
Recombinant human BMP-2 accelerates the migration of bone marrow mesenchymal stem cells via the CDC42/PAK1/LIMK1 pathway in vitro and in vivo
Shuhao
Liu†
 a,
Yang
Liu†
bc,
Libo
Jiang†
a,
Zheng
Li
a,
Soomin
Lee
a,
Changsheng
Liu
bc,
Jing
Wang
*bc and
Jian
Zhang
*a
a,
Yang
Liu†
bc,
Libo
Jiang†
a,
Zheng
Li
a,
Soomin
Lee
a,
Changsheng
Liu
bc,
Jing
Wang
*bc and
Jian
Zhang
*a
aDepartment of Orthopedic Surgery, Zhongshan Hospital, Fudan University, Shanghai, 200030 People's Republic of China. E-mail: zhang.jian@zs-hospital.sh.cn; Tel: +86-21-64041990
bThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, 200237 People's Republic of China. E-mail: biomatwj@163.com; Tel: +86-21-64251308
cEngineering Research Center for Biomedical Materials of Ministry of Education, East China University of Science and Technology, Shanghai, 200237, People's Republic of China
First published on 24th November 2018
Abstract
Biomaterials are widely used for bone regeneration and fracture repair. The migration of bone marrow mesenchymal stem cells (BMSCs) into bone defect sites or material implantation sites, and their differentiation into osteoblasts, is central to the fracture healing process, and the directional migration of BMSCs depends on cytokines or chemokines at the defect site. BMP-2 can stimulate the migration of a variety of cells, but it remains unclear whether BMSC migration can be induced. To provide evidence for BMP-2-induced BMSC migration, we tested the cytoskeletal changes and migration ability of BMSCs after treatment with recombinant human BMP-2 (rhBMP-2). We also explored the recruitment of BMSCs from the circulatory system using a collagen sponge incorporating rhBMP-2 that was implanted in vivo. Furthermore, to understand the mechanism underlying this migration, we investigated the effect of rhBMP-2 on migration-related signal pathways. Here, we found that, rhBMP-2 treatment significantly increased the migration of BMSCs in vitro via activation of the CDC42/PAK1/LIMK1 pathway, and that this migration could be blocked by silencing CDC42. In vivo, collagen sponge material loaded with rhBMP-2 could recruit BMSCs injected into the circulatory system. Moreover, inhibition using the small interfering RNA for CDC42 led to a significant decrease in the number of BMSCs within the material. In conclusion, our data prove that rhBMP-2 can accelerate BMSC migration via the CDC42/PAK1/LIMK1 pathway both in vivo and in vitro, and therefore provides a foundation for further understanding and application of rhBMP-2-incorporated materials by enhancing BMSC recruitment.
Introduction
A bone defect is a serious medical condition caused by a fracture, tumor, or severe bone infection. Given the limitations of traditional methods for treating bone defects, such as autogenous bone grafts, which result in an additional wound, and bone xenografts, which stimulate undesirable immunological responses, the use of biomaterials to fill large bone defects is an attractive and feasible option.1 After implantation, the recruitment of sufficient numbers of bone marrow mesenchymal stem cells (BMSCs) to defect sites is the cytological basis of bone repair and an essential prerequisite for this process.2 However, factors that mediate BMSC recruitment and migration mechanisms by materials are rarely investigated.Early features of cell migration include cell polarization and pseudopodia formation caused by actin polymerization.3,4 Many factors participate in the regulation of signal transduction leading to actin skeleton reorganization during these processes.5 CDC42, a small GTPase of the Rho family, plays a role in cell polarization and migration by regulating cytoskeleton structure and membrane transportation.6 Depending on upstream signaling, CDC42 can exist in either active (GTP-bound) or inactive (GDP-bound) states.7 Activated CDC42 can transmit signals to downstream effectors, such as the p21-activated kinases (PAK), WASP and ACK. PAK activation leads to reorganization of peripheral actin through phosphorylation of LIM kinase 1 (LIMK1).8 Subsequently, activated LIMK1 causes inactivation of cofilin and stabilization of actin filaments.8 During cell migration, both activated PAK and inactivated cofilin are key to changes in the cytoskeleton architecture and their maintenance.9,10
Bone morphogenetic protein 2 (BMP-2) is a highly significant molecule involved in bone formation. BMP signaling pathways function in osteoblast differentiation and the synthesis and secretion of the extracellular matrix.11 In addition, BMP-2 can stimulate the migration of a variety of cells, such as C2C12 cells,12 osteoblasts,13 and tumor cells;14 however, few studies have investigated whether BMSC migration can be induced. Different signals have been shown to act on certain cell types, including SDF-1/CXCR4,15,16 p38/MK2/Hsp25,17 and CDC42-related pathways;12,18,19 however, the mechanism underlying how this induces BMSC migration remains unclear; few studies have investigated the mechanism of cell migration triggered by BMP-2 in vivo.
Collagen is a common biomaterial for drug delivery. The combination of recombinant human BMP-2 (rhBMP-2) with a collagen sponge matrix has proven to be a very promising therapeutic in a variety of applications.11 Previous research has proven that the binding of rhBMP-2 to collagen sponges in vivo was not affected by sponge characteristics such as crosslinking, pH, and collagen mass.20 To better reflect rhBMP-2-induced migration, we used the collagen sponge as a carrier in this study.
Although the migration of cells has been widely studied, along with the mechanisms underlying this process, it remains unknown as to whether BMSC migration can be induced by BMP-2 and if so, which mechanisms are involved. In this study, we explored the molecular mechanism by which BMP-2 promotes the migration of BMSCs in vitro and investigated its effects on migration with in vivo experiments using collagen sponge incorporating rhBMP-2. The CDC42 pathway on BMSC recruitment was specifically investigated.
