminated poly(2-methacryloyloxyethyl phosphorylcholine) (DOX-pMPC)
Conjugating DOX to the terminal hydrazide group of NH2NH-pMPC was performed according to a previously published methodology.18,45 It was synthesized by mixing NH2NH-pMPC (150 mg) and DOX hydrochloride (12.5 mg) in dry methanol (5 mL). To this solution, 20 μL of acetic acid and 100 mg of anhydrous sodium sulfate were added. The mixture was stirred at room temperature in dark for 2 days, and the product was isolated by extensive dialysis against ultrapure water (MWCO 3.5 kDa) for 2 days. It was then freeze-dried to yield wine colored powder DOX-pMPC (yield: 67%). DOX content was measured by determining absorbance at 520 nm, recorded on a BioTek Synergy H4 plate-reader with background correction.
Preparation of gelatin nanoparticles (GNPs)
Gelatin nanoparticles were prepared using a previously reported two-step desolvation method with slight modifica-tion.46 Typically, 1.25 g of GelA was dissolved in 25 mL of ultrapure water and slowly heated to 50 °C. Then, 25 mL acetone was rapidly added to the solution with gentle shaking. After centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min, a white color supernatant was removed and the gel-like precipitate was dissolved in 25 mL of ultrapure water by slowly heating to 50 °C. Next, the pH was adjusted to 3.0 using hydrochloric acid, and the solution was constant stirred at 40 °C during a slow addition of acetone (75 mL, less than 3 mL min−1). The obtained ivory solution was immediately mixed with 0.1 mL of 50% glutaraldehyde under stirring for another 1 h, followed by overnight incubation at room temperature. The GNPs stock solution was preserved at −20 °C for further studies.
000 rpm for 15 min, a white color supernatant was removed and the gel-like precipitate was dissolved in 25 mL of ultrapure water by slowly heating to 50 °C. Next, the pH was adjusted to 3.0 using hydrochloric acid, and the solution was constant stirred at 40 °C during a slow addition of acetone (75 mL, less than 3 mL min−1). The obtained ivory solution was immediately mixed with 0.1 mL of 50% glutaraldehyde under stirring for another 1 h, followed by overnight incubation at room temperature. The GNPs stock solution was preserved at −20 °C for further studies.
Preparation of cationic gelatin nanoparticles ((+)nGNPs)
Before preparation of (+)nGNPs, GNPs stock solution was dialyzed against phosphate buffer (50 mM, pH 7.4) to replace the acetone completely, and the gelatin concentration was determined by BCA protein quantification analysis. Cationic gelatin nanoparticles were prepared by a previously reported surface in situ polymerization method.47–49 GNPs were first conjugated with NAS to attach acryloyl groups onto their surfaces. In a typical procedure, NAS (1%, w/w, in DMSO) was added into the GNPs solution (2 mg mL−1) with a mass ratio of GNPs:NAS = 30:1. Conjugation was achieved by keeping the reaction at 4 °C for 2 h. The solution was then thoroughly re-dialyzed against phosphate buffer (50 mM, pH 7.4) to remove any unreacted NAS. The obtained acryloylated GNPs were then modified with the cationic monomer APm by an in situ polymerization method. APm was first prepared as 10% (w/w) stock solution in ultrapure water and its pH was adjusted to neutral. Then, APm was added into the dilute solution of acryloylated GNPs (2 mg mL−1) at a mass ratio of GNPs:APm = 1:2. Polymerization was initiated by the addition of APS and TEMED (mass ratio APm:APS:TEMED = 5:1:1) and kept at 4 °C under constant stirring for 1 h. After polymerization, the unreacted monomer and non-bonded polymers were removed by ultrafilter centrifugation with 100 kDa molecular weight cut-off (MWCO). The obtained (+)nGNPs solution was re-diluted to 2 mg mL−1 using 1× PBS and preserved at 4 °C for further studies. For agarose gel electrophoresis and flow cytometry experiments, gelatin nanoparticles and cationic gelatin nanoparticles labelled with fluorescent probe FITC, termed as FGNPs and (+)nFGNPs, were also prepared under the same conditions.
Preparation of core–shell nanoparticles (+)nGNPs@DOX-pMPC
(+)nGNPs@DOX-pMPC was prepared by decorating DOX-pMPC onto the (+)nGNPs surface via EDC/NHS conjugation between the carboxyl groups of (+)nGNPs and the amino group of DOX. To furthest avoid the undesirable internal conjugation between the carboxyl groups and amino groups of pAPm short chain, 10 mg of EDC was added into 1 mL of (+)nGNPs (2 mg mL−1) and stirred at 4 °C for 2 h in a typical procedure. After that, DOX-pMPC with different mass ratios to (+)nGNPs were added, followed by thorough mixing. 5.4 mg of NHS was then added into the mixture and the reaction was allowed to proceed for another 4 h. The process of encapsulation was evaluated by agarose gel electrophoresis. Finally, the solution was dialyzed (MWCO: 14 kDa) against 1× PBS for 2 days to give (+)nGNPs@DOX-pMPC. As a positive control, acryloylated GNPs directly conjugated with DOX (DOX-GNPs) was also prepared under the same conditions.
Gelatin quantitative analysis
The gelatin content in nanoparticles was determined by bicinchoninic acid (BCA) colorimetric method. In a typical procedure, a tartrate buffer (pH 11.25) containing 25 mM BCA, 3.2 nM CuSO4, and appropriately diluted nanoparticles solutions were incubated at 60 °C for 1 h. Next, the solutions were cooled to room temperature, and absorbance readings at 562nm were determined with a UV-Vis spectrometer. Gelatin solutions with known concentration were used as standards.
