
Differentiating human pluripotent stem cells into vascular smooth muscle cells in three dimensional thermoreversible hydrogels†
Haishuang
Lin
 a,
Qian
Du
b,
Qiang
Li
ac,
Ou
Wang
ac,
Zhanqi
Wang
d,
Kan
Liu
b,
Leonard
Akert
a,
Chi
Zhang
b,
Soonkyu
Chung
e,
Bin
Duan
f and
Yuguo
Lei
*abfg
a,
Qian
Du
b,
Qiang
Li
ac,
Ou
Wang
ac,
Zhanqi
Wang
d,
Kan
Liu
b,
Leonard
Akert
a,
Chi
Zhang
b,
Soonkyu
Chung
e,
Bin
Duan
f and
Yuguo
Lei
*abfg
aDepartment of Chemical and Biomolecular Engineering, University of Nebraska-Lincoln, Nebraska, USA. E-mail: ylei14@unl.edu; Fax: +1 402-472-6989; Tel: +1 402-472-5313
bDepartment of Biological Systems Engineering, University of Nebraska-Lincoln, Nebraska, USA
cBiomedical Engineering Program, University of Nebraska-Lincoln, Nebraska, USA
dDepartment of Vascular Surgery, Beijing Anzhen Hospital of Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Diseases, Beijing, China
eDepartment of Nutrition and Health Sciences, University of Nebraska-Lincoln, Lincoln, NE, USA
fMary and Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, Nebraska, USA
gFred & Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, Nebraska, USA
First published on 22nd November 2018
Abstract
Vascular smooth muscle cells (VSMCs) are of great value and are needed in large quantities for tissue engineering, drug screening, disease modeling and cell-based therapies. However, getting high quantity VSMCs remains a challenge. Here, we report a method for the scalable manufacturing of VSMCs from human pluripotent stem cells (hPSCs). hPSCs are expanded and differentiated into VSMCs in a three dimensional (3D) thermoreversible hydrogel. The hydrogel not only acts as a 3D scaffold for cells to grow, but also protects cells from hydrodynamic stresses in the culture vessel and prevents cells from excessive aggregation. Together, the hydrogel creates a cell-friendly microenvironment, leading to high culture efficiency. We show that VSMCs can be generated in 10 days with high viability (>90%), high purity (>80%) and high yield (∼2.0 × 107 cells per mL hydrogel) in the hydrogel scaffold. The generated VSMCs have normal functions. Genome-wide gene expression analysis shows VSMCs made in the hydrogel (i.e. 3D-VSMCs) have higher expression of genes related to vasculature development and glycolysis compared to VSMCs made in the conventional 2D cultures (i.e. 2D-VSMCs), while 2D-VSMCs have higher expression of genes related to cell proliferation. This simple, defined and efficient method is scalable for manufacturing hPSC-VSMCs for various biomedical applications.
Introduction
Vascular smooth muscle cells (VSMCs) are major components of blood vessels.1 They play important roles in the blood vessel development, function and maintenance.2 Dysfunction of VSMCs leads to various diseases.3,4 VSMCs are thus of great value and needed in large numbers for drug discovery,5 disease modeling,6 tissue engineering7,8 and cell therapies.9–11 However, it is very difficult to obtain sufficient quantities of primary VSMCs from fetal or adult human tissues.1,12,13 Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs)14 and induced pluripotent stem cells (iPSCs),15,16 provide a potential solution to this challenge.17 hPSCs have unlimited proliferation capability and can be expanded in vitro to generate large numbers of cells. They have the potential to differentiate into all somatic cell types.18,19 Additionally, cells derived from patient-specific iPSCs have the patient's genetic information and can faithfully model many human diseases.16,20 They induce no or minimal immune response in the patient.21 During the past decade, protocols for efficiently differentiating hPSCs into VSMCs have been developed. For instance, Sinha and colleagues have generated origin-specific VSMC subtypes from hPSCs such as neuroectodermal VSMCs, lateral plate mesodermal VSMCs and paraxial mesodermal VSMCs.22–24 Gerecht and colleagues have developed protocols for making both synthetic and contractile VSMCs from hPSCs.25–29 VSMCs have two typical phenotypes.30 VSMCs with contractile phenotype are at a quiescent and functional state, which is important for regulating normal vessel tone and blood flow.31 VSMCs with synthetic phenotype are at a proliferative state, and plays a detrimental role in human blood vessels, such as in conditions of atherosclerosis and hypertension.32,33 VSMCs have high plasticity and the two phenotypes can be switched under certain conditions.34 Recently, Patsch and colleagues reported an efficient protocol that could differentiate hPSCs into VSMCs in 6 days in 2D culturing.35 This protocol is simple and quick, thus is very appealing for making VSMCs for various biomedical application, especially for clinical applications.Although hPSCs derived VSMCs (i.e. hPSC-VSMCs) can be readily made at small scales in laboratories for basic research, producing hPSC-VSMCs at large scales for various biomedical applications is still a challenge. The current two dimensional (2D) cell culturing methods (e.g. culturing cells on 2D surfaces such as cell culturing flasks) have low cell yield and are considered only suitable for preparing small numbers of cells.36,37 3D suspension culturing methods (e.g. culturing cells in agitated medium in stirred-tank bioreactors) have been widely studied to scale up the cell production with encouraging results. However, they also have limitations.19,36–41 hPSCs, which have strong cell-to-cell and cell-to-matrix adhesions, frequently aggregate to form large cellular agglomerates in 3D suspension culturing.36 It is well known that the diffusion limit in human tissue is about 400 μm, and the mass transport in cellular agglomerates larger than 400 μm (in diameter) becomes limited. The impaired mass transport results in slow cell growth, significant apoptosis and uncontrolled differentiation.36 Agitating the culture can enhance the medium mixing and mass transport and reduce cell agglomeration. However, agitation generates significant, negative and uncontrollable hydrodynamic stresses (e.g. shear force) that induce large cell death.36,42,43 As a result, low cell viability, growth and volumetric yield are typical in 3D suspension culturing.19 For instance, hPSCs typically expanded about 4-fold per 4 days per passage to yield ∼2.0 × 106 cells per mL. These cells only occupy ∼0.4% of the bioreactor volume.38–41 In summary, producing hPSC-VSMCs in large scales is still a challenge.
To address this challenge, we previously developed a method for expanding hPSCs in 3D thermoreversible hydrogel (Mebiol™ Gel) based on PNIPAAm-PEG polymers.19 The hydrogel scaffold not only provides 3D spaces for cells to grow, but also protects cells from hydrodynamic stresses in the culture vessel and prevents cells from excessive agglomeration. Together, the hydrogel creates a cell-friendly microenvironment that leads to high culture efficiency. We have shown that hPSCs can be cultured for long-term and multiple passages with a high cell viability (e.g. >90%), growth rate (e.g. 20-fold per 5 days), yield (e.g. 2.0 × 107 cells per mL) and purity (>99%), all of which offer large advances over the 3D suspension cultures.19,44,45 In addition to the high efficiency, this method is scalable, defined and cost-effective. It has high potential to address the challenge of producing hPSCs and their derivatives at large scales. With these exciting results, we ask if hPSCs can be efficiently differentiated into VSMCs in the hydrogel scaffold to make this method for the scalable manufacturing of hPSC-VSMCs. In this paper, we show that hPSCs including hESCs and iPSCs can be differentiated into VSMCs with high efficiency (>80%), high viability (>90%) and high yield (2.0 × 107 cells per mL hydrogel). These VSMCs are functional as determined by in vitro and in vivo assays. Genome-wide gene expression analysis show VSMCs made in the hydrogel (i.e. 3D-VSMCs) have higher expression of genes related to vasculature development and glycolysis compared to VSMCs made in the conventional 2D cultures (i.e. 2D-VSMCs), while 2D-VSMCs have higher expression of genes related to cell proliferation. As a result of these studies, we have developed a scalable bioprocess for making high quality VSMCs with high volumetric yield, high viability and high purity.
Experimental
Routine cell culture
H9 hESCs (#WA09, WiCell) were purchased from WiCell Research Institute. Fib-iPSCs (iPSCs reprogrammed from fibroblasts46) were obtained from Human Embryonic Stem cell core, Harvard Medical School. They were maintained in Matrigel coated plates (#354277, BD Biosciences) in Essential 8™ medium (E8, #A1517001, Invitrogen) passaged every 4 days with 0.5 mM EDTA (#AM9260G, Invitrogen).19 Their expression of pluripotency markers, capability to form teratomas and karyotypes, and bacterial or mycoplasma contamination were checked regularly. HUVECs (#00191027, Lonza) were obtained from Lonza.Culturing hPSCs in 3D PNIPAAm-PEG hydrogels
Single hPSCs were mixed with 10% PNIPAAm-PEG solution on ice and cast on tissue culture plate and incubated at 37 °C for 10 minutes to form hydrogels.19 Cells were cultured in E8 medium containing 10 μM ROCK inhibitor (RI, Y-27632, #Y5301, LC Laboratories) with daily medium change. To passage cells, hydrogel was dissolved with ice-cold PBS (5 minutes). Cell spheroids were collected and incubated in Accutase at 37 °C for 10 minutes and dissociated into single cells.VSMC differentiation
For 2D VSMC differentiation, single hPSCs were plated on Matrigel-coated plate at a density of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2. After 24 hours, differentiation was initiated. The differentiation N2B27 medium consists of 1:1 mixture of DMEM/F12 (#SH30004.04, HyClone) and Neurobasal medium (#21103049, Life Technologies) supplemented with N2 (#17502048, Life Technologies) and B27 minus vitamin A (#12587010, Life Technologies). 8 μM CHIR99021 (#C6556, LC laboratories) and 25 ng mL−1 BMP4 (#314BP010, R&D Systems) were added for 3 days. After 3 days, the differentiation medium was replaced by VSMC induction medium consisting of N2B27 medium supplemented with 10 ng mL−1 PDGF-BB (#100-14B, PeproTech) and 2 ng mL−1 ActivinA (#338-AC, R&D Systems). Medium was changed daily. VSMCs were harvested for analysis on day 6. For 3D VSMC differentiation, single hPSCs were expanded for 5 days before differentiation, then followed 2D differentiation protocol. Cells were harvested for analysis on day 10.