Materials and methods
Study design
BMSCs were cultured and treated with rhBMP-2 to examine the activation of cell migration and the potential mechanism of action by using small interfering RNA (siRNA) technology, wound-healing assays, transwell migration assays, western blot analysis, and implantation. For implantation, a collagen sponge was chosen as the carrier of rhBMP-2. The characteristics of these materials were tested by scanning electron microscopy (SEM), release study, and live/dead cell viability assay. To examine the migration of BMSCs in vivo, 72 rats were divided into three groups: the control group (n = 24) were implanted with collagen sponges alone and BMSCs injected; the other two groups were planted with rhBMP-2-treated collagen sponges and injected with either BMSCs (n = 24) or BMSCs treated with CDC42 siRNA (n = 24). Eight rats from each group were sacrificed 3, 7, and 14 days after implantation. After paraffin embedding and sectioning, immunofluorescence staining was used to determine cell migration. Twenty 6-week-old nude mice were implanted in the back with materials with (n = 10) or without (n = 10) rhBMP-2, followed by an injection of fluorescently labelled BMSCs into the left ventricle. At different times, the migration of mesenchymal stem cells in vivo was observed by bioluminescence imaging.All specific-pathogen-free-level rats and nude mice were provided with standard water and food in the Lab Animal Center of Zhongshan Hospital, Fudan University. This study was approved by the Animal Ethics Committee of Zhongshan Hospital (No. DF371).
Cell isolation and identification
Four-week-old Sprague–Dawley (SD) rats were euthanized by anesthetic overdose and immersed in 75% ethanol for 5–10 min. Then, they were transferred to a clean bench, the diaphysis of each femur and tibia dissected, and the surrounding soft tissue scraped off. Bone marrow was collected by flushing the medullary cavities using phosphate-buffered saline (PBS) and filtered through a 70 μm filter to remove tissue fragments and fat. After centrifugation for 5 min at 200g, collected cells were resuspended in SD Rat Mesenchymal Stem Cell Medium (Cyagen Biosciences, Guangzhou, China) and cultured at a density of 1 × 109 cells per mL in a humidified incubator at 37 °C with 5% CO2. When they reached 80% confluence, cells were detached from cell culture vessels with 0.25% Trypsin-EDTA (Gibco) for 2 min and sub-cultured. Third (P3) or fourth (P4) passage cells were used for subsequent experiments.The method of cell identification was described in our previous study.21 Briefly, mesenchymal stem cell surface makers were evaluated by flow cytometry. P3 cells (2 × 106 mL−1) were collected and incubated with 5 μL of fluorescein isothiocyanate-coupled monoclonal antibodies against CD34, CD45, CD73, and CD90, as well as phycoerythrin-coupled monoclonal antibodies against CD105 and human leukocyte antigen (HLA)-DR. All antibodies used for identification were purchased from eBioscience, San Diego, USA. Multilineage differentiation and staining assays with P3 cells were conducted following the protocols for each kit, with induction medium for osteogenic, adipogenic, and chondrogenic differentiation (Cyagen Biosciences, Guangzhou, China).
Fluorescent staining with CM-Dil and rhodamine-phalloidin
P3 BMSCs were collected using 0.25% Trypsin-EDTA, washed twice with PBS, then suspended in D-PBS at 1 × 107 cells per mL. Next, cells were incubated with the fluorescent dye, chlormethylbenzamido-1,1-dioctadecyl-3,3,3′3′-tetramethylin-docarbocyamine (CM-Dil, Invitrogen, Ltd, Eugene, USA) at a final concentration of 5 μM. After 5 min incubation at 37 °C, followed by 15 min at 4 °C, labeled cells were collected by centrifugation, washed, and cultured in fresh medium. When they reached 75% confluence, they were observed under a fluorescence microscope and the labeling rate calculated.For rhodamine-phalloidin labeling (Yeasen, Shanghai, China), P4 BMSCs were seeded in 15 mm glass-bottomed cell culture dishes (NEST Biotechnology, Wuxi, China), and cultured to 50% confluence. Cells were fixed with 4% formaldehyde in PBS for 10 min, washed twice, and 0.5% Triton X-100 (Beyotime, Hangzhou, China) used for cell permeabilization (5 min). Cells were then washed three times for 10 min each and covered with rhodamine-phalloidin dye (working concentration, 100 nM). After 30 min of incubation in the dark at room temperature, cells were washed again and covered with 100 nM 4′,6-diamidino-2-phenylindole (DAPI; Beyotime, Hangzhou, China) for 1 min. Photomicrographs of cytoskeletons were captured under a laser scanning confocal microscope (Leica TCS SP8) at 400× magnification.