DOX loading content and in vitro drug release study of DOX-pMPC
The drug loading content (DLC) of DOX-GNPs and (+)nGNPs@DOX-pMPC was measured by determining the absorbance of sample solutions at 520 nm using linear standard plots. DLC was calculated according to the following formula:
| DLC (wt%) = (weight of drug/weight of nanoparticles) × 100% |
According to the relatively stable amide linkage between DOX and nanoparticles, there is no great significance to the investigation of DOX release behaviours of (+)nGNPs@DOX-pMPC and DOX-GNPs under simulated acidic conditions. Thereby, only the release profile of DOX-pMPC was evaluated using dialysis method as follows: 0.4 mL of DOX-pMPC solution (1 mg mL−1) was transferred to dialysis tubing (MWCO, 3.5 kDa), and then immersed into 25 mL of phosphate buffer solution (50 mM) at pH 6.8 or 7.4. With mild shaking at 37 °C, 0.2 mL of the external solution was aliquoted and replaced with 0.2 mL of a fresh solution at predetermined time intervals. The amount of released DOX was determined using fluorescence at the wavelength of λex = 480 nm/λem = 580 nm.
Nonspecific protein adsorption assay in vitro
Normal mouse serum (protein concentration ∼40 mg mL−1) was diluted 4 × 103 fold with coating buffer solution (NaHCO3/Na2CO3, pH 9.5). Diluted mouse serum solution was then transferred into a 96-well enzyme linked immunosorbent assay (ELISA) plate at 200 μL per well and incubated overnight at 4 °C for sufficient coating. Afterwards, the solution was replaced by 100 μL PBS containing the FITC-labelled nanoparticles with identical gelatin concentrations, followed by mildly shaking at 37 °C for 2 h. The supernatant was then discarded; subsequently, 100 μL of PBS was added to each well to dissolve the adsorptive samples. Finally, the fluorescent intensity (λem = 525 nm) was determined by BioTek Synergy H4 plate-reader to calculate the adsorptive ratio of samples. Working curve was also plotted by determining the fluorescence of a series of GNPs samples with different concentrations.
Cell culture
HeLa cells (cell line of human uterine cervix carcinoma) were cultured in Dulbecco's Modified Eagle's medium (DMEM) in a humidified atmosphere containing 5% CO2 at 37 °C. DMEM was supplied with 10% FBS, penicillin (50 units per mL), and streptomycin (50 units per mL).
Cellular internalization study
The internalization of nanoparticles by HeLa cells was investigated by both flow cytometry (FCM) and confocal laser scanning microscopy (CLSM). For flow cytometry study, 24-well plates were used for the seeding of HeLa cells with a density of 1.0 × 105 per well in 1 mL DMEM. After incubation for 24 h, the culture medium was discarded and replaced by the fresh neutral medium (pH 7.4) or weak acidic medium (pH 6.8) treated with 2 M HCl. The cells were then treated with FITC-labelled samples (GNPs, (+)nGNPs, (+)nGNPs@DOX-pMPC) with the same gelatin concentration and incubated at 37 °C. The untreated cells were used as control. At different time intervals, after removal of culture medium, the cells were rinsed with PBS three times. Subsequently, the cells were collected from the plates were treated with trypsin, centrifuged (1000 rpm, 5 min) and rinsed with PBS three times. Finally, the cells were suspended in 0.5 mL of PBS for analysis. Measurements were performed on a BD LSRFortessa flow cytometer and data for 1.0 × 104 gated events were collected. For CLSM study, HeLa cells were seeded in 12-well plates at a density of 5.0 × 104 per well in 1 mL DMEM and incubated overnight (a clean coverslip was put on the bottom of each well). Next, the culture medium was discarded and replaced by a fresh neutral medium (pH 7.4) or weak acidic medium (pH 6.8) treated with 2 M HCl. The cells were then incubated with samples with the same DOX concentration for 30 min and 2 h at 37 °C. At specific time intervals, the culture medium was removed and the cells were rinsed with cold PBS three times. 4% formaldehyde was used to fix the cells for 30 min with subsequent rinsing with PBS three times. The cells were then stained with Hoechst 33342 for 10 min followed by another wash with PBS. Finally, the resulting slides were mounted and imaged on a LEICA TCS SP8 fluorescence microscope.
In vitro cytotoxicity study
The relative cytotoxicity of (+)nGNPs@DOX-pMPC against cultured HeLa cells was evaluated through MTT assay. For comparison, cytotoxicity of DOX and DOX-GNPs was also investigated. HeLa cells with a density of 8 × 103 per well in 200 μL of DMEM were seeded on 96-well plates and incubated overnight. Next, the culture medium was removed and replaced with 200 μL of fresh medium containing samples with different concentrations. The cells were incubated for another 72 h. Afterwards, 20 μL of 5 mg mL−1 MTT assay stock solution in sterilized PBS was added to each well. After 4 h incubation, the medium containing unreacted MTT was removed carefully, followed by the addition of 200 μL of DMSO per well to dissolve the blue formazan crystals. Finally, the absorbance at 490 nm was recorded on a plate-reader to calculate the ratio of viable cells. For the apoptosis study, HeLa cells were seeded in 6-well plates at a density of 5.0 × 105 cells per well in 2 mL DMEM and incubated overnight, followed by treatment with DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC with identical DOX concentration of 20 μM at 37 °C for 24 h. HeLa cells without treatment were used as control. For quantitative measurement of apoptosis, both floating and attached cells were collected and rinsed with cold PBS three times, according to the manufacturer's instructions. Next, the cells were stained with Alexa Fluor 488 annexin V and propidium iodide (PI) for apoptosis analysis, and the data for 1.0 × 104 gated events was analyzed.
Animals and tumor models
All animal experiments were conducted in agreement with the guidelines of the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University in agreement with the guidelines of Association for Assessment and Accreditation of Laboratory Animal Care. Male SD rats (≈200 g) and nude mice (six weeks of age) were supplied by the Chinese Academy of Sciences (Shanghai). The nude mice were injected subcutaneously in the right flank region with 200 μL of cell suspension containing 4 × 106 HeLa cells. The tumor was allowed to grow to ≈100 mm3 for antitumor experiments.