000 cells per cm2. After 24 hours, differentiation was initiated. The differentiation N2B27 medium consists of 1:1 mixture of DMEM/F12 (#SH30004.04, HyClone) and Neurobasal medium (#21103049, Life Technologies) supplemented with N2 (#17502048, Life Technologies) and B27 minus vitamin A (#12587010, Life Technologies). 8 μM CHIR99021 (#C6556, LC laboratories) and 25 ng mL−1 BMP4 (#314BP010, R&D Systems) were added for 3 days. After 3 days, the differentiation medium was replaced by VSMC induction medium consisting of N2B27 medium supplemented with 10 ng mL−1 PDGF-BB (#100-14B, PeproTech) and 2 ng mL−1 ActivinA (#338-AC, R&D Systems). Medium was changed daily. VSMCs were harvested for analysis on day 6. For 3D VSMC differentiation, single hPSCs were expanded for 5 days before differentiation, then followed 2D differentiation protocol. Cells were harvested for analysis on day 10.
Suspension culturing in the bioreactor
The prototype bioreactor consists of a pump for medium perfusion, an oxygen-permeable plastic bag for stocking medium and a closed container (e.g. a 50 mL conical tube with a septa cap). On day 0, single hPSCs (∼4.0 × 106 cells) were mixed with 4 mL 10% PNIPAAm-PEG solution at 4 °C, and were injected into room temperature E8 medium in the container. Fibrous hydrogels (with diameter <1.0 mm) were instantly formed. Cells were cultured in E8 medium for 5 days, followed by additional 5 days of VSMC differentiation medium. Medium was continuously perfused. On day 10, hydrogel scaffolds were liquefied by perfusing ice-cold PBS. Spheroids were pelleted by centrifugation. Spheroids were dissociated into single cells through incubating in Accutase at 37 °C for 10 minutes. The bioprocess was repeated 2 times.Immunocytochemistry and flow cytometry
For 2D immunostaining, the 2D cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 20 minutes, permeabilized with 0.25% Triton X-100 for 30 minutes, and blocked with 5% donkey serum for 1 hour before incubating with primary antibodies (Table S1†) at 4 °C overnight. After extensive washing, secondary antibodies (Table S1†) and 10 μM 4′,6-diamidino-2-phenylindole (DAPI) in 2% BSA were added and incubated at room temperature for 4 hours. Cells were washed with PBS for 3 times before imaging with A1 confocal microscope.For 3D immunostaining (spheroids), spheroids were fixed with 4% PFA at room temperature for 30 minutes, and then incubated with PBS + 0.25% Triton X-100 + 5% (vol/vol) goat serum + primary antibodies at 4 °C for 48 hours. After extensive washing, secondary antibodies in 2% BSA were added and incubated at 4 °C for 24 hours. Cells were washed with PBS for three times before imaging with a confocal microscope.
For flow cytometry analysis, the harvested cells were dissociated into single cells with Accutase, then fixed with 4% PFA at room temperature for 20 minutes. Single cells were stained with primary antibodies (Table S1†) at 4 °C overnight. After 3 times washing with 1% BSA in PBS, secondary antibodies were added and incubated at room temperature for 2 hours. Cells were washed with 1% BSA in PBS and analyzed using Cytek flow cytometry.
RNA extraction, cDNA synthesis and quantitative real-time PCR
Total RNAs for qRT-PCR and RNA sequencing were extracted from undifferentiated hPSCs, day 6 2D-VSMCs and day 10 3D-VSMCs using Trizol (#15596018, Invitrogen), according to the manufacturer's instructions. Reverse transcription is done with the Maxima First Strand cDNA Synthesis Kit (#K1642, Life Technologies). Quantitative real-time PCR was carried out in an Eppendorf MasterCycler RealPlex4 (ThermoFisher Scientific) using the Power SYBR Green PCR Master Mix (#4367659, ThermoFisher), according to the manufacturer's instructions. The data were normalized to the endogenous GAPDH. Primer sequence were listed in Table S2.†Embryoid body (EB) differentiation
hPSCs were suspended in DMEM containing 20% FBS (vol/vol) and 10 μM β-mercaptoethanol in low adhesion plate for 6 days. The cell masses were then transferred into plates coated with 0.1% gelatin and cultured in the same medium for another 6 days, followed by fixation and staining as described above.Teratoma formation in vivo
The animal experiments were carried out following the protocols approved by the University of Nebraska–Lincoln Animal Care and Use Committee. 1.0 × 106 hPSCs were suspended in 25 μL PBS + 25 μL Matrigel (BD Biosciences) and injected subcutaneously at the back of the neck of the NOD-SCID mice (female, age 7 weeks, Charles River Laboratory). Three mice (two teratomas per mouse) were used for teratoma assay for each hPSC line. Teratoma was harvested when its size reached 2 cm. Teratoma was fixed with 4% PFA for 48 hours, dehydrated with 70%, 95% and 100% ethanol sequentially, and de-fated with xylene for 2 hours before embedded in paraffin. 10 μm thick section was cut and stained with hematoxylin and eosin. The structures from all 3 germ layers were identified by trained specialist.Co-culture assay of HUVECs and VSMCs
200 μL of Matrigel was added into each well of a 12-well plate and incubated for 30 minutes at 37 °C to allow forming a thin layer of hydrogel. For the functional tube formation and association assays in vitro, the HUVECs and the hPSC-VSMCs were prestained with DiI (red) and DiO (green) (#V22889, Vybrant multicolor cell-labeling kit, Fisher Scientific), respectively, according to the manufacturer's instructions. Cells were grown in EGM-2 medium supplemented with 50 ng mL−1 VEGF-A. For the experiment, 2.0 × 104 HUVECs per cm2 and 2.0 × 104 hPSC-VSMCs per cm2 were co-cultured for 24 hours in the incubator. Cells were fixed with 2% PFA for 10 minutes and analyzed using Zeiss fluorescence microscope.Fibronectin production assay
After 5 days differentiation, 2D-VSMCs and 3D-VSMCs were seeded in N2B27 containing 10 ng mL−1 PDGF-BB at 40000 cells per cm2 on gelatin-coated wells. After 24 hours, the medium was changed to N2B27 with 10 ng mL−1 PDGF-BB supplemented with DMSO or 2.5 ng mL−1 TGF-β. After 24 hours, cells were washed with PBS, fixed with 4% PFA for 10 minutes at room temperature, and immunofluorescence staining of deposited Fibronectin (Table S1†) was performed.
Contraction assay
2D-VSMCs and 3D-VSMCs were seeded in N2B27 containing 2 ng mL−1 ActivinA and 2 μg mL−1 at 40000 cells per cm2 on Collagen-coated wells. After 48 hours, they were prestained with 2.5 μM Fluo-4 AM (#50018, Biotium) at 37 °C for 1 hour. Contraction was induced by treating the cells with 100 μM carbachol (#2810, Tocris). Contraction images of VSMCs were acquired by Zeiss fluorescence microscope. The fluorescence intensity of intracellular calcium flux and change of cell surface area was assessed by ImageJ software.