Transfection of CDC42 siRNA
siRNA targeting the rat CDC42 gene (5′-AAAGACUCCUUUCUUGCUUGU-3′) and control gene siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were provided by Sangon Biotech (Shanghai, China). P3 BMSCs were seeded in six-well plates in 2 mL per well in growth medium without antibiotics and cultured to 70% confluence. Before transfection, 5 μL of lipofectamine 2000 (Invitrogen, Ltd, USA) and 1 nmol siRNA were each separately added to 250 μL Opti-MEM (Invitrogen, Ltd, USA), mixed and incubated for 15 min at room temperature. Then, the two solutions were combined and mixed for another 15 min to allow complexes to form. Complexes were then added to cells in each well and the plates were gently agitated. After 16 h at 37 °C in a humidified incubator, total proteins were extracted from cells and gene knockdown evaluated by western blotting (see below).Cell proliferation assay
To investigate the influence of rhBMP-2 on the proliferation ability of BMSCs, 2 × 103 P4 cells were cultured in 96-well plate for 24 h. Then, Cell Counting Kit-8 solution (CCK8; Dojindo, Tokyo, Japan) was added at different times after incubation with or without rhBMP-2. After incubation for another 2 h, the optical density (OD) values at 450 nm were measured with a micro-plate reader (Bio-Rad, Hercules, USA).Wound-healing assays and transwell migration assays
Wound-healing assays were performed as described previously.21 Briefly, P4 BMSCs were cultured to 90% confluence in six-well plates and a p200 pipette tip used to scratch a clear area in the cell monolayer. After washing with PBS, photomicrographs of the same area were taken before and after incubation in serum-free medium for 24 h. The migration area was measured and calculated using Image J software. Quantitative analysis of the invaded area was then performed using four photographed fields.For transwell migration assays, the upper chambers of transwell plates were seeded with 2 × 104 P4 cells in 200 μL serum-free medium per well and the lower chambers filled with 600 μL medium containing 0.5% fetal bovine serum (FBS), with or without 100 ng mL−1 rhBMP-2. After incubation in a humidified incubator at 37 °C, 5% CO2 for 24 h, migrated cells on the underside of membranes were fixed and stained with 0.1% crystal violet. After washing with distilled water, four pictures of each chamber were randomly taken in a 200× microscope field and the total number of migrating BMSCs calculated.
Western blotting
P4 BMSCs were detached by scraping, washed, and lysed on ice. Samples (20 μg of protein each) were then resolved by electrophoresis and transferred to polyvinylidene fluoride membranes. After blocking, membranes were incubated overnight at 4 °C with GAPDH, CDC42, p-LMIK1/LIMK1, p-PAK1/PAK1, and p-cofilin/cofilin primary antibodies followed by incubation with a horseradish peroxidase-conjugated secondary antibody for 1 h. All these antibodies were purchased from Cell Signaling Technology, Danvers, USA. Signals were visualized using Chemiluminescence HRP Substrate (Millipore, Billerica, USA) and a Luminescent Imaging Workstation (Tanon, Shanghai, China).GST pull-down assay
Cdc42 activation was tested with a Cdc42 Pull-down Activation Assay Biochem Kit from Cytoskeleton, Denver, USA. P4 BMSCs were seeded and expanded to 90% confluence in Petri dishes. After 4 h of starvation treatment, the cells were pretreated with 100 ng mL−1 rhBMP-2 for 0, 5, 15, 30, 60, and 120 min prior to collection on ice with cell lysis buffer and protease inhibitor cocktail. After centrifugation for 1 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, supernatants were collected and 20 μL reserved for protein quantitation by BCA assay and total CDC42 protein quantitation. After adjustment to 0.5 mg mL−1, protein samples were added to centrifuge tubes containing 10 μg of PAK-PBD beads. The tubes were shaken at 4 °C for 1 h, centrifuged for 1 min to collect the beads, and supernatants discarded. Next, 2× Laemmli sample buffer was added to the beads, which were boiled for 2 min, and used for subsequent western blot experiments. Cell lysates treated with GTPγs and GDP were used as positive and negative controls, respectively. His-CDC42 protein was also used as a control.
000g, supernatants were collected and 20 μL reserved for protein quantitation by BCA assay and total CDC42 protein quantitation. After adjustment to 0.5 mg mL−1, protein samples were added to centrifuge tubes containing 10 μg of PAK-PBD beads. The tubes were shaken at 4 °C for 1 h, centrifuged for 1 min to collect the beads, and supernatants discarded. Next, 2× Laemmli sample buffer was added to the beads, which were boiled for 2 min, and used for subsequent western blot experiments. Cell lysates treated with GTPγs and GDP were used as positive and negative controls, respectively. His-CDC42 protein was also used as a control.
Material preparation and SEM
RhBMP-2 was provided by Shanghai Rebone Co., Ltd (Shanghai, China). Materials were produced by the State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, China. Collagen sponges (0.5 × 0.5 × 0.5 cm3) were loaded with 2 μg rhBMP-2, which comprised the experimental group, while untreated collagen sponges were used as controls. BMSCs were added to both groups to evaluate the contacts between the collagen sponge material and BMSCs in vitro by SEM.Materials were placed in suspensions of P4 BMSCs (1 × 106 cells per mL in medium with 10% FBS) and incubated for 24 h. As the experimental group, BMSCs were treated with 100 ng mL−1 rhBMP-2 for 1 or 24 h before collection. After fixation, materials were examined by SEM and images captured.
Release study and live/dead cell viability assay
To evaluate the release behavior of the collagen sponge material, 500 μg of rhBMP-2 were loaded onto collagen sponges (0.5 × 0.5 × 0.5 cm3). Then, materials were dispersed into 1 mL PBS in a 24-well plate, and shocked on an orbital shaker (200 rpm) at 37 °C. At 1, 3, 6, 12, 24, 48, and 96 h, 0.5 mL of the supernatant was collected and 0.5 mL of PBS was added to the wells. The rhBMP-2 released into the supernatant was measured by a Bradford protein assay kit from Beyotime, Shanghai, China.The viability of BMSCs on scaffolds was evaluated using a Live/Dead Cell Imaging Kit (Life Technologies, Carlsbad, USA). Then, 1 × 106 cells per mL of P4 BMSCs were planted onto the collagen sponge material and incubated for 24 h with or without 100 ng mL−1 rhBMP-2. The constructs were then washed twice with PBS and added to the working solution for 15 min at 25 °C. The materials were then observed under a confocal laser scanning microscope (NikonA1R; Nikon, Japan). Four random fields were selected and the number of live or dead cells was counted at 200× magnification.