Pharmacokinetics and biodistribution
For pharmacokinetics study, SD rats (≈200 g) were randomly divided into three groups (n = 3). PBS solutions of DOX·HCl, DOX-GNPs and (+)nGNPs@DOX-pMPC were intravenously injected through tail vein at a drug dose of 8 mg kg−1. Blood samples (0.1 mL) were collected via eye puncture at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 10 h, 24 h and 48 h after injection. Serum samples were collected by centrifugation at 3000 rpm for 10 min and stored at −20 °C until analysis. Each serum sample (50 μL) was diluted to 200 μL with PBS and analyzed using the fluorescence intensity of DOX (λex = 480 nm, λem = 580 nm). The signal value of samples collected at the first time point was set as 100%. Pharmacokinetics parameters, AUC and T1/2, were calculated by integration and the equation T1/2 = 0.693/k, where k was obtained from the slopes of logC-time plots.
For biodistribution study, HeLa tumor-bearing nude mice were intravenously injected via tail vein with free DOX, DOX-GNPs and (+)nGNPs@DOX-GNPs in PBS at a drug dose of 8 mg kg−1. Mice were sacrificed at 15 min, 2 h and 6 h post-administration (n = 3 at each time point), and the heart, liver, spleen, lung, kidney and tumor were collected. Tissue samples were rinsed with PBS, blotted using paper towel, weighed and stored at −80 °C before being homogenized. The homogenates were collected and diluted to by the same amount with PBS, and then examined for fluorescence intensity of DOX (λex = 480 nm, λem = 580 nm) directly.
In vivo anticancer study and histochemical analysis
The HeLa tumor-bearing nude mice were randomly divided into 4 groups (n = 3 per group) and intravenously injected with PBS, free DOX·HCl, DOX-GNPs and (+)nGNPs@DOX-pMPC via the tail vein at a drug dose of 6 mg kg−1 every 3 days for 22 days. The lengths and widths of the tumors and body weights of the mice were measured before every injection. Tumor volume (V) was calculated using the formula
| V (mm3) = 1/2 × length (mm) × width (mm)2 |
After 22 days of treatment, the mice were sacrificed and the main organs were sectioned into small pieces, fixed in 10% formalin, and embedded in paraffin for H&E staining. Tumors were separated for TUNNEL assays, which were performed with the one-step TUNEL fluorescent kit. All experimental procedures were in accordance to the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± standard deviation. Statistical significance was determined using Student's T-test to evaluate statistical significance, which was set at p < 0.05 for all experiments.
Results and discussion
Preparation and characterization of core–shell nanoparticles (+)nGNPs@DOX-pMPC
For the purpose of endowing the materials with both anti-biofouling ability and high cellular internalization, the nanomedicine in this work is designed with separable cationic core and zwitterionic shell ((+)nGNPs@DOX-pMPC) fabricated by gelatin and pMPC, respectively. The preparation route of (+)nGNPs@DOX-pMPC, including the synthesis of DOX-pMPC, preparation of (+)nGNPs and the final conjugation of these two components, is schematically illustrated in Scheme 1. Zwitterionic polymer pMPC was synthesized via one-pot RAFT polymerization (Scheme 2), and CTP and ACVA were used as chain transfer reagent and initiator, respectively. The reserved terminus carboxyl group of pMPC chain was then converted into hydrazide via a reaction with hydrazine, with sulphuric acid as a catalyst, to obtain NH2NH-pMPC. Afterwards, DOX was conjugated onto the polymer tail end with a reaction between hydrazide and the carbonyl group in the DOX structure to afford the product DOX-pMPC. 1H NMR spectra and the corresponding H atoms are shown in Fig. 1A. The multiple peaks ranging from 7.42 to 7.86 ppm are assigned to the characteristic protons of ArH from CTP moieties, suggesting that the functional terminal groups are well preserved during polymerization. After reaction with hydrazine, a new signal appears at 3.86 ppm corresponding to the protons of hydrazide group of NH2NH-pMPC. The signals of DOX in the sample of DOX-pMPC tend to be inconspicuous due to the shielding effect in aqueous solution.17,18,50 Moreover, no apparent change in the main structure of pMPC backbone was observed throughout the whole reaction. As shown in Fig. 1B, the characteristic peak of hydrazide group at 3.86 ppm disappears after conjugation with DOX, indicating successful synthesis of the objective polymer DOX-pMPC.
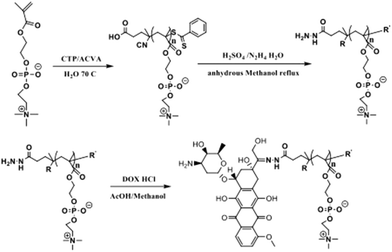 |
| | Scheme 2 Schematic illustration of the synthesis of pMPC, NH2NH-pMPC and DOX-pMPC. | |
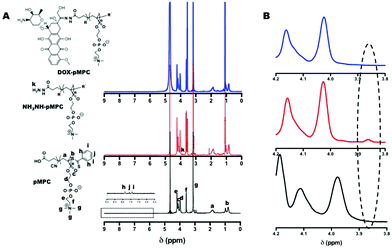 |
| | Fig. 1 (A) 1H NMR spectra of pMPC, NH2NH-pMPC and DOX-pMPC; (B) enlarged 1H NMR spectra in the range of chemical shift between 3.8–4.2 ppm. | |
The molecular weight of pMPC was measured by GPC and the data is summarized in Table 1. The representative GPC profiles are shown in Fig. S1 (ESI†). The polymer has a relatively unimodal elution peak with narrow molecular weight distribution (PDI 1.38). For comparison, pMPC with higher molecular weight (pMPChigh) was also synthesized by decreasing the molar ratio of MPC/CTP, resulting in a clear shift toward shorter elution time of the GPC trace. In addition, when compared with the free drug, the polymer chain pMPChigh conjugated with DOX cannot penetrate the ultra filter membrane (MWCO = 10 kDa). DOX-pMPC clearly stays in the inner ultra filter tube after centrifugation (Fig. S2†), thus further confirming the successful conjugation of DOX onto the polymer chain. Since reactivity of the terminus group is significantly affected by the polymer molecular weight, pMPC with lower molecular weight is preferentially chosen for further studies in this work.