Glycolysis
Cell glycolysis were analyzed using the Glycolysis Assay Kit (cat # 600450, Cayman Chemical) according to the manufacturer's instruction. Briefly, on day 5 of the differentiation, 10 μL cell culture medium was collected and mixed with 90 μL assay buffer. Then, 100 μL reaction solution was added to each well, and the plate was incubated with gentle shaking for 30 minutes at room temperature. A plate reader was used to read the absorbance at 490 nm.Matrigel plug assay
Animal procedures were performed in accordance with an IACUC-approved protocol reviewed by the University of Nebraska–Lincoln Animal Care and Use Committee. The female 6–8 weeks old SCID mice (Charles River Laboratory) were used. HUVECs and hPSC-VSMCs were added to the Matrigel mixture to a final concentration of 10 million cells per mL. The Matrigel mixture (300 μL) was then immediately engrafted subcutaneously into the dorsal flank of the mouse. Two implants were engrafted per animal. Implants were recovered after 14 days, then the implants were excised. They were fixed in 4% PFA. Hematoxylin & eosin staining, and immunostaining was performed to analyze the tube formation potential in vivo. Three mice were used for each experiment group.RNA sequencing and data analysis
Total RNA of day 6 VSMCs cultured in 2D and day 10 VSMCs cultured in 3D PEG hydrogel, were prepared with RNeasy mini kit (cat # 74104 QIAGEN) according to the manufacturer's instruction. Libraries were prepared with TruSeq Stranded mRNA Library Prep Kit and sequenced with Illumina NextSeq 500. 20 million 75 bp paired-end reads were generated for each sample. The reads were trimmed to make sure the average quality score was larger than 30 and the minimum length was 40 bp. All trimmed short reads were mapped to the human Genome (version hg19) using TopHat, allowing up to two base mismatches per read. Reads mapped to multiple locations were discarded. Numbers of reads in genes were counted by the HTSeq-count tool using corresponding human gene annotations and the “union” resolution mode. For normalization and pair-wise comparisons, differentially expressed genes were identified by using DEseq to analyze the numbers of reads aligned to genes. The thresholds for differential expression were set at fold-change >2 and adjusted P-values <0.001 for the null hypothesis.PCA analysis
Principle Component Analysis (PCA) was implemented with R function prcomp and the 2D PCA plot was produced with R package rgl. For PCA analysis, genes with zero reads were discarded and a natural log transformation was applied to the number of reads for each gene. The proportion of total variance suggested that those two PCs (PC1 48.7% and PC2 25.4%) are sufficient to describe the major changing pattern of all samples.Heatmap for gene expression
For each pathway, the expression values of genes in the given pathway were extracted and, to further stabilize the variance, a natural log transformation was applied to normalized read numbers for genes. The heatmap of expression profiles for genes in a given pathway was plotted with R package heatmap. In the heatmap, dark red indicates high expression while blue represents low expression.Statistical analysis
The data are presented as the mean ± standard deviation (SD) from three biological replicates. We used an unpaired t-test to compare two groups and one-way ANOVA to compare more than two groups. All data were processed using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA).Data availability
The final processed data and raw fastq files were submitted to Gene Expression Omnibus (GEO) with the accession number GSE99776 and GSE109683. All other data supporting the findings of this study are available within the paper and its ESI.†Results
Starting materials
We use both hESCs and iPSCs reprogramed from human dermal fibroblast (Fib-iPSCs)46 for this paper. They have the typical pluripotent stem cell morphologies when cultured on Matrigel-coated plates (Fig. S1a and e†). Both express high levels of the pluripotency markers such as OCT4, NANOG, alkaline phosphatase (ALP) and SSEA-4 (Fig. S1b and f†). They differentiate into all the three germ layer cells such as the NESTIN+ ectodermal, α-SMA+ mesodermal and HNF-3β+ endodermal cells in the embryoid body assay (Fig. S1c and g†). After injecting into the immunodeficient mice, both result in teratomas containing all the three germ layer tissues (Fig. S1d and h†). They have normal karyotypes,19 and they can be efficiently differentiated into high purity somatic cell types such as neural stem cells,47 neurons,47 endothelial cells48 and cardiomyocytes19 using directed differentiation. These results show our starting cells are pluripotent stem cells.Culturing cells in 3D thermoreversible PNIPAAm-PEG hydrogels
The 10% PNIPAAm-PEG polymer solution is liquid at low temperature (e.g. at 4 °C) and forms elastic hydrogel at high temperature (e.g. above 22 °C) (Mebiol™ Gel). The polymer is synthetic, defined and non-toxic to cells.19,38,44,45,47,49 Single cells can be mixed with the solution on ice, heated to form hydrogels and culture the cells at 37 °C in the hydrogel. To harvest cells, the hydrogel can be liquefied by adding ice cold PBS buffer. This method is simple and scalable and has no harm to cells. Single hPSCs in the hydrogel scaffold expand clonally into uniform spheroids in 5 days (Fig. S2a and d†) with minimal cell death (Fig. S2c and f†). Cells in the spheroids express high levels of the pluripotency markers such as OCT4, NANOG, alkaline phosphatase (ALP) and SSEA-4 (Fig. S2b and e†). When seeded at 1.0 × 106 cells per mL, cells expand about 20-fold to generate 2.0 × 107 cells per mL of hydrogel on day 5 (Fig. S2g and h†). As shown in our previous publication, hPSCs could be cultured for long-term in this hydrogel without detectable genetic and phenotypic changes.19 After long-term culture, hPSCs retained their pluripotency as shown by their ability to form all three germ layer cells in the embryoid body assay, and to form teratomas containing all the three germ layer tissues in the teratoma assay.19Differentiating hPSCs into VSMCs using 2D culturing