Muscle pouch and subcutaneous transplantation models
The muscle pouch model was generated using 6-week-old SD rats. After the animals were anesthetized and sterilized, 15 mm of the skin in the sural inner side was cut. Then, we opened the intermuscular space and exposed the peroneal muscle pocket. The collagen sponge materials described above were then implanted into these pouches. Then, fluorescent (CM-Dil) labeled BMSCs were injected into the heart (left ventricle).Nude mice were used for live imaging because of the poor imaging performance of SD rats. After being anesthetized and sterilized, a 10 mm incision was made and a pocket was created by bluntly separating subcutaneous tissue in the back. The material was then implanted into the created pocket, followed by injection of fluorescently labelled BMSCs.
Statistical analysis
All experiments were performed in triplicate, at least. Data are expressed as mean ± SD. Statistical comparisons were carried out using one-way analysis of variance (ANOVA) using SPSS software, version 21 (IBM Corp., Chicago, IL, USA). P < 0.05 was considered to be statistically significant.Results
Culture and identification of BMSCs
Following culture to P3, staining of cell surface markers followed by flow cytometry, showed that the expression levels of the BMSC-associated surface antigens, CD73, CD90, and CD105, were >99%, while the hematopoietic stem/progenitor cell marker, CD34, the common leukocyte antigen, CD45, and the immune cell antigen, HLA-DR, were present at <1% (Fig. 1A). Subsequently, the ability of BMSCs to differentiate was determined. After osteogenic induction, Alizarin Red staining revealed a large number of calcium nodules. After adipogenic induction, lipid vacuoles were visualized by oil red O stainin; chondrogenesis induction generated cartilage pellets, which were then fixed, sliced and stained with Alcian blue to identify proteoglycans (Fig. 1B).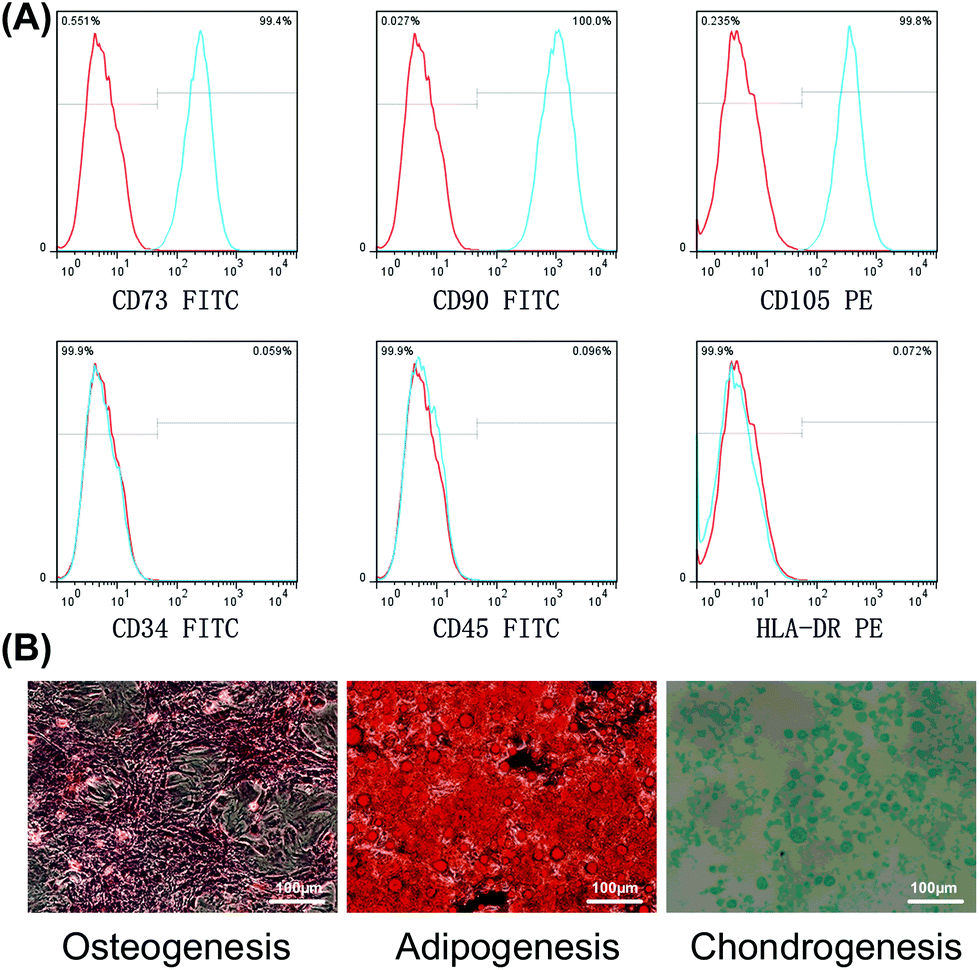 | ||
| Fig. 1 Identification of BMSCs. (a) Identification of immunophenotypic profiles, showing that CD73, CD90, and CD105 positive cells comprised up to 99% of the total population of cells, while CD34, CD45, and HLA-DR positive cells made up <1%. (b) Identification of multilineage differentiation ability within 21 days of induction. | ||