Table 1 GPC characterization data of pMPC and pMPChigh
| Polymers |
M
n, GPC (g mol−1) |
M
w, GPC (g mol−1) |
PDI |
| pMPC |
6519.4 |
9017.5 |
1.38 |
| pMPChigh |
8759.7 |
12448.3 |
1.42 |
Native electronegative GNPs scarcely penetrate the cell membrane due to the homologue of gelatin and protein and has very low cellular internalized ratio.51,52 Thereby, a popular choice for increasing this internalization ratio has been nanomedicine surface modification using cationic materials that have strong electrostatic interactions with the cytomembrane. Accordingly, GNPs are firstly prepared by a two-step desolvation method with subsequent surface in situ polymerization using the cationic monomer APm to obtain electropositive (+)nGNPs. A high density of positive charge is known to be beneficial to the cellular internalization. However, an increased density of pAPm can also significantly hinder the subsequent conjugation of zwitterionic polymers. Hence, the polymerization of APm is carefully regulated to impart a weak electropositive charge to the nanoparticles for facilitating their uptake by cancer cells. Consequently, DOX-pMPC with different mass ratios were covalently attached onto the surface (+)nGNPs via EDC/NHS conjugation. According to the TEM images shown in Fig. 2A and B, (+)nGNPs exhibit core–shell-like spherical morphology and their size increased compared with native GNPs. The results are also validated by DLS measurement. As shown in Fig. 2C–F, the size of nanoparticles increased from 157 nm to 231 nm after the two step modification and is consistent with TEM results. To investigate the decladding features of peripheral pMPC shell, (+)nGNPs@DOX-pMPC was incubated under simulated weak acidic conditions of pH 6.0 for 6 h and the resultant particle size was then monitored by DLS. As expected, the size decreases back to 171 nm (close to the original state), indicating an effective removal of the zwitterionic polymer shell. Moreover, zeta potential of (+)nGNPs@DOX-pMPC shifts from 8.4 mV to 0.8 mV and then returns back to 6.8 mV after acidic treatment, which is in accordance with the DLS results. Agarose gel electrophoresis was performed to further confirm the results. After cationic modification, the surface charge of GNPs is reversed from negative to weakly positive (Fig. 3A). During the preparation of (+)nGNPs@DOX-pMPC, with increased mass ratio of DOX-pMPC/(+)nGNPs, it is clearly observed that the samples are gradually compressed in the pore (Fig. 3B), suggesting that the positive charges are shielded by the outer pMPC shell. Based on these results, mass ratio of DOX-pMPC/(+)nGNPs is defined as 40:1 for further studies. In addition, upon incubation at pH 6.0 for 6 h, the nanoparticles re-escape and migrate to the cathode. The results are consistent with DLS data and further corroborate the successful preparation of (+)nGNPs@DOX-pMPC.
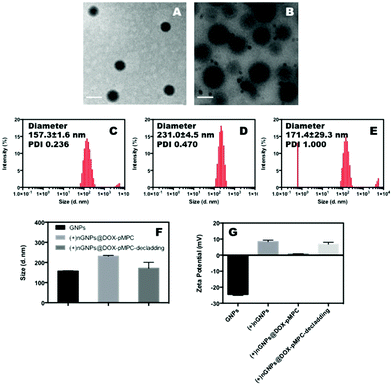 |
| | Fig. 2 TEM images of (A) GNPs and (B) (+)nGNPs@DOX-pMPC (scale bars represent 200 nm); size distribution of (C) GNPs, (D) (+)nGNPs@DOX-pMPC and (E) (+)nGNPs@DOX-pMPC incubated at pH 6.0 for 6 h; (F) histogram of sizes of GNPs, (+)nGNPs@DOX-pMPC and de-cladded (+)nGNPs@DOX-pMPC; (G) zeta potentials of GNPs, (+)nGNPs, (+)nGNPs @DOX-pMPC and de-cladded (+)nGNPs@DOX-pMPC. Data are presented as average ± standard deviation (n = 3). | |
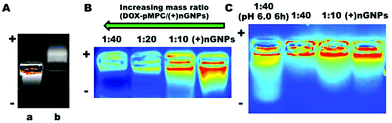 |
| | Fig. 3 Agarose gel electrophoresis fluorescent images of FITC labeled gelatin nanoparticles. (A) a. (+)nGNPs, b. GNPs; (B) (+)nGNPs@DOX-pMPC with different mass ratios; (C) de-cladded (+)nGNPs@DOX-pMPC after treatment at pH 6.0 for 6 h. | |
In vitro drug release of DOX-pMPC
Fast release of anticancer drug from the drug delivery system is a critical factor for the efficacy of cancer therapy.53 In this study, the anticancer drug DOX acts as not only a bioactive constituent but also as the essential connecting molecule between the zwitterionic polymer and gelatin nanoparticles. Thus, the hydrolysis rate of conjugated acyl hydrazine bond is of great significance. To investigate the decladding behavior of nanoparticles in tumor microenvironment, the in vitro release behavior of DOX-pMPC was evaluated by dialysis under neutral (pH 7.4) and acidic (pH 6.8) conditions at 37 °C. As shown in Fig. 4A, compared with the sample treated in neutral conditions, DOX-pMPC exhibits faster release behavior in the weakly acidic environment, indicating more efficient hydrolysis of acyl hydrazone bond. There is approximately 29.3% DOX released at pH 6.8 after 48 h. In comparison, 12.7% DOX release is observed at pH 7.4, suggesting a higher stability of DOX-pMPC under physiological conditions.