We decide to use the protocol described in Patsch et al.35 to make VSMCs in this study due to its simplicity and high efficiency (Fig. 1a). We successfully repeat this protocol with our hPSCs in 2D culture (Fig. 1a, b and Fig. S3a†). Immunostaining show majority of cells on day 6 express VSMC markers including SM22A and α-SMA (Fig. 1c and Fig. S3b†). Flow cytometry analysis shows ∼84% of the cells are SM22A+/α-SMA+ VSMCs (Fig. 1d and Fig. S3c†). We find ∼10% VE-Cadherin+ endothelial cells (Fig. 1e, g and Fig. S3d, f†), but no OCT4+ or NANOG+ undifferentiated hPSCs (Fig. 1f, g and Fig. S3e, f†). Our results are very close to the original report,35 showing that the differentiation protocol is robust and can be applied to different hPSC lines in different laboratories. We term VSMCs made in 2D culturing as 2D-VSMCs.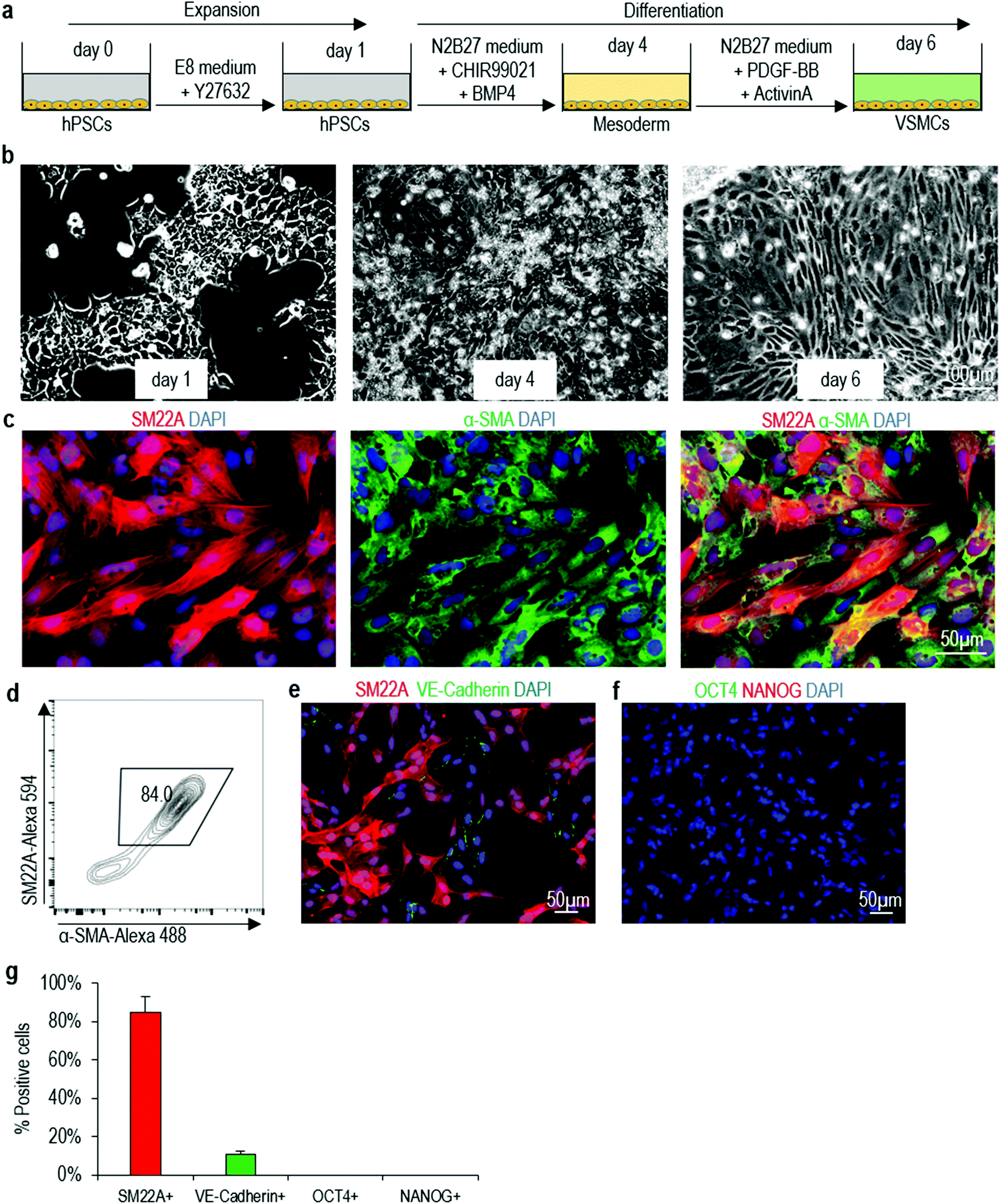 | ||
| Fig. 1 Differentiating of H9 hESCs into VSMCs in 2D cultures. (a) The differentiation protocol. (b) Phase images of day 1, 4 and 6 cells. (c, d) Immunostaining and flow cytometry analysis of VSMCs markers SM22A and α-SMA on day 6 cells. (e–g) The day 6 cells contained ∼10% VE-Cadherin+ endothelial cells (e, g), but no OCT4+/NANOG+ undifferentiated H9 hESCs (f, g). Data are represented as mean ± SD of three biological replicates (n = 3). | ||
Differentiating hPSCs into VSMCs in 3D thermoreversible PNIPAAm-PEG hydrogels
We then study if the protocol can be applied to make VSMCs in the 3D thermoreversible hydrogel (Fig. 2a). Cells in 3D aggregates face different local microenvironment as these in 2D monolayer culture. The microenvironment can have significant influence on hPSCs differentiation. For instance, when differentiating hPSCs into cardiomyocytes, much lower concentration of CHIR99021 (a small molecule agonist for the canonical WNT signaling) was needed in 3D culture50,51 than in 2D culture.52,53 On our own hands, we find many protocols in the literature, which can efficiently differentiate hPSCs into specific somatic cell types in 2D cultures, cannot efficiently differentiate hPSC aggregates in the hydrogel scaffold. For the VSMC differentiation, single hPSCs are encapsulated into the gel and expanded for 5 days to generated hPSC spheroids with diameter around 150 μm (i.e. the expansion phase). On day 5, the differentiation is initiated, and cells are harvested for analysis on day 10 (i.e. the differentiation phase) (Fig. 2b). Live/dead staining detects very few dead cells on day 10 (Fig. 2c and d). Immunostaining and confocal imaging show majority of cells in the day 10 spheroids are positive for VSMC markers SM22A and α-SMA (Fig. 2e). Flow cytometry analysis find about 84.4% of the cells are SM22A+ and α-SMA+ (Fig. 2f). About 1.7 × 107 cells and 2.0 × 107 cells are produced in each milliliter of hydrogel on day 5 and 10, respectively (Fig. 2g), indicating cells do not expand significantly during the differentiation phase. About 20 cells are generated from one input hPSC on day 10. When the day 10 VSMC spheroids are dissociated into single cells and plated on Matrigel-coated plate at high density overnight, they attach to the plate to form a monolayer. Immunostaining and image-based quantification show that majority of the cells are SM22A+/α-SMA+ VSMCs (Fig. 2h), ∼10% of cells are VE-Cadherin+ endothelial cells (Fig. 2i and k), and there are no undifferentiated OCT4+ or NANOG+ H9 hESCs (Fig. 2j and k). The differentiation can be repeated with Fib-iPSCs, generating similar outcomes (Fig. S4†). The differentiation efficiency in 3D hydrogel and 2D culturing are close. We term VSMCs made in 3D hydrogel as 3D-VSMCs.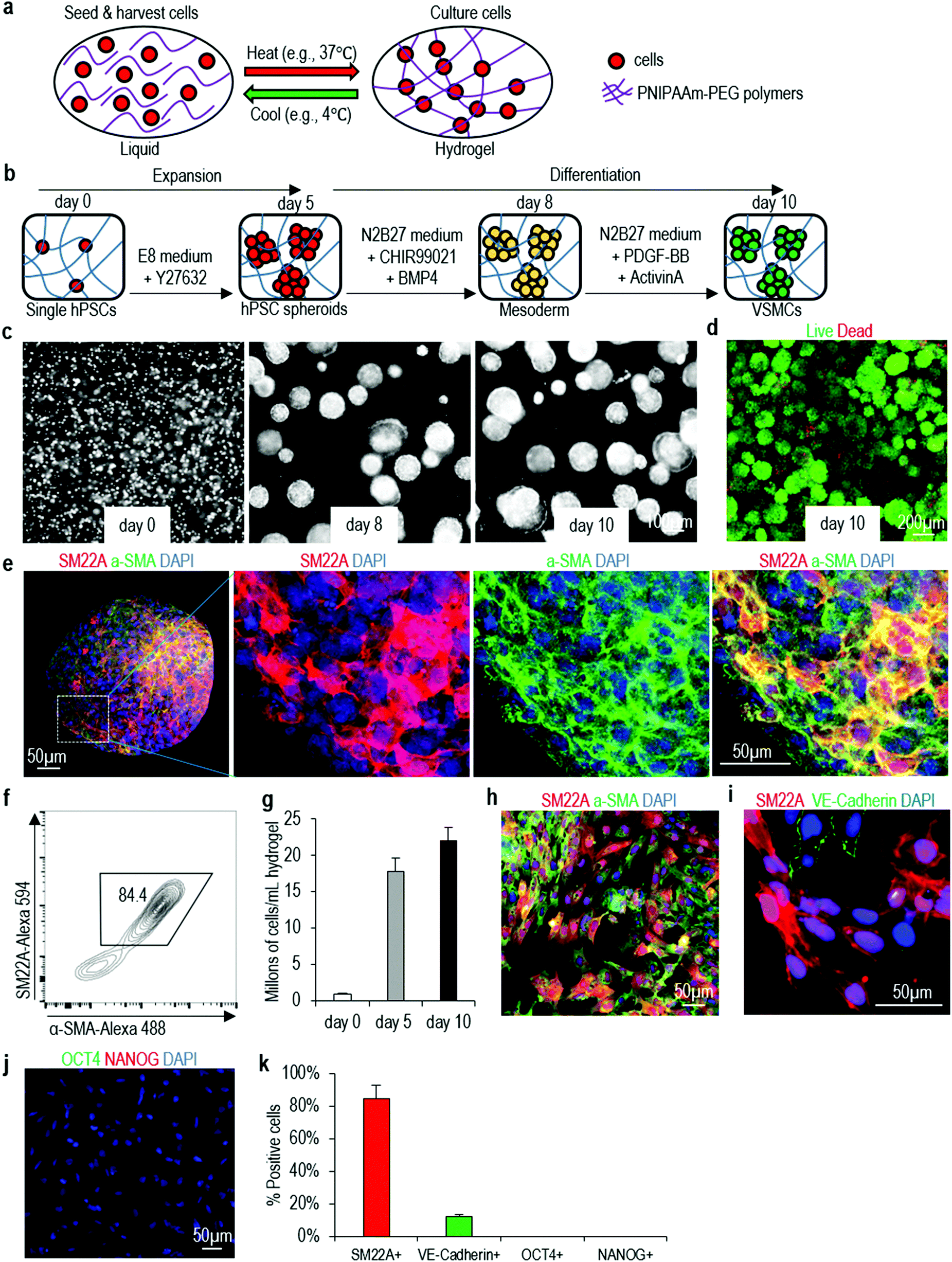 | ||
| Fig. 2 Differentiating H9 hESCs into VSMCs in 3D thermoreversible hydrogel. (a) Illustration of culturing cells in thermoreversible hydrogels. (b) The differentiation protocol. (c) Phase images of day 0, 8 and 10 cells. (d) Live/dead cell staining of harvested day 10 cells. (e, f) Immunostaining and flow cytometry analysis of VSMCs markers SM22A and α-SMA on day 10 spheroids. (g) When seeded at 1 × 106 cells per mL, ∼2 × 107 VSMCs per mL hydrogel were produced on day 10. Data are represented as mean ± SD of three biological replicates (n = 3). (h–k) When the day 10 spheroids were dissociated and plated on 2D surface overnight, immunostaining showed majority cells were SM22A+/α-SMA+ VSMCs (h), and ∼10% cells were VE-Cadherin+ endothelial cells (i, k), but no undifferentiated OCT4+/NANOG+ H9 hESCs (j, k). Data are represented as mean ± SD of three biological replicates (n = 3). | ||
Properties of 3D-VSMCs and 2D-VSMCs
Our culture system provides cells a 3D microenvironment. We ask if 3D-VSMCs and 2D-VSMCs are similar in phenotypes, functions and gene expression. The fibronectin deposition assay show they have similar fibronectin productions in response to TGF-β stimulation (Fig. 3a–c). When co-cultured with human umbilical vein endothelial cells (HUVECs), both VSMCs attach to the tubular network formed by HUVECs (Fig. 3d). 3D-VSMCs contract slightly more in response to carbachol treatment than 2D-VSMCs (Fig. 3e and f), however, calcium imaging reveals that carbachol induces a similar level of intracellular calcium between 3D-VSMCs and 2D-VSMCs (Fig. 3g). These results may be explained by the different sensitivity of the contraction experiment and the calcium imaging experiment.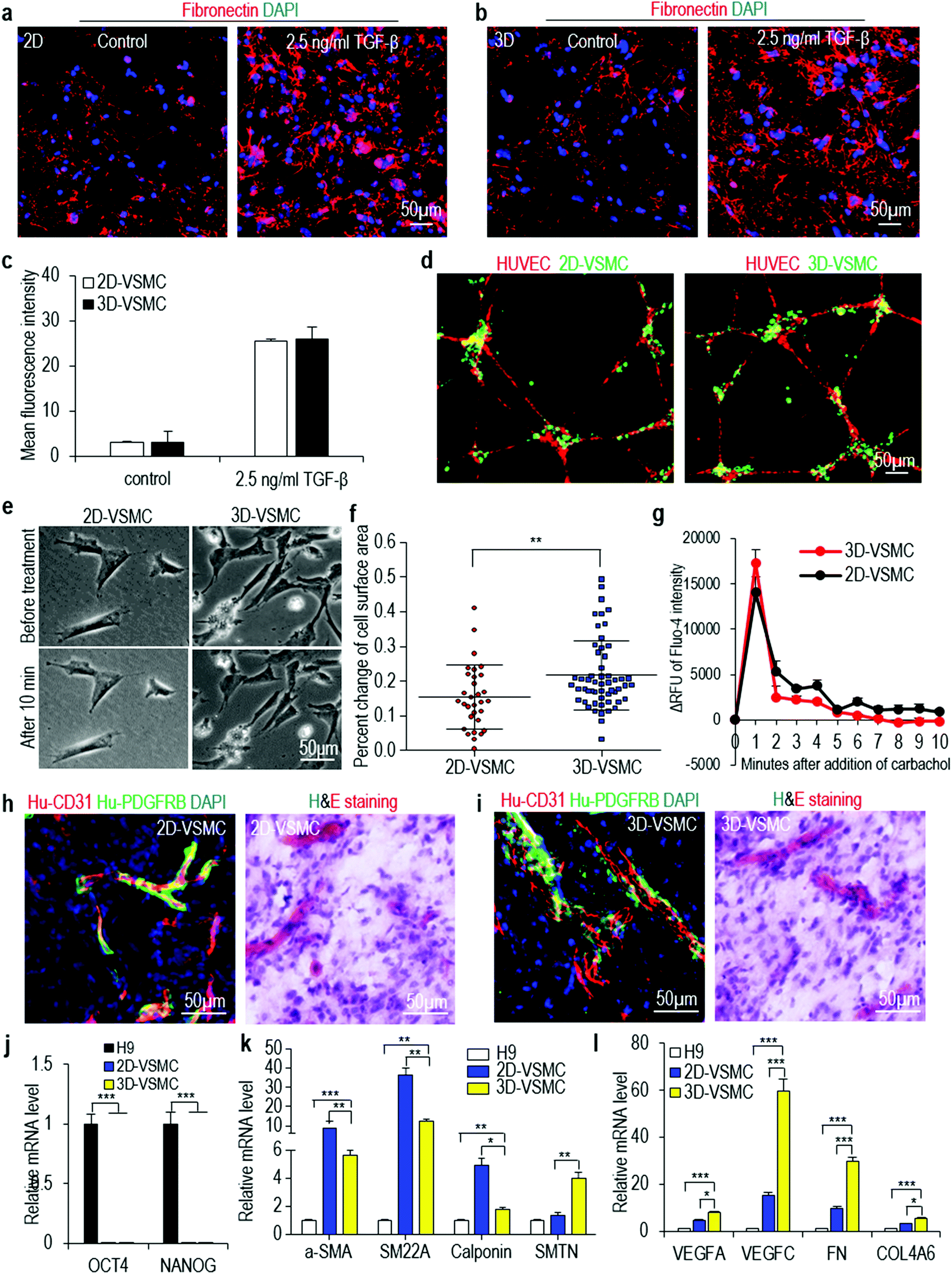 | ||