Proliferation ability and rhBMP-2-induced cell migration in vitro
To examine the proliferation ability of BMSCs after activation with rhBMP-2, the CCK-8 assay was used to create a growth curve. Compared with the control group, the ODs of the other three groups revealed similar proliferation abilities. No significant differences were observed at any of the times analyzed (Fig. 2A).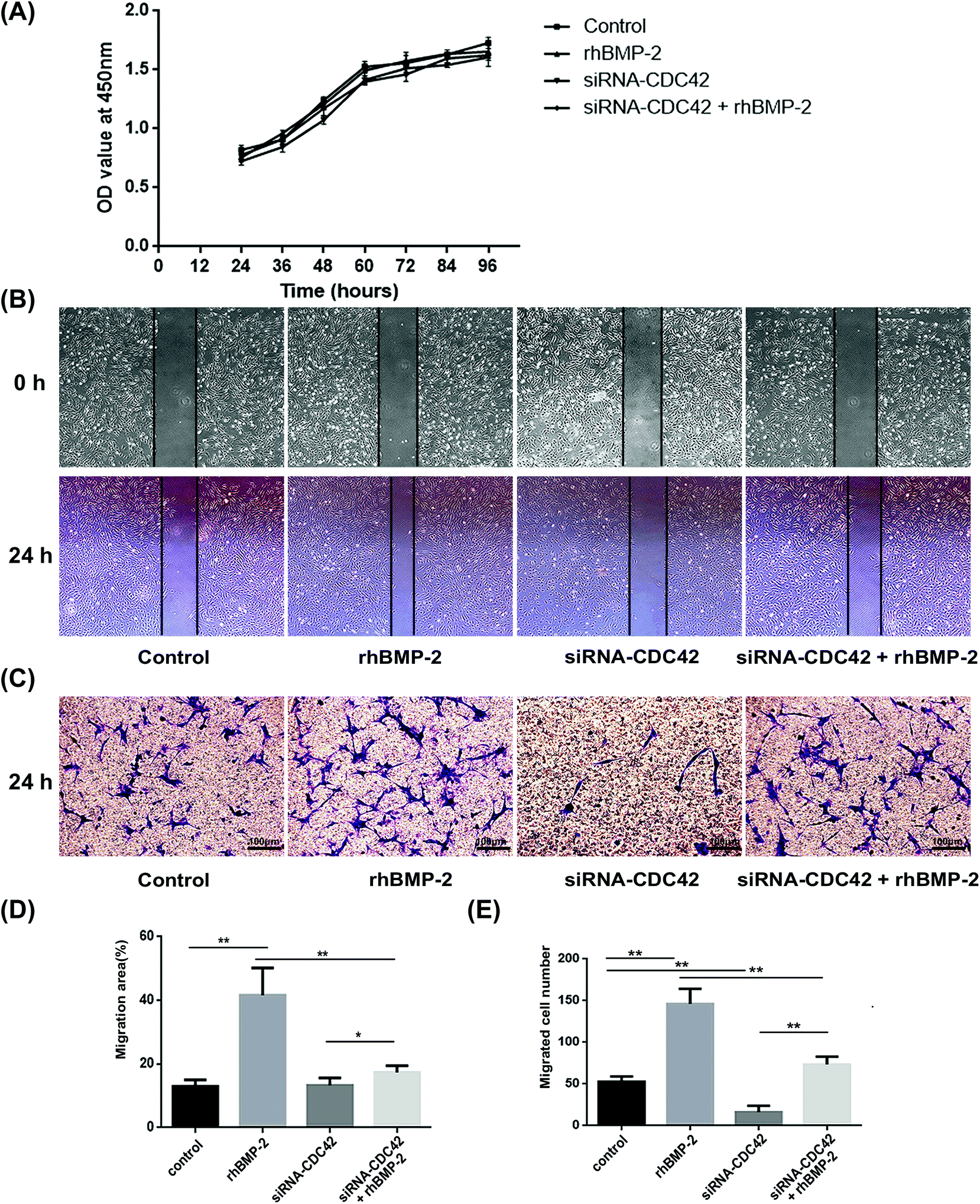 | ||
| Fig. 2 Proliferation ability and rhBMP-2-induced cell migration in vitro. (a) Proliferation assays showed that no significant differences were observed among the control group, rhBMP-2 group, siRNA-CDC42-treated BMSCs group and the rhBMP-2 + siRNA-CDC42-treated BMSCs group at all time points n = 4. (b) Wound-healing assays showing that scratched wounds closer further when cells were treated with rhBMP-2. (c) Typical representative images showing the different efficiencies of migration in transwell assays. (d) Quantitative data illustrating the differences among groups in migration areas. The migration area (%) = (wound area at 0 h − wound area at 24 h)/wound area at 0 h × 100%. *P < 0.05, **P < 0.01, n = 6. (e) Differences in the numbers of migrated cells among groups. **P < 0.01, n = 6. | ||
In wound-healing assays, BMSCs underwent significant migration after treatment with rhBMP-2 for 24 h, whereas siRNA targeting CDC42 decreased their migration, as shown in Fig. 2B. The migration areas under different treatment conditions were then quantified (Fig. 2D). Transwell assays demonstrated that the total number of cells migrating to the lower compartment 24 h after rhBMP-2 treatment differed significantly among groups; the numbers of migrated cells in each treatment group are presented in Fig. 2C and E.
To examine the formation of pseudopods by BMSCs after activation with rhBMP-2, we used laser confocal microscopy and photographed phalloidin-labeled cells. In the rhBMP-2 treated group, the number of cells with actin filament accumulation increased significantly, while CDC42 siRNA treatment inhibited the formation of cortical actin protrusions (Fig. 3). In conclusion, the migration of BMSCs in vitro was enhanced by rhBMP-2.