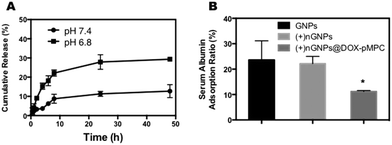 |
| | Fig. 4 (A) Cumulative release profile of DOX from DOX-pMPC at pH 7.4 and pH 6.8; (B) in vitro nonspecific protein adsorption of GNPs, (+)nGNPs and (+)nGNPs@DOX-pMPC. Data are presented as average ± standard deviation (n = 3) (statistical significance level is *p < 0.05). | |
Nonspecific protein adsorption study
As mentioned before, exogenetic nanoparticles tend to be recognized by biological species, such as complement proteins, when they are introduced into blood circulation, resulting in rapid elimination by macrophages and reticuloendothelial organs. Therefore, a simulated nonspecific protein adsorption study was carried out to evaluate the anti-biofouling characteristics of (+)nGNPs@DOX-pMPC. The results showed that (+)nGNPs@DOX-pMPC can considerably resist adsorption by mouse serum albumin compared with both GNPs (23.6 ± 7.6%) and (+)nGNPs (22.2 ± 2.9%), and the adsorption ratio of (+)nGNPs@DOX-pMPC at 11.2 ± 0.4% (Fig. 4B) is significantly different. It is inferred that the cationic gelatin core can be effectively protected by the superhydrophilic outer pMPC layer.
Cellular internalization studies
Cellular internalization was first investigated by flow cytometry analysis. HeLa cells were treated with FITC-labeled nanoparticles for predetermined time intervals, and cellular uptake was expressed as the increase of cellular fluorescent intensity. Since gelatin behaves like protein, native GNPs show a high repulsion to the cell membrane. As shown in Fig. 5, there is no observable internalization of GNPs by HeLa cells over time. In contrast, over 40% of (+)nGNPs has been internalized in HeLa cells within 2 h, exhibiting much higher affinity to the cell membrane.
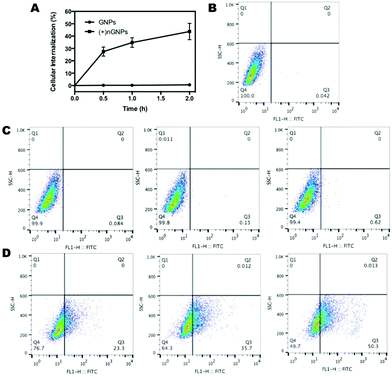 |
| | Fig. 5 Cellular internalization of GNPs and (+)nGNPs by flow cytometry analysis. HeLa cells are incubated with the samples for different times (30 min, 1 h, and 2 h). (A) Cellular internalization ratio-time plots; FACS profiles of (B) blank control, (C) GNPs and (D) (+)nGNPs at different time intervals. Quantitative data are presented as average ± standard deviation (n = 3). | |
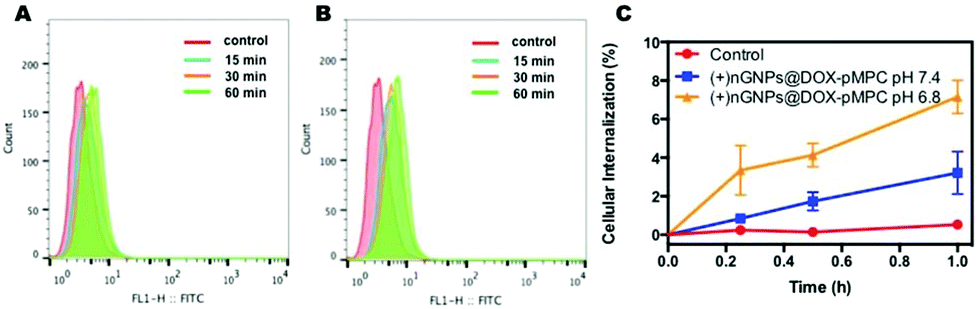
After modification by DOX-pMPC, as shown in Fig. 6, (+)nGNPs@DOX-pMPC exhibit distinctly lower uptake rate by HeLa cells with internalized ratio of 3.2% within 1 h, demonstrating that the outer pMPC shell can noticeably restrain the internalization of inner cationic core by HeLa cells. However, when incubated in the weakly acidic conditions (pH 6.8) that simulate tumor microenvironment, (+)nGNPs@DOX-pMPC show an obviously higher cellular affiliative behavior (internalized ratio of ∼7.2% within 1 h), indicating a gradual removal of the outer pMPC shell accompanied with more exposure of the positive charges. Upon increasing the incubation time to 4 h, there is a more significant difference between these two conditions: cellular internalization ratio is ∼20.1% at pH 7.4 and ∼32.6% at pH 6.8 (Fig. S3†).
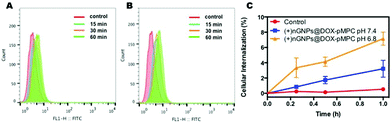 |
| | Fig. 6 Flow cytometry histogram profiles of HeLa cells incubated with (A) (+)nGNPs@DOX-pMPC at pH 7.4 and (B) (+)nGNPs@DOX-pMPC at pH 6.8; (C) quantitative plots of the cellular internalization ratio. DOX-GNPs was used as control. Data are presented as average ± standard deviation (n = 3). | |
CLSM was also performed for further investigation of the cellular uptake behaviours of (+)nGNPs@DOX-pMPC. After culturing HeLa cells with the samples for predetermined time, the nuclei were stained with Hoechst 33342 for observation by CLSM (Fig. 7). Compared with the quick location of free DOX at nuclei, there is no obvious DOX fluorescence for both tested nanoparticles when observed at the initial 30 min. Nevertheless, relatively strong red fluorescence of (+)nGNPs@DOX-pMPC is observed in the cytoplasm after 2 h of incubation, which is consistent with the flow cytometry results. In comparison, the signal of DOX-GNPs still cannot be detected, indicating poor cellular internalization capacity of the native gelatin nanoparticles.