| Fig. 3 Properties of VSMCs made in 3D hydrogel (3D-VSMCs) and 2D culture (2D-VSMCs). (a–c) Fibronectin production after 24 hours of 2.5 ng mL−1 TGF-β treatment. Data are represented as mean ± SD of three biological replicates (n = 3). (d) Co-culture of VSMCs and HUVECs. (e, f) 2D-VSMCs and 3D-VSMCs contracted in response to carbachol treatment. Data are represented as mean ± SD. **P < 0.01. (g) The relative fluorescence unit (ΔRFU) of Fluo-4 loaded VSMCs over 10 minutes after adding carbachol. Data are represented as mean ± SD of three biological replicates (n = 3). (h, i) When VSMCs and HUVECs were co-transplanted subcutaneously, they formed nice vascular structures. (j–l) qRT-PCR analyses of 2D-VSMCs, 3D-VSMCs and H9 hESCs for pluripotency markers OCT4 and NANOG, synthetic VSMC markers α-SMA, SM22A and Calponin, and contractile VSMC marker SMTN, and other genes related to VSMCs including growth factors VEGFA and VEGFC, and ECM genes FN and COL4A. Data are represented as mean ± SD of three biological replicates (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. | ||
To test if these VSMCs can contribute to the blood vessel formation in vivo, we subcutaneously inject VSMCs and HUVECs with a Matrigel matrix into the immunodeficient mice for 2 weeks. Hematoxylin & eosin staining and immunostaining shows both 3D-VSMCs and 2D-VSMCs attach to the blood vessels in vivo (Fig. 3h and i). We use qRT-PCR to quantitatively analyze the expression of pluripotency markers (OCT4 and NANOG), and a few genes important to VSMCs including their markers (α-SMA, SM22A, Calponin), their secreted growth factors (VEGFA, VEGFB and VEGFC) and their extracellular matrices (ECMs) (FN and COL4A). Both VSMCs have very low expression level of pluripotency markers (Fig. 3j). 2D-VSMCs have higher expression of synthetic VSMC markers α-SMA, SM22A and Calponin (Fig. 3k), and 3D-VSMCs have higher expression of the contractile VSMC marker Smoothelin (SMTN) (Fig. 3k). 3D-VSMCs also have enhanced expression of the growth factors and ECM genes compared to 2D-VSMCs (Fig. 3l). Similar results are found for Fib-iPSCs derived VSMCs (Fig. S5†). These results indicate that 2D-VSMCs are closer to a synthetic phenotype compared to 3D-VSMCs, and 3D-VSMCs upregulate ECM components, a frequently observed phenomenon in 3D differentiation.54
Transcriptome analysis of 3D-VSMCs and 2D-VSMCs
The above qRT-PCR findings drive us to study the genome-wide gene expression difference between 3D-VSMCs and 2D-VSMCs. We sequence the mRNAs of undifferentiated H9s, H9-derived 3D-VSMCs and 2D-VSMCs (3 biological replicates for each). Hierarchical clustering analysis shows 3D-VSMCs and 2D-VSMCs cluster closely and are very different from H9s (Fig. 4a). The genome-wide gene expression profile correlation coefficients between 3D-VSMCs and 2D-VSMCs are >0.79, indicating that they have similar global gene expressions (Fig. 4b and c). However, the separation of 3D-VSMCs and 2D-VSMCs in PC2 of the Principal component analysis (PCA) indicates these cells have some differences in gene expression (Fig. 4d), which drives us to perform differential gene expression analysis.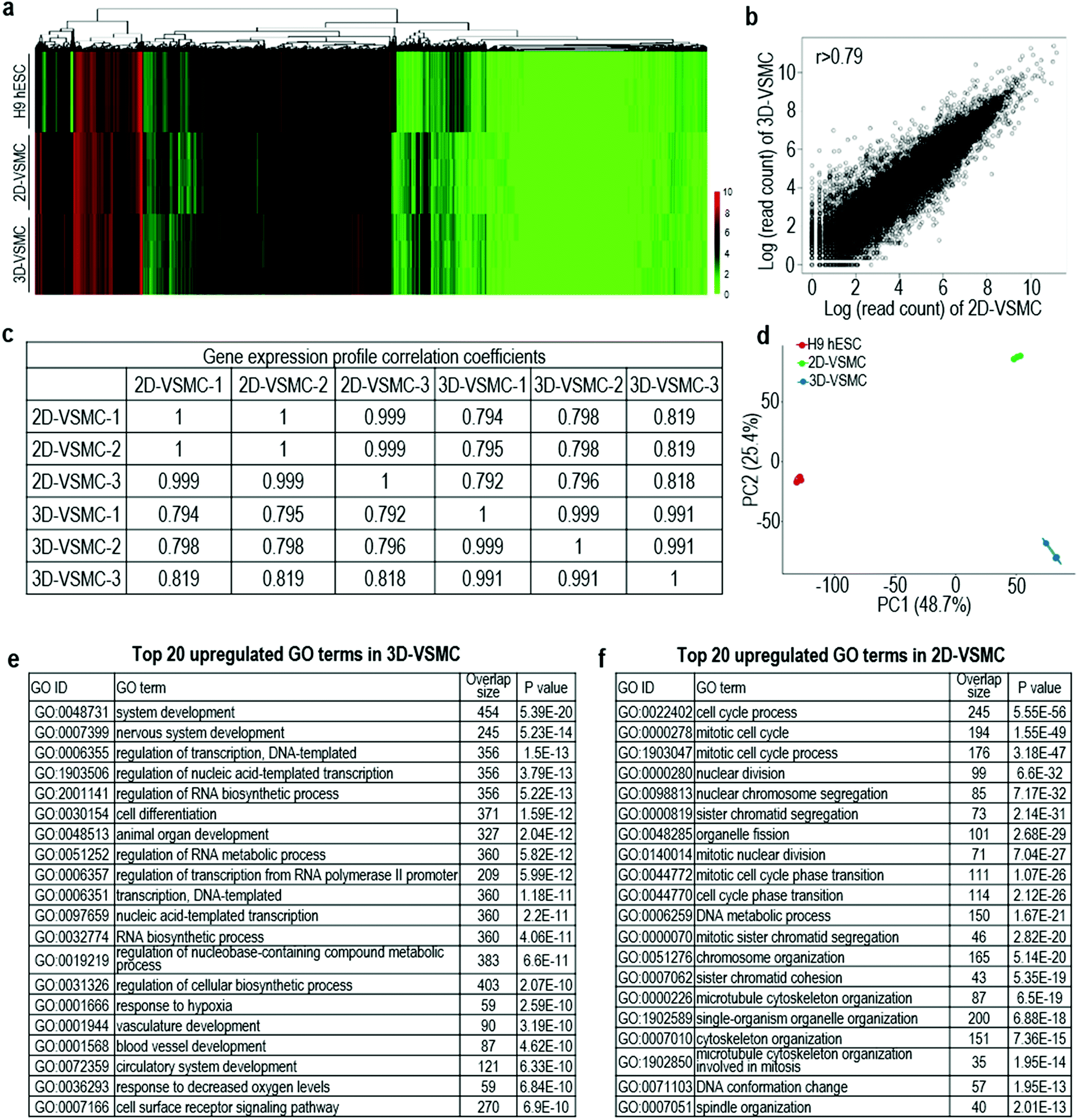 | ||
| Fig. 4 Whole transcriptome analysis 3D-VSMCs and 2D-VSMCs derived from H9s. Global heat map (a) and principal component analysis (PCA) (b) of 3D-VSMCs and 2D-VSMCs. The global gene expression correlation coefficients (c) and scatterplot in log scale (d) of gene expression between 3D-VSMCs and 2D-VSMCs. Three biological replicates are used for each sample. (e, f) Top 20 GO terms of upregulated genes in 3D-VSMCs and 2D-VSMCs. | ||
Differential gene expression analysis identifies 1307 genes upregulated in 3D-VSMCs, and 1267 genes upregulated in 2D-VSMCs. The top GO terms upregulated in 3D-VSMCs are related to vasculature development, transcription and RNA biosynthesis (Fig. 4e). The top GO terms upregulated in 2D-VSMCs are related to cell cycle process (Fig. 4f). Detailed gene expression analysis with the RNA-Seq data shows 3D-VSMCs and 2D-VSMCs express different extracellular matrix components. For instance, 3D-VSMCs have higher expression of COL15A1, COL20A1, COL21A1, COL7A1, COL25A1, COL11A2, COL16A1, COL12A1, and COL22A1 collagens, LAMB3 and LAMC2 laminins, ITGA10, ITGA2, ITGB3, ITGA11, ITGAX, ITGA3, ITGB6 and ITGA6 integrins, and MMP9, MMP1 and MMP16 proteases (Fig. 5a–d). While 2D-VSMCs have higher expression of COL10A1, COL2A1, COL6A6, COL6A5, COL4A1, COL4A2, COL18A1 and COL4A5 collagens, LAMA4 and LAMA1 laminins, and MMP11, MMP19 proteases and TIMP2, TIMP3 protease inhibitors (Fig. 5a–d). 3D-VSMCs and 2D-VSMCs express different secreted factors. For instance, 3D-VSMCs have higher expression of CXCL16, CXCL10, IL8, IL1B, IGF1, MMP9, TNFRSF10C, IGFBP2, CDH5, FGF10, FGF7, CCL7, ANG, VEGFA, CCL4, VEGFB, ENG and CXCL5, while 2D-VSMCs have higher expression of TGFA, IL6, CTGF, ANGPT1, HGF, IL18, FGF1, ANGPT2, NGF, HBEGF, CCL2, PIGF, PDGFA, TIMP2 and IL4 (Fig. 5e). We find 3D-VSMCs express many genes related to vasculature development such as SOX17, EMCN, LMO2, NODAL, FOXC1, ROBO4, CDH5, DLL4, SOX18, ECSCR, NOS2, RUNX1, HEY1, VEGFA, RBPJ, NOTCH4, APOLD1 and MYO1E (Fig. 5f), and have high expression of genes related to glycolysis (Fig. 5g). While 2D-VSMCs express many genes related to cell cycle and proliferation at high level (Fig. 5h). The global gene expression analyses indicate 2D-VSMCs have higher proliferation and 3D-VSMCs have more glycolysis. These results are confirmed using functional proliferation (Fig. 6a and b) and glycolysis assays (Fig. 6c and d). We also show both 3D-VSMCs and 2D-VSMCs can be cultured for multiple passages without losing their markers using the conventional 2D cultures (Fig. 6e).