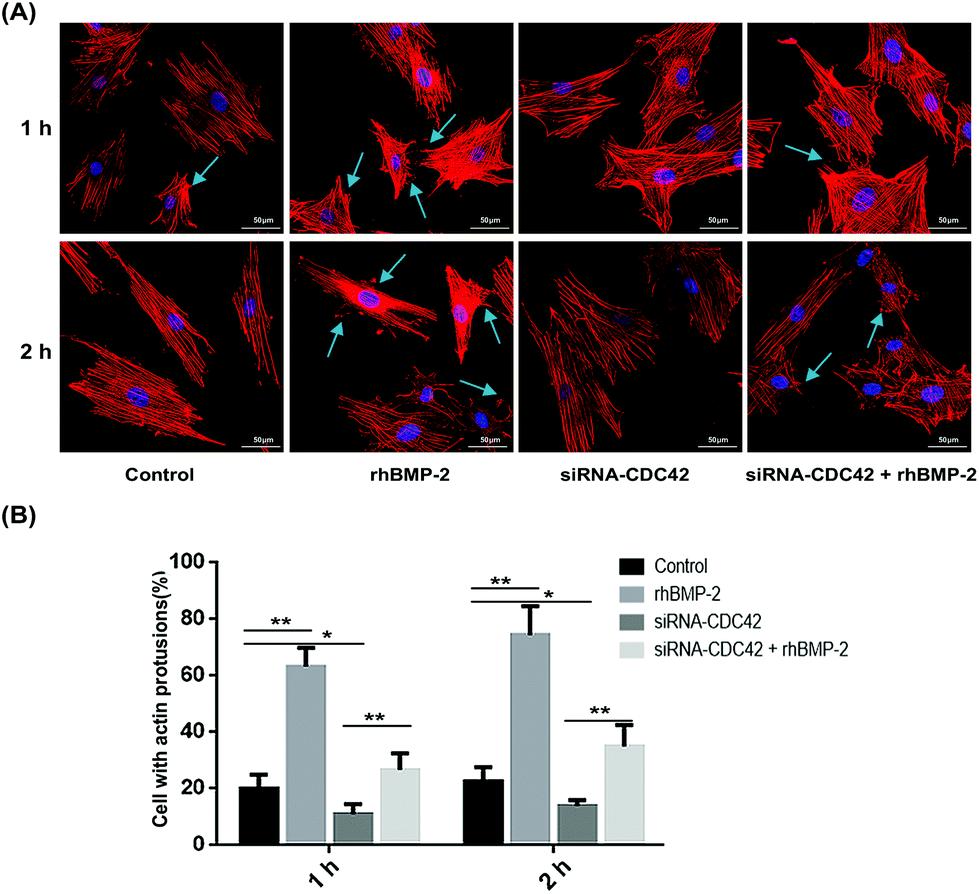 | ||
| Fig. 3 Rhbmp2-induced actin reorganization. (a) The number of actin cortical protrusions were significantly increased in BMP-2-treated cells; however, treatment with siRNA-CDC42 led to a decrease in protrusions. Arrows indicate actin cortical protrusions. (b) The proportion of cells with actin cortical protrusions in different groups. BMSCs showing cortical actin protrusions were all defined as positive cells. No less than 50 cells were counted for each experiment. *P < 0.05, **P < 0.01, n = 4. | ||
Cell SEM morphology
SEM showed that mesenchymal stem cells adhered well to collagen sponges and that the numbers of actin cortical protrusions increased significantly over time with rhBMP-2 treatment. After treatment with rhBMP-2 for 1 h, some of the cells developed filopodia. After 24 h of treatment, large numbers of filopodia were visible (Fig. 4). | ||
| Fig. 4 Sem images (a) morphology of a collagen sponge under low-power magnification. (b) Collagen sponge without rhBMP-2 treatment showing BMSCs with fewer actin cortical protrusions. (c) Cortical actin protrusions appeared after treatment with rhBMP-2 for 1 h. (d) The number of cortical actin protrusions increased significantly after treatment with rhBMP-2 for 24 h. | ||
Release behavior and cell viability in materials
The cumulative release of rhBMP-2 is shown in Fig. 5A; approximately 90% was released during the first 12 h, and almost 98% by 96 h. In general, rhBMP-2 was released from the collagen sponge relatively fast.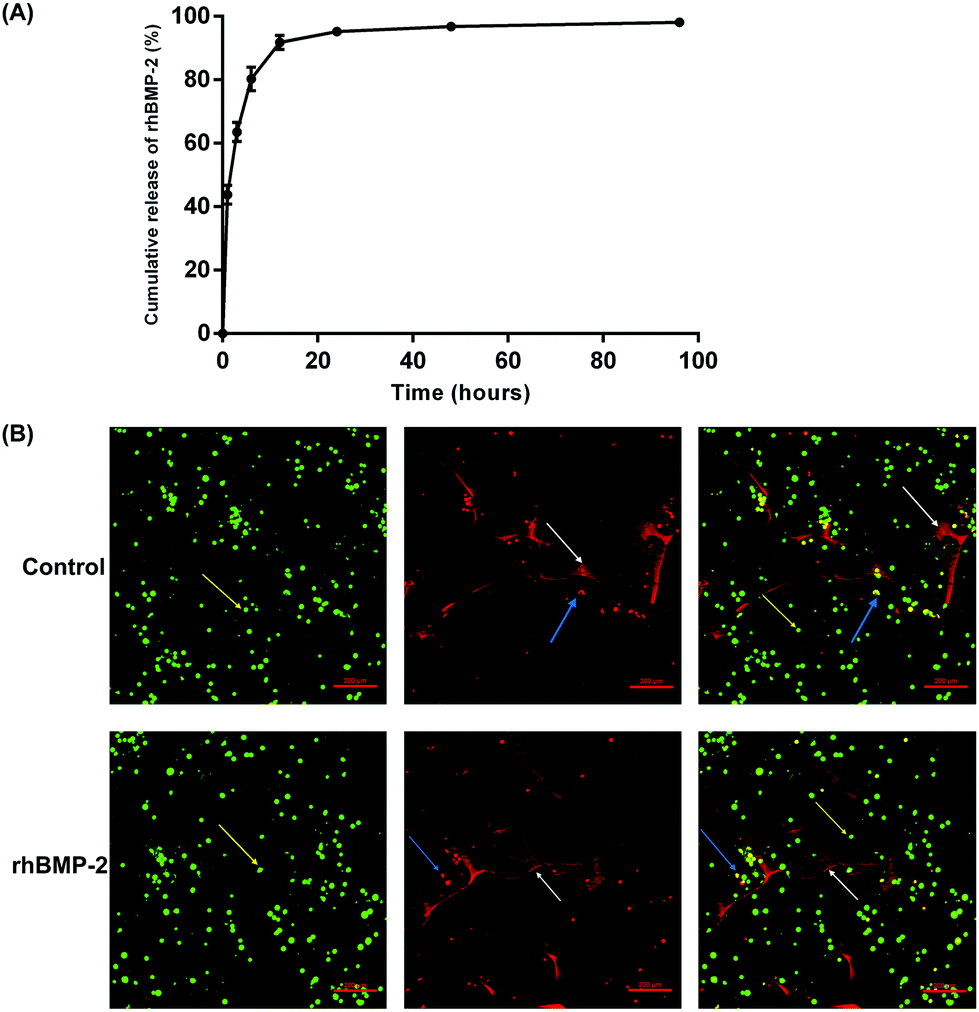 | ||