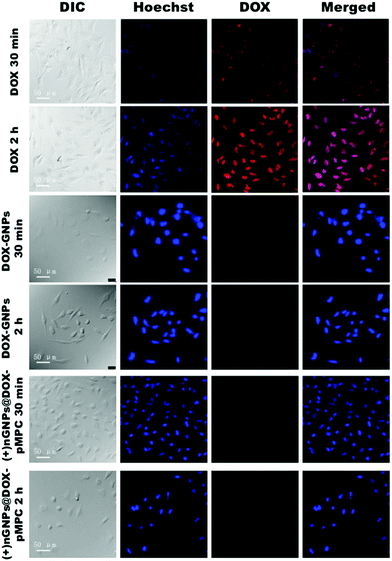 |
| | Fig. 7 CLSM images of HeLa cells incubated with DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC at different time intervals (30 min and 2 h) at pH 6.8. Cell nuclei were stained with Hoechst 33342. | |
In vitro cytotoxicity studies
The proliferation inhibition by (+)nGNPs@DOX-pMPC was evaluated by MTT assay, with free DOX and DOX-GNPs as positive controls. HeLa cells were treated with the samples at different DOX concentrations from 0.5 to 50 μM. As displayed in Fig. 8a, after 72 h incubation, (+)nGNPs@DOX-pMPC exhibits similar cytotoxicity as free DOX, especially at high concentrations (≥10 μM). This indicates sufficient delivery of DOX by (+)nGNPs into the cancer cells, further confirming the delamination of this core–shell nanoparticles. In comparison to (+)nGNPs@DOX-pMPC, the cytotoxicity of DOX-GNPs is distinctly lower, suggesting the stagnation of these nanoparticles in the extracellular matrix. Apoptosis assay was also performed using flow cytometry to further evaluate the cell-prohibiting efficacy. As shown in Fig. 8B–E, after 24 h incubation at DOX concentration of 20 μM, (+)nGNPs@DOX-pMPC resulted in total cells apoptosis of 81.5%, with much higher efficacy than that of DOX-GNPs (35.5%) and approximately close to that of free DOX (90.9%). The results are in accordance with MTT assay, revealing the core–shell nanoparticles to be a promising candidate for cancer therapy.
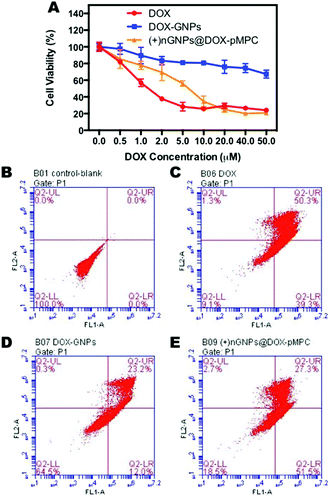 |
| | Fig. 8 (A) In vitro cytotoxicity of DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC against HeLa cells for 72 h. The data are presented as average ± standard error (n = 6). Flow cytometric analysis for apoptosis of HeLa cells treated with (B) negative control; (C) DOX; (D) DOX-GNPs and (E) (+)nGNPs@DOX-pMPC for 24 h. Lower left, living cells; lower right, early apoptotic cells; upper right, late apoptotic cells; upper left, necrotic cells. Inserted numbers in the profiles indicate the percentage of cells present in the corresponding quadrant. | |
Pharmacokinetics, biodistribution and anticancer activities studies
To evaluate the anti-biofouling properties of (+)nGNPs@DOX-pMPC in vivo, pharmacokinetic studies were conducted by intravenous injection of the nanoparticles in SD rats (∼200 g) at a drug dose of 8 mg kg−1. Rats treated with DOX and DOX-GNPs were also investigated as comparisons. As shown in Fig. 9A, compared with the rapid clearance of DOX and DOX-GNPs in vivo (signals of free DOX and DOX-GNPs decreased to 15.5% and 30.2% at 2 h, respectively), the fluorescence signal of (+)nGNPs@DOX-pMPC decreased slowly (72.6% at 2 h) in a time-dependent manner and there was still 16.9% signal remaining at 10 h post-injection. Furthermore, Table S1† shows a comparison of free drug and the nanoparticles. Compared with free DOX and DOX-GNPs, (+)nGNPs@DOX-pMPC exhibit higher T1/2 (∼5.69 h) and much higher AUC (279.3 μg mL−1 h−1), indicating its relatively high stability in vivo and bioavailability of the system. The results showed that these core–shell nanoparticles had good bioavailability and prolonged circulation time, which was by virtue of the peripheral hydrated pMPC layer in an aqueous solution.
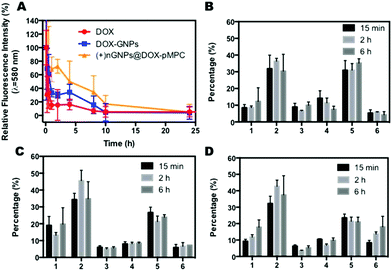 |
| | Fig. 9 (A) Representative plasma fluorescence-time profiles of DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC (λem = 580 nm). Biodistribution of (B) DOX, (C) DOX-GNPs and (D) (+)nGNPs@ DOX-pMPC (1. heart, 2. liver, 3. spleen, 4. lung, 5. kidney, and 6. tumor). Data are presented as mean ± standard error (n = 3). | |
To examine the biodistribution of (+)nGNPs@DOX-pMPC, HeLa tumor bearing mice were sacrificed after intravenous administration of the nanoparticles at different time intervals. Mice treated with DOX and DOX-GNPs were also investigated. As shown in Fig. 9B–D, possibly due to its relatively large size, (+)nGNPs@DOX-pMPC accumulated in liver during a short time period. Even then, in contrast to the free DOX, these nanoparticles exhibited time-dependent accumulation in tumor sites, indicating their considerable bioavailability.