 | ||
| Fig. 5 Differential gene expression analysis of H9s, and 3D-VSMCs and 2D-VSMCs derived from H9s. (a–h) Heat map and log2 (expression level in 3D-VSMCs/expression level in 2D-VSMCs) analysis of genes related to extracellular matrices (a–d), VSMC secretome (e), vasculature development (f), glycolysis (g), and cell cycle (h). | ||
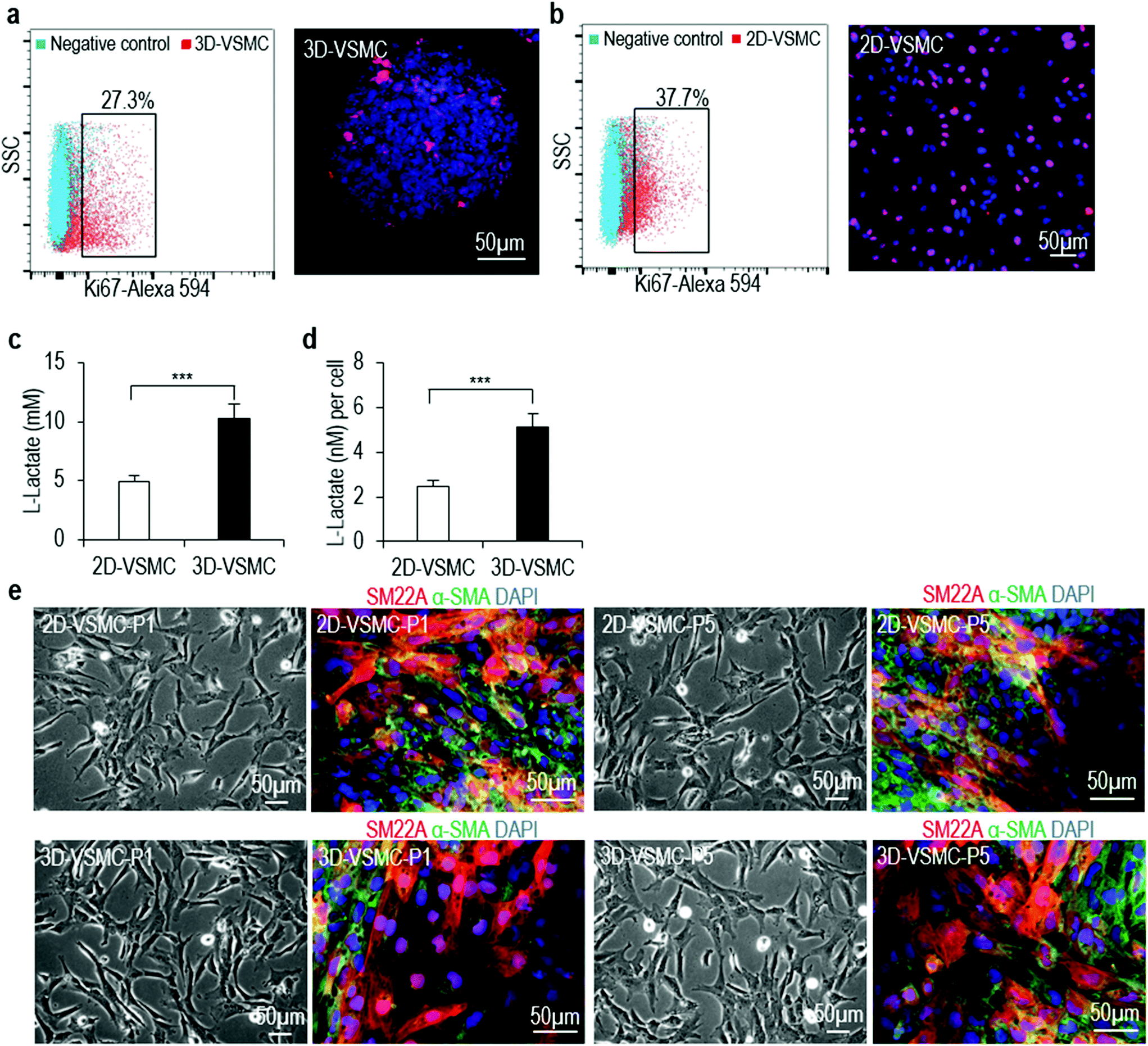 | ||
| Fig. 6 Functional comparison of 3D-VSMCs and 2D-VSMCs. (a, b) Immunostaining and flow cytometry of Ki67 marker showed 2D-VSMCs had more proliferating cells than 3D-VSMCs. (c, d) Glycolysis analysis showed 3D-VSMCs produced more L-lactates than 2D-VSMCs. Data are represented as mean ± SD of three biological replicates (n = 3). ***P < 0.001. (e) Phase and immunostaining images of 2D-VSMCs at passage 1 (P1) and 5 (P5), and 3D-VSMCs at passage 1 (P1) and 5 (P5) after 3D-VSMCs were plated on 2D plates. | ||
Scalable production of hPSC-VSMCs using the hydrogel scaffold in a bioreactor
In the above experiments (Fig. 1–5), we cast the hydrogel on cell culture plate as a thin layer (e.g. less than 500 μm thickness) for culturing cells. Theoretically, this method can be used to produce large numbers of VSMCs through casting gels on a large surface area. However, a better way to utilize the hydrogel to produce large-scale cells is to process them into fibers that can be suspended in the culture medium. We process the PNIPAAm-PEG hydrogel into small fibers (with cells encapsulated in the fibers) that are suspended in the cell culture medium. hPSCs are expanded for 5 days and differentiated for 5 days (Fig. 7a). With 4 mL of hydrogel scaffold contained in a conical tube, we are able to make about 8.0 × 107 VSMCs within in 10 days (Fig. 7b). Live/dead cell staining does not detect significant cell death (Fig. 7c). Immunostaining and flow cytometry analysis show 86.0% of the product are VSMCs (Fig. 7d and e). The produced VSMCs contract and increase their intracellular calcium levels in response to carbachol treatment (Fig. 7f and g). When co-transplanted with HUVECs subcutaneously, they contribute to the new formed blood vessels (Fig. 7h and i). qRT-PCR analyses show 3D-VSMCs made in the bioreactor have similar expression levels of VSMC markers to 3D-VSMCs made in the tissue culture plates (Fig. 7j). This bioreactor can be further scaled up for producing VSMCs in the future.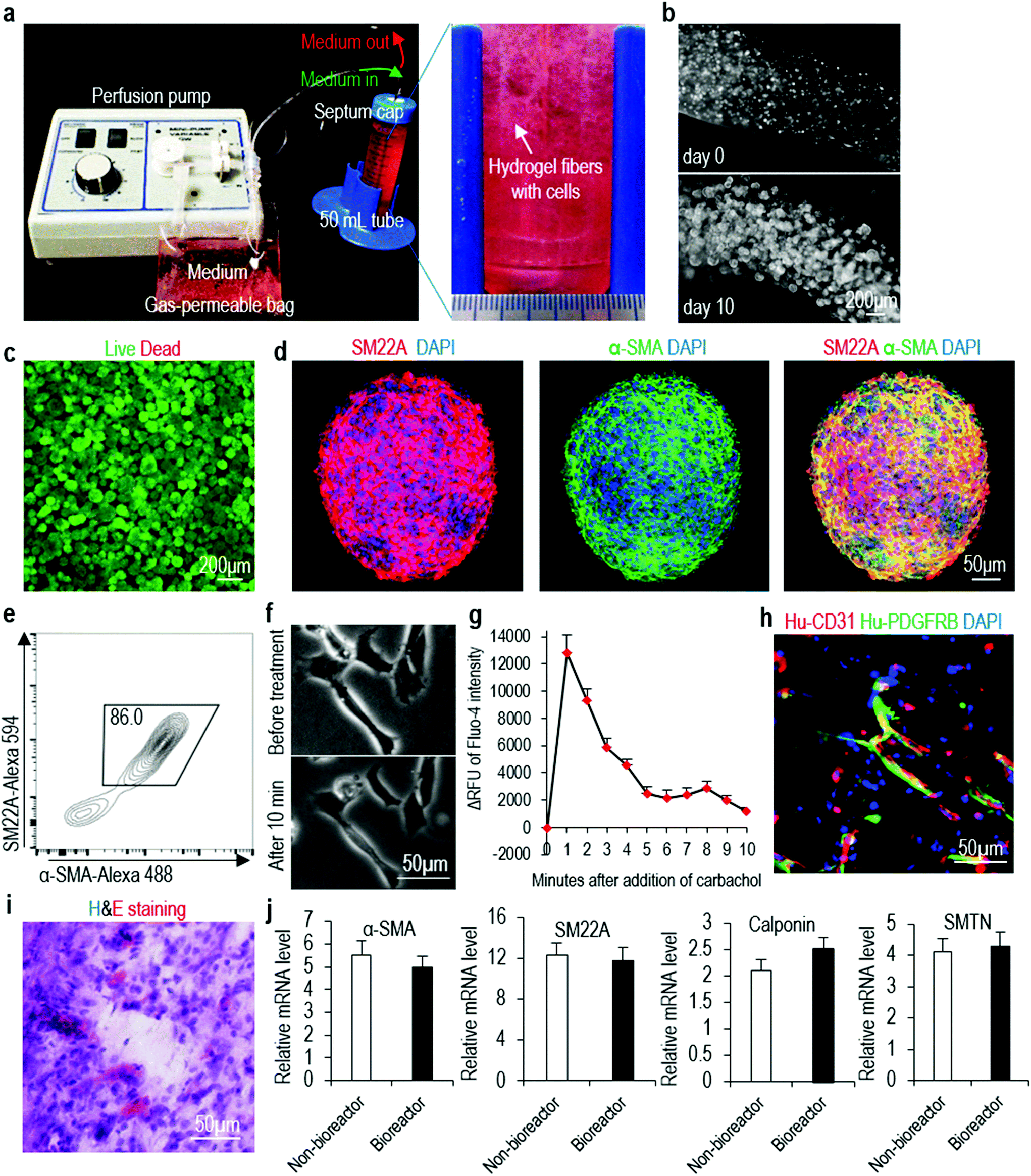 | ||
| Fig. 7 Manufacturing VSMCs in a bioreactor. (a, b) The bioreactor consists of a pump for medium perfusion, an oxygen-permeable plastic bag for stocking medium and a closed container (e.g. a 50 mL conical tube), where hydrogel fibers with cells were suspended. Medium was continuously perfused. (c–e) Live/dead cell staining, immunostaining and flow cytometry analysis of day 10 cells. (f) VSMCs made in the bioreactor contracted in response to carbachol treatment. (g) The relative fluorescence unit (ΔRFU) of Fluo-4 loaded VSMCs made in the bioreactor after adding carbachol. Data are represented as mean ± SD of three biological replicates (n = 3). (h, i) Immunostaining and H&E staining showed they formed nice vascular structures, when co-transplanted with HUVECs. (j) qRT-PCR analyses showed 3D-VSMCs made in the bioreactor had similar expression levels of VSMC markers to 3D-VSMCs made in the tissue culture plates (non-bioreactor). Data are represented as mean ± SD of three biological replicates (n = 3). | ||
Discussion
Manufacturing hPSCs and their derivatives in large scales is still very challenging. The conventional 2D culturing is labor-, time-, space- and reagent-consuming, and not considered suitable for preparing large-scale cells.36 3D suspension culturing has been widely studied to scale up the production.36,42,43 However, cellular agglomeration and hydrodynamic stresses are significant problems.36,42,43 Agglomeration causes transport problems.36,42,43 Agitation (stirring or shaking) generates complicate hydrodynamic conditions including the medium flow direction, velocity, pressure, shear force and chemical environment. These conditions vary spatially and temporally, generating critical stresses near vessel wall and impeller tip that induce significant cell death and phenotype changes.19,36–40,42,43,55 Additionally, the hydrodynamic conditions are sensitive to many factors including the medium viscosity, agitation rate and the bioreactor configuration such as the impeller geometry, size & position, vessel geometry & size, positions of probes for pH, temperature, oxygen.54 How to precisely control these conditions and how they influence the behaviors of different cell types are still not well-understood.42,43,54 These knowledge gaps and technology limitations lead to large culture variations between batches as well as difficulty in scaling up using 3D suspension culturing.50,51 To our best knowledge, the largest 3D suspension culture volume that has been demonstrated for hPSCs is <10 liters.56Using the thermoreversible hydrogel scaffold for expanding and differentiating hPSCs can overcome the limitations of both 2D culturing and 3D suspension culturing. The hydrogel provides 3D spaces for cells to grow. The scaffold also isolates hydrodynamic stresses from the cells. Additionally, the scaffold only allows clonal expansion without merging of the neighboring spheroids, addressing the cellular aggregation problem. Eliminating these negative factors lead to significantly enhanced culture efficiency. VSMCs can be produced with high viability, high purity (>80%) and high yield (∼2.0 × 107 cells per mL hydrogel) within 10 days. Additionally, the synthetic hydrogel is chemically-defined. Due to its thermoreversible nature, seeding and harvesting cells are simple. The system can be readily adapted to multiple scales-from the laboratory and towards the clinic-to support research in cell therapies, tissue engineering, and high throughput drug discovery with hPSC-VSMCs. For instance, ∼50 mL hydrogel would be sufficient to produce ∼109 VSMCs for preclinical animal studies, and a bioreactor with ∼5 L of hydrogel could yield ∼1011 VSMCs for clinical studies. The ability to move a single culture system through multiple scales may aid clinical development.