| Fig. 5 Release behavior and cell viability in materials. (a) Cumulative release of rhBMP-2 from a collagen sponge. (b) Typical representative images showing the viability of BMSCs. The viable cells are green and indicated by yellow arrows. The non-viable cells are red and indicated by blue arrows. The white arrows indicate the collagen sponge. Images on the right show a combination of the two images to the left. | ||
Live/dead cell imaging was used to investigate the viability of BMSCs on rhBMP-2-loaded collagen sponges. After culture within materials for 24 h, live BMSCs were stained green, while dead cells were stained red. The percentage of viable BMSCs planted on the rhBMP-2-loaded collagen sponge was 89.33 ± 2.56% (n = 6), while the proportion of viable cells planted on the collagen sponge was 90.24 ± 2.13% (n = 6). No differences in viability were observed between the control group and the rhBMP-2-treated group (Fig. 5B).
RhBMP-2 induced cell migration in vivo
To observe BMSC migration in rhBMP-2-loaded collagen sponges at different times, the proportion of stained cells was calculated following fluorescence microscope. Over 2 weeks, the number of migrating BMSCs increased over time, while the number of cells migrating after siRNA treatment was significantly lower than that for the treated controls (Fig. 6A). At day 3, approximately 20% of cells in the materials were positive, then this increased to almost 27% by day 7 in the rhBMP-2-treated group. Compared with the control group, significant increases were observed at each time point. For the group, in which BMSCs were silenced by CDC42 siRNA, the number of cells was far lower than in the group with injected BMSCs (Fig. 6B).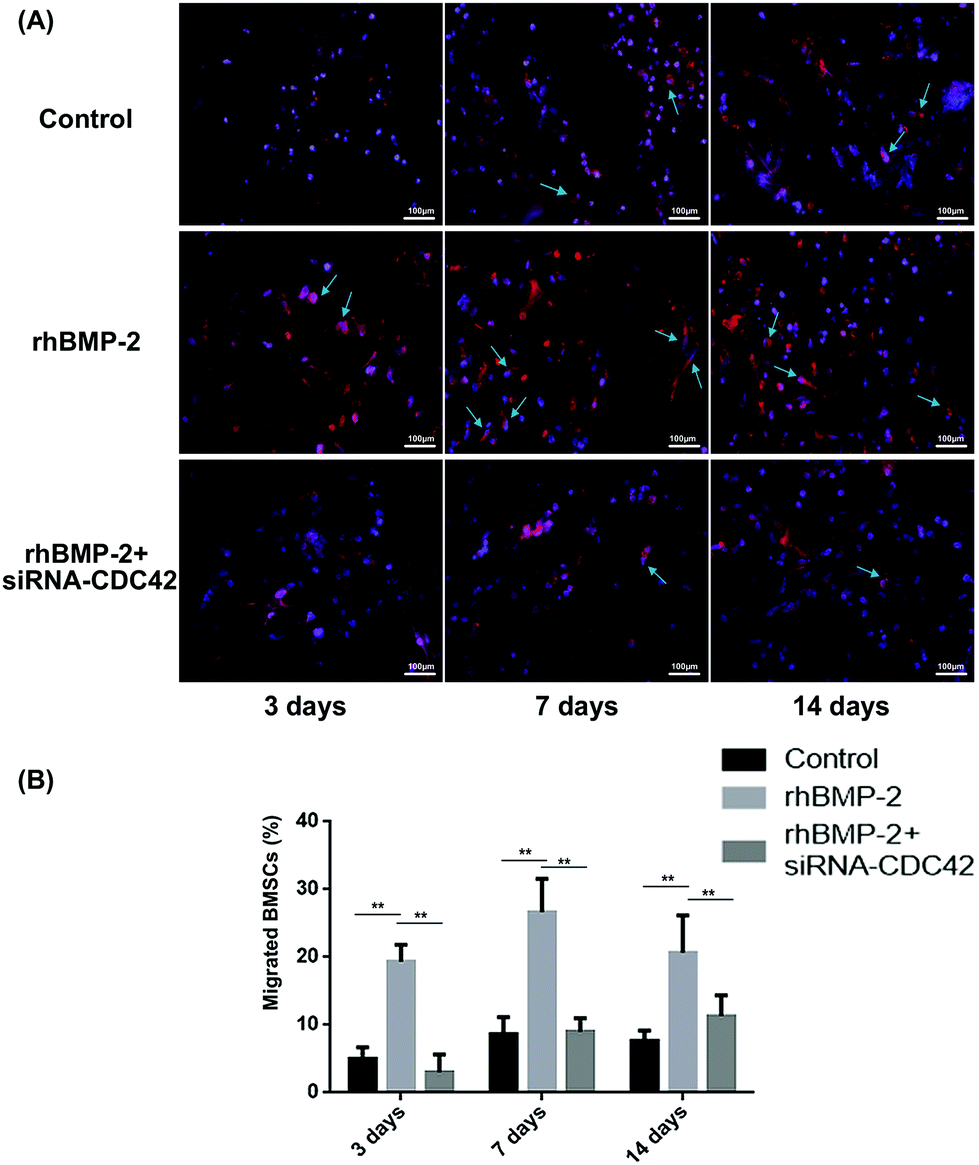 | ||