Moreover, for the investigation of in vivo anticancer activity of the nanoparticles, HeLa tumor-bearing mice were intravenously injected with PBS, free DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC at a drug dose of 6 mg kg−1. As shown in Fig. 10A, (+)nGNPs@DOX-pMPC exhibited significantly higher anticancer efficacy than DOX-GNPs. The difference in therapeutic effects between free DOX and nanomedicine might be attributed to the relatively large size of the nanoparticles. The results of body weight measurements (Fig. 10B) showed that there was no obvious weight change for mice treated with (+)nGNPs@DOX-pMPC, while a severe weight loss was recorded in the DOX treated samples, further validating the good biocompatibility of this core–shell nanoparticles. We also utilized TUNEL staining to study the anticancer activities in different experimental groups. As expected in Fig. 10C–F, the tumor tissues can hardly be stained in the PBS groups, while apparent apoptosis signals were observed in samples treated with DOX and (+)nGNPs@DOX-pMPC, indicating similar anticancer activities of (+)nGNPs@DOX-pMPC and the free drug. Additionally, as shown in Fig. S4,† when compared with the samples treated with free DOX, the major organs (heart, liver, spleen, lung and kidney) of samples treated with (+)nGNPs@DOX-pMPC exhibited less cell necrosis or apoptosis, further confirming its low-toxicity to normal tissues.
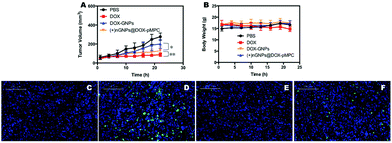 |
| | Fig. 10
In vivo anticancer activities. (A) Tumor growth plots and (B) body weight changes for HeLa cells tumor-bearing mice (treatment with PBS, DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC) (n = 3); (C)–(F) TUNEL histology section analysis of tumor slices from mice treated with PBS, DOX, DOX-GNPs and (+)nGNPs@DOX-pMPC after 22 days. | |
Conclusions
In summary, we have fabricated a type of nanoparticles with core–shell structure termed as (+)nGNPs@DOX-pMPC, which possess both superior resistance to biofouling and high cellular internalization ratio. Benefitting from the surface decorated zwitterions pMPC, the encapsulated cationic gelatin core can be efficiently rescued from non-specific protein adsorption (decreased from 23.6% to 11.2%), making long-acting cancer therapy possible. On the other hand, the peripheral polymer shell can be easily separated from the core by hydrolysis of the linking acyl hydrazone bond under weakly acidic conditions, which is a typical physiological tumor microenvironment. Thus, the embedded positive charge is re-exposed and the uptake of nanoparticles by cancer cells occurs, thereby considerably enhancing their therapeutic efficacy. Based on the combination of benefits mentioned above, it is expected that these novel core–shell nanoparticles (+)nGNPs@DOX-pMPC may provide a potential platform for long-acting and high-efficient cancer therapy.
Statement of ethics
All experiments procedures were conducted in conformity with NIH guidelines (Publication No. 85-23, revised 1996) and approved by Animal Care and Use Committee of Shanghai Jiao Tong University.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from National Natural Science Foundation of China (51690151, 51503122, 51473093), the National Basic Research Program of China (2015CB931801), Shanghai Rising-Star Program (17QC1401100), Shanghai Municipal Government (18JC1410800), Natural Science Foundation of Liaoning Province of China (2015020439), Shanghai Jiao Tong University (15X110070008, 16X100100019, 17X100040070) and Innovation Fund (IFPM2016B009) from Joint Research Center for Precision Medicine set up by Shanghai Jiao Tong University & Affiliated Sixth People's Hospital South Campus (Fengxian Central Hospital).
References
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353 CrossRef CAS PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748 CrossRef CAS PubMed.
- Y. Wang, P. Huang, M. Hu, W. Huang, X. Zhu and D. Yan, Bioconjugate Chem., 2016, 27, 2722 CrossRef CAS PubMed.
- X. Wu, L. Zhou, Y. Su and C.-M. Dong, Polym. Chem., 2015, 38, 6857 RSC.
- A. Jhaveri, P. Deshpande and V. Torchilin, J. Controlled Release, 2014, 190, 352 CrossRef CAS PubMed.
- K. Zhu, G. Liu, J. Hu and S. Liu, Biomacromolecules, 2017, 18, 2571 CrossRef CAS PubMed.
- Y. Liu, J. Li and Y. Lu, Adv. Drug Delivery Rev., 2015, 90, 24 CrossRef CAS PubMed.
- P. Zhang, F. Sun, C. Tsao, S. Liu, P. Jain, A. Sinclair, H. C. Hung, T. Bai, K. Wu and S. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046 CrossRef CAS PubMed.
- L. Zhang, Y. Liu, G. Liu, D. Xu, S. Liang, X. Zhu, Y. Lu and H. Wang, Nano Res., 2016, 9, 2424 CrossRef CAS.
- S. Liang, Y. Liu, X. Jin, G. Liu, J. Wen, L. Zhang, J. Li, X. Yuan, I. S. Y. Chen, W. Chen, H. Wang, L. Shi, X. Zhu and Y. Lu, Nano Res., 2016, 9, 1022 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS PubMed.
- J. Hu, X. Wang, Y. Qian, Y. Yu, Y. Jiang, G. Zhang and S. Liu, Macromolecules, 2015, 48, 5959 CrossRef CAS.
- D. Narayanan, M. G. Geena, H. Lakshmi, M. Koyakutty, S. Nair and D. Menon, Nanomedicine, 2013, 9, 818 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- J. W. Maina, J. J. Richardson, R. Chandrawati, K. Kempe, M. P. van Koeverden and F. Caruso, Langmuir, 2015, 31, 7776 CrossRef CAS PubMed.
- Z. Cao, L. Zhang and S. Jiang, Langmuir, 2012, 28, 11625 CrossRef CAS PubMed.
- G.-Y. Liu, L.-P. Lv, C.-J. Chen, X.-S. Liu, X.-F. Hu and J. Ji, Soft Matter, 2011, 7, 6629 RSC.
- X. Chen, S. S. Parelkar, E. Henchey, S. Schneider and T. Emrick, Bioconjugate Chem., 2012, 23, 1753 CrossRef CAS PubMed.
- C. Pegoraro, D. Cecchin, L. S. Gracia, N. Warren, J. Madsen, S. P. Armes, A. Lewis, S. Macneil and G. Battaglia, Cancer Lett., 2013, 334, 328 CrossRef CAS PubMed.