Our research show the differentiation efficiency in 2D culture and 3D hydrogel are similar (Fig. 1 and 2). Phenotype assays and genome-wide gene expression analysis show 3D-VSMCs and 2D-VSMCs are similar. Both of them have the typical VSMC phenotypes (Fig. 3). Both contribute to blood vessel formation in vitro and in vivo (Fig. 3d, h and i). However, they do have some differences in the global gene expression. 3D-VSMCs have higher expression of genes related to vasculature development and extracellular matrix components (Fig. 4e and 5a–d). These results agree with recent research on organoids, which shows a 3D microenvironment is required for tissue formation,57–59 as well as our own research showing directed differentiation of hPSCs in 3D culture promotes the expression of ECM components and tissue morphogenesis.54,60 Thus, our method provides a valuable tool for studying VSMC development in 3D. Since VSMCs in our culture system are made in a 3D environment, which is more physiologically relevant, we hypothesize our cells will have better functions in vivo. This hypothesis should be tested in the future using sensitive assays and disease models.
Our results show 2D-VSMCs are closer to synthetic phenotype. For instance, they have higher proliferation (Fig. 5h, 6a and b) and higher expression of synthetic VSMC genes (Fig. 3k). The stiff substrate of 2D culturing induces cell proliferation is a frequently observed phenomenon.61–63 A recent research shows stiff substrate switches the contractile VSMCs to synthetic VSMCs with more proliferation and migration both in vitro and in vivo.64 Our findings agree well with their results. From this point of view, our method provides a valuable tool for researchers for preparing VSMCs without having the stiff substrate-induced phenotypes. This may lead to novel scientific discovery.
Conclusion
In summary, we have develop a scalable method for manufacturing VSMCs from hPSCs with high viability, high purity (>80%) and high yield (∼2.0 × 107 cells per mL of hydrogel). The method will make VSMCs broadly available and affordable for various applications.Author contributions
Y.L. and H.L. conceived the idea, designed the experiments and wrote the manuscript. L.A. built the bioreactor. H.L., Q.L., O.W. performed experiments. H.L., Q.D., K. L. and C.Z. analyzed the data. S.C., Z.W. and B.D. revised the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially funded by the UNL-UNMC Science Engineering and Medicine (SEM) initiative grant. Confocal microscope imaging was done in the Morrison Microscopy Core Research Facility at University of Nebraska, Lincoln. Drs You Zhou assisted the confocal imaging. Flow cytometry was done in the Morrison center, the Flow Cytometry core, University of Nebraska, Lincoln with the assistance of Dirk Anderson. RNA-sequencing was done at the UNMC deep sequencing core.References
- G. K. Owens, M. S. Kumar and B. R. Wamhoff, Physiol. Rev., 2004, 84, 767–801 CrossRef CAS PubMed.
- P. Carmeliet, Nat. Med., 2000, 6, 389–395 CrossRef CAS PubMed.
- M. Marchand, E. K. Anderson, S. M. Phadnis, M. T. Longaker, J. P. Cooke, B. Chen and R. A. Reijo Pera, Stem Cells Transl. Med., 2014, 3, 91–97 CrossRef CAS PubMed.
- K. E. Porter and K. Riches, Clin. Sci., 2013, 125, 167–182 CrossRef CAS PubMed.
- N. M. Biel, K. E. Santostefano, B. B. DiVita, N. El Rouby, S. D. Carrasquilla, C. Simmons, M. Nakanishi, R. M. Cooper-DeHoff, J. A. Johnson and N. Terada, Stem Cells Transl. Med., 2015, 4, 1380–1390 CrossRef PubMed.
- H. Ji, H. S. Kim, H.-W. Kim and K. W. Leong, Curr. Opin. Biomed. Eng., 2017, 1, 38–44 CrossRef PubMed.
- L. S. Ferreira, S. Gerecht, H. F. Shieh, N. Watson, M. A. Rupnick, S. M. Dallabrida, G. Vunjak-novakovic and R. Langer, Circ. Res., 2007, 101, 286–294 CrossRef CAS PubMed.
- S. Levenberg, Curr. Opin. Biotechnol., 2005, 16, 516–523 CrossRef CAS PubMed.
- L. Ye, Y. H. Chang, Q. Xiong, P. Zhang, L. Zhang, P. Somasundaram, M. Lepley, C. Swingen, L. Su, J. S. Wendel, J. Guo, A. Jang, D. Rosenbush, L. Greder, J. R. Dutton, J. Zhang, T. J. Kamp, D. S. Kaufman, Y. Ge and J. Zhang, Cell Stem Cell, 2014, 15, 750–761 CrossRef CAS PubMed.
- X. Hong, A. Margariti, A. Le Bras, L. Jacquet, W. Kong, Y. Hu and Q. Xu, Sci. Rep., 2017, 7, 5590 CrossRef PubMed.
- E. Ruiz, A. Gordillo-Moscoso, E. Padilla, S. Redondo, E. Rodriguez, F. Reguillo, A. M. Briones, C. van Breemen, E. Okon and T. Tejerina, Diabetes, 2006, 55, 1243–1251 CrossRef CAS PubMed.
- C. Cheung, A. S. Bernardo, R. A. Pedersen and S. Sinha, Nat. Protoc., 2014, 9, 929–938 CrossRef CAS PubMed.
- M. Poh, M. Boyer, A. Solan, S. L. Dahl, D. Pedrotty, S. S. Banik, J. A. McKee, R. Y. Klinger, C. M. Counter and L. E. Niklason, Lancet, 2005, 365, 2122–2124 CrossRef.
- J. A. Thomson, J. Itskovitz-eldor, S. S. Shapiro, M. A. Waknitz, J. J. Swiergiel, V. S. Marshall and J. M. Jones, Science, 1998, 282, 1145–1147 CrossRef CAS PubMed.
- J. Yu, M. A. Vodyanik, K. Smuga-otto, J. Antosiewicz-bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS PubMed.
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, Cell, 2007, 131, 861–872 CrossRef CAS PubMed.
- S. Levenberg, J. Zoldan, Y. Basevitch and R. Langer, Blood, 2007, 110, 806–814 CrossRef CAS PubMed.
- G. Chen, D. R. Gulbranson, Z. Hou, J. M. Bolin, V. Ruotti, M. D. Probasco, K. Smuga-Otto, S. E. Howden, N. R. Diol, N. E. Propson, R. Wagner, G. O. Lee, J. Antosiewicz-Bourget, J. M. C. Teng and J. A. Thomson, Nat. Methods, 2011, 8, 424–429 CrossRef CAS PubMed.