| Fig. 6 rhBMP-2 induced cell migration in vivo. (a) Typical representative photographs captured by fluorescence microscopy. The arrows indicate CM-Dil-stained BMSCs in materials implanted. (b) The proportion of migrated BMSCs. Four random fields at 200× magnification were captured and cells stained red were deemed to be positive cells. **P < 0.01, n = 8. | ||
In addition, we also performed in vivo imaging experiments using nude mice, in which a collagen sponge was implanted into the backs of mice, and their left ventricles injected with fluorescent-stained BMSCs at different times. In the control group, although many BMSCs survived, there was no significant migration to the material implants (Fig. 7A). Significant numbers of BMSCs accumulated around the implanted material incorporating rhBMP-2 after implantation for 7 days, and they stayed alive for at least 14 days (Fig. 7C and D).
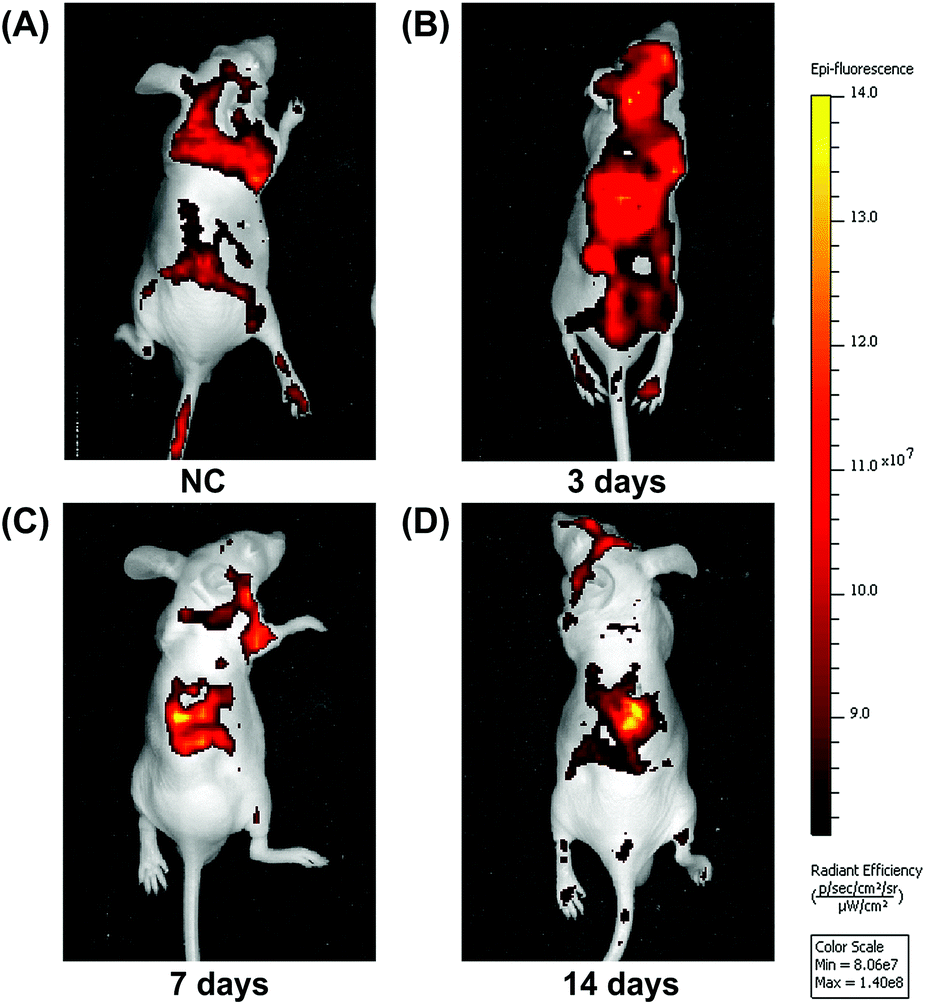 | ||
| Fig. 7 In vivo imaging experiments. (a) As a control, collagen sponges without rhBMP-2 treatment did not recruit BMSCs after implantation for 7 days. (b) BMSCs did not accumulate to rhBMP-2 treated collagen sponges by day 3. (c) The accumulation of BMSCs increased significantly in rhBMP-2 treated collagen sponges at day 7. (d) BMSCs accumulated and stayed alive in rhBMP-2 treated collagen sponges after implantation in the back of nude mice for 14 days. | ||
RhBMP-2 induced CDC42 activation
The CDC42 gene was inhibited using siRNA; knockdown efficiency is illustrated in Fig. 8A. The expression of GTP-CDC42 in BMSCs was examined at different times (0–120 min) after rhBMP-2 treatment. With no