- E. Ellis, K. Zhang, Q. Lin, E. Ye, A. Poma, G. Battaglia, X. J. Loh and T.-C. Lee, J. Mater. Chem. B, 2017, 5, 4421 RSC.
- H. Jiang, D. Chen, D. Guo, N. Wang, Y. Su, X. Jin, G. Tong and X. Zhu, Biomater. Sci., 2017, 5, 686 RSC.
- J. Zhao, Z. Qin, J. Wu, L. Li, Q. Jin and J. Ji, Biomater. Sci., 2017, 6, 200 RSC.
- L. Ye, Y. Zhang, B. Yang, X. Zhou, J. Li, Z. Qin, D. Dong, Y. Cui and F. Yao, ACS Appl. Mater. Interfaces, 2016, 8, 4385 CrossRef CAS PubMed.
- J. Wu, Y. Lin, H. Li, Q. Jin and J. Ji, J. Colloid Interface Sci., 2017, 485, 251 CrossRef CAS PubMed.
- J.-G. Piao, F. Gao, Y. Li, L. Yu, D. Liu, Z.-B. Tan, Y. Xiong, L. Yang and Y.-Z. You, Nano Res., 2018, 11, 3193 CrossRef CAS.
- X. Zhang, D. Xu, X. Jin, G. Liu, S. Liang, H. Wang, W. Chen, X. Zhu and Y. Lu, J. Controlled Release, 2017, 255, 54 CrossRef CAS PubMed.
- D. Chen, Y. Huang, S. Xu, H. Jiang, J. Wu, X. Jin and X. Zhu, Macromol. Biosci., 2017, 1700174 CrossRef PubMed.
- S. Liang, X. Jin, Y. Ma, J. Guo and H. Wang, RSC Adv., 2015, 5, 88560 RSC.
- W. Li, Q. Liu, P. Zhang and L. Liu, Acta Biomater., 2016, 40, 254 CrossRef CAS PubMed.
- H. Ou, T. Cheng, Y. Zhang, J. Liu, Y. Ding, J. Zhen, W. Shen, Y. Xu, W. Yang, P. Niu, J. Liu, Y. An, Y. Liu and L. Shi, Acta Biomater., 2018, 65, 339 CrossRef CAS PubMed.
- J. Lu, H. Jia, L. Guo, G. Zhang, Y. Cao, H. Yan and K. Liu, Eur. Polym. J., 2015, 66, 376 CrossRef CAS.
- H. Li, J. Du, J. Liu, X. Du, S. Shen and Y. Zhu, ACS Nano, 2016, 10, 6753 CrossRef CAS PubMed.
- H. Li, J. Du, X. Du, C. Xu, C. Sun and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- X. Xu, P. Saw, W. Tao, Y. Li, X. Ji and M. Yu, Nano Lett., 2017, 17, 4427–4435 CrossRef CAS PubMed.
- Y. Yuan, C. Mao, X. Du, J. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed.
- R. Mo and Z. Gu, Mater. Today, 2016, 19, 274 CrossRef CAS.
- J. Liu, Z. Luo, J. Zhang, T. Luo, J. Zhou, X. Zhao and K. Cai, Biomaterials, 2016, 83, 51 CrossRef CAS PubMed.
- K. Han, J. Zhang, W. Zhang, S. Wang, L. Xu, C. Zhang, X. Zhang and H. Han, ACS Nano, 2017, 11, 3178 CrossRef CAS PubMed.
- W. Cheng, H. Cheng, S. Wan, X. Zhang and M. Yin, Chem. Mater., 2017, 29, 4218 CrossRef CAS.
- Y. Han, J. Li, M. Zan, S. Luo, Z. Ge and S. Liu, Polym. Chem., 2014, 5, 3707 RSC.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452 CrossRef CAS PubMed.
- P. R. Sarika and R. J. Nirmala, Mater. Sci. Eng., C, 2016, 65, 331 CrossRef CAS PubMed.
- J. Zhang, J. Li, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2017, 5, 245 RSC.
- F. Palace-Berl, K. F. Pasqualoto, S. D. Jorge, B. Zingales, R. R. Zorzi, M. N. Silva, A. K. Ferreira, R. A. de Azevedo, S. F. Teixeira and L. C. Tavares, Eur. J. Med. Chem., 2015, 96, 330 CrossRef CAS PubMed.
- H. Wang, F. Xu, D. Li, X. Liu, Q. Jin and J. Ji, Polym. Chem., 2013, 4, 2004 RSC.
- T. G. Shutava, S. S. Balkundi, P. Vangala, J. J. Steffan, R. L. Bigelow, J. A. Cardelli, D. P. O'Neal and Y. M. Lvov, ACS Nano, 2009, 3, 1877 CrossRef CAS PubMed.
- Y. Liu, J. Du, M. Yan, M. Y. Lau, J. Hu, H. Han, O. O. Yang, S. Liang, W. Wei, H. Wang, J. Li, X. Zhu, L. Shi, W. Chen, C. Ji and Y. Lu, Nat. Nanotechnol., 2013, 8, 187 CrossRef CAS PubMed.
- M. Yan, M. Liang, J. Wen, Y. Liu, Y. Lu and I. S. Chen, J. Am. Chem. Soc., 2012, 134, 13542 CrossRef CAS PubMed.
- N. Wang, X. Jin, D. Guo, G. Tong and X. Zhu, Biomacromolecules, 2017, 18, 461 CrossRef CAS PubMed.
- G. Liu, Q. Jin, X. Liu, L. Lv, C. Chen and J. Ji, Soft Matter, 2011, 7, 662 RSC.
- J. Li, L. Zhang, Y. Liu, J. Wen, D. Wu, D. Xu, T. Segura, J. Jin, Y. Lu and H. Wang, Chem. Commun., 2016, 52, 13608 RSC.
- S. Karthikeyan, S. L. Hoti and N. R. Prasad, Biomed. Pharmacother., 2015, 70, 274 CrossRef CAS PubMed.
- P. Sun, N. Wang, X. Jin and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 36675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm00788h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Huangyong
Jiang
a,
Huangyong
Jiang