- Y. Lei and D. V. Schaffer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E5039–E5048 CrossRef CAS PubMed.
- K. Okita, Y. Matsumura, Y. Sato, A. Okada, A. Morizane, S. Okamoto, H. Hong, M. Nakagawa, K. Tanabe, K. Tezuka, T. Shibata, T. Kunisada, M. Takahashi, J. Takahashi, H. Saji and S. Yamanaka, Nat. Methods, 2011, 8, 409–412 CrossRef CAS PubMed.
- P. A. Lalit, D. J. Hei, A. N. Raval and T. J. Kamp, Circ. Res., 2014, 114, 1328–1345 CrossRef CAS PubMed.
- L. Raphel, A. Talasila, C. Cheung and S. Sinha, PLoS One, 2012, 7, e44052 CrossRef CAS PubMed.
- S. Sinha, M. H. Hoofnagle, P. A. Kingston, M. E. Mccanna and G. K. Owens, Am. J. Physiol.: Cell Physiol., 2004, 287, C1560–C1568 CrossRef CAS PubMed.
- C. Cheung, A. S. Bernardo, M. W. B. Trotter, R. A. Pedersen and S. Sinha, Nat. Biotechnol., 2012, 30, 165–173 CrossRef CAS PubMed.
- E. Vo, D. Hanjaya-Putra, Y. Zha, S. Kusuma and S. Gerecht, Stem Cell Rev. Rep., 2010, 6, 237–247 CrossRef PubMed.
- S. Gerecht, L. S. Ferreira and R. Langer, Methods Mol. Biol., 2010, 584, 333–354 CrossRef CAS PubMed.
- M. Wanjare, N. Agarwal and S. Gerecht, Am. J. Physiol.: Cell Physiol., 2015, 309, C271–C281 CrossRef CAS PubMed.
- J. H. Eoh, N. Shen, J. A. Burke, S. Hinderer, Z. Xia, K. Schenke-Layland and S. Gerecht, Acta Biomater., 2017, 52, 49–59 CrossRef CAS PubMed.
- D. Pei, J. Xu, Q. Zhuang, H.-F. Tse and M. A. Esteban, Adv. Biochem. Eng./Biotechnol., 2010, 123, 127–141 CAS.
- S. Ayoubi, S. P. Sheikh and T. V. Eskildsen, Cardiovasc. Res., 2017, 113, 1282–1293 CrossRef CAS PubMed.
- M. R. Alexander and G. K. Owens, Annu. Rev. Physiol., 2012, 74, 13–40 CrossRef CAS PubMed.
- S. F. Louis and P. Zahradka, Exp. Clin. Cardiol., 2010, 15, e75–e85 CAS.
- P. Lacolley, V. Regnault, A. Nicoletti, Z. Li and J. B. Michel, Cardiovasc. Res., 2012, 95, 194–204 CrossRef CAS PubMed.
- M. W. Majesky and C. L. Mummery, Nat. Biotechnol., 2012, 30, 152–154 CrossRef CAS PubMed.
- C. Patsch, L. Challet-Meylan, E. C. Thoma, E. Urich, T. Heckel, J. F. O'Sullivan, S. J. Grainger, F. G. Kapp, L. Sun, K. Christensen, Y. Xia, M. H. C. Florido, W. He, W. Pan, M. Prummer, C. R. Warren, R. Jakob-Roetne, U. Certa, R. Jagasia, P.-O. Freskgård, I. Adatto, D. Kling, P. Huang, L. I. Zon, E. L. Chaikof, R. E. Gerszten, M. Graf, R. Iacone and C. a. Cowan, Nat. Cell Biol., 2015, 17, 994–1003 CrossRef CAS PubMed.
- C. Kropp, D. Massai and R. Zweigerdt, Process Biochem., 2017, 59, 244–254 CrossRef CAS.
- M. J. Jenkins and S. S. Farid, Biotechnol. J., 2015, 10, 83–95 CrossRef CAS PubMed.
- Y. Lei, D. Jeong, J. Xiao and D. V. Schaffer, Cell. Mol. Bioeng., 2014, 7, 172–183 CrossRef CAS PubMed.
- D. Steiner, H. Khaner, M. Cohen, S. Even-Ram, Y. Gil, P. Itsykson, T. Turetsky, M. Idelson, E. Aizenman, R. Ram, Y. Berman-Zaken and B. Reubinoff, Nat. Biotechnol., 2010, 28, 361–364 CrossRef CAS PubMed.
- M. Serra, C. Brito, C. Correia and P. M. Alves, Trends Biotechnol., 2012, 30, 350–358 CrossRef CAS PubMed.
- F. M. Wurm, Nat. Biotechnol., 2004, 22, 1393–1398 CrossRef CAS PubMed.
- M. A. Kinney, C. Y. Sargent and T. C. Mcdevitt, Tissue Eng., Part B, 2011, 17, 249–262 CrossRef PubMed.
- K. M. Fridley, M. A. Kinney and T. C. Mcdevitt, Stem Cell Res. Ther., 2012, 3, 45 CrossRef CAS PubMed.
- H. Lin, Q. Li and Y. Lei, Sci. Rep., 2017, 7, 40191 CrossRef CAS PubMed.
- Q. Li, H. Lin, O. Wang, X. Qiu, S. Kidambi, L. P. Deleyrolle, B. A. Reynolds and Y. Lei, Sci. Rep., 2016, 6, 31915 CrossRef CAS PubMed.
- I. H. Park, R. Zhao, J. A. West, A. Yabuuchi, H. Huo, T. A. Ince, P. H. Lerou, M. W. Lensch and G. Q. Daley, Nature, 2008, 451, 141–146 CrossRef CAS PubMed.
- H. Lin, Q. Li and Y. Lei, Biofabrication, 2017, 9, 025007 CrossRef PubMed.
- D. Qi, S. Wu, H. Lin, M. A. Kuss, Y. Lei, A. Krasnoslobodtsev, S. Ahmed, C. Zhang, H. J. Kim, P. Jiang and B. Duan, ACS Appl. Mater. Interfaces, 2018, 10, 21825–21835 CrossRef CAS PubMed.
- Q. Li, Q. Wang, O. Wang, K. Shao, H. Lin and Y. Lei, PLoS One, 2018, 13, e0190364 CrossRef PubMed.
- M. Jara-avaca, H. Kempf, R. Olmer, C. Kropp, M. Ru, D. Robles-diaz, A. Franke, D. A. Elliott, D. Wojciechowski, M. Fischer, A. R. Lara, G. Kensah, I. Gruh, A. Haverich, U. Martin and R. Zweigerdt, Stem Cell Rep., 2014, 3, 1132–1146 CrossRef PubMed.
- V. C. Chen, J. Ye, P. Shukla, G. Hua, D. Chen, Z. Lin, J. Liu, J. Chai, J. Gold, J. Wu, D. Hsu and L. A. Couture, Stem Cell Res., 2015, 15, 365–375 CrossRef CAS PubMed.
- X. Lian, C. Hsiao, G. Wilson, K. Zhu, L. B. Hazeltine, S. M. Azarin, K. K. Raval, J. Zhang, T. J. Kamp and S. P. Palecek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1848–E1857 CrossRef CAS PubMed.
- M. Z. Chow, K. R. Boheler and R. A. Li, Stem Cell Res. Ther., 2013, 4, 97 CrossRef PubMed.
- H. Lin, Q. Du, Q. Li, O. Wang, Z. Wang, N. Sahu, C. Elowsky, K. Liu, C. Zhang, S. Chung, B. Duan and Y. Lei, Stem Cell Rep., 2018, 11, 454–469 CrossRef CAS PubMed.
- M. Ismadi, P. Gupta, A. Fouras, P. Verma and S. Jadhav, PLoS One, 2014, 9, e106493 CrossRef PubMed.
- H. Kempf, B. Andree and R. Zweigerdt, Adv. Drug Delivery Rev., 2016, 96, 18–30 CrossRef CAS PubMed.
- N. Hattori, Mov. Disord., 2014, 29, 185 CrossRef PubMed.
- J. Jo, Y. Xiao, A. X. Sun, E. Cukuroglu, H. D. Tran, J. Göke, Z. Y. Tan, T. Y. Saw, C. P. Tan, H. Lokman, Y. Lee, D. Kim, H. S. Ko, S. O. Kim, J. H. Park, N. J. Cho, T. M. Hyde, J. E. Kleinman, J. H. Shin, D. R. Weinberger, E. K. Tan, H. S. Je and H. H. Ng, Cell Stem Cell, 2016, 19, 248–257 CrossRef CAS PubMed.
- Y. Takahashi, S. Sato, Y. Kurashima, T. Yamamoto, S. Kurokawa, Y. Yuki, N. Takemura, S. Uematsu, C. Lai, M. Otsu, H. Matsuno, H. Osawa, T. Mizushima, J. Nishimura, M. Hayashi and T. Yamaguchi, Stem Cell Rep., 2018, 10, 314–328 CrossRef CAS PubMed.
- H. Lin, Q. Du, Q. Li, O. Wang, Z. Wang, K. Liu, C. Elowsky, C. Zhang and Y. Lei, ACS Appl. Mater. Interfaces, 2018, 10, 29238–29250 CrossRef CAS PubMed.
- J. H. Chamley, G. R. Campbell, J. D. McConnell and U. Gröschel-Stewart, Cell Tissue Res., 1977, 177, 503–522 CAS.
- R. M. Wazen, S. Kuroda, C. Nishio, K. Sellin, J. B. Brunski and A. Nanci, Biomaterials, 2013, 34, 677–684 CrossRef PubMed.
- S. B. H. Timraz, R. Rezgui, S. M. Boularaoui and J. C. M. Teo, Procedia Eng., 2015, 110, 29–36 CrossRef CAS.
- S. Xie, T. Zhang, J. Wang, F. Zhao, Y. Zhang, W. Yao, S. Sik, Y. Yeh, W. Pang, L. Zheng, Y. Fan, W. Kong, X. Wang, J. Chiu and J. Zhou, Biomaterials, 2018, 155, 203–216 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01128a |
| This journal is © The Royal Society of Chemistry 2019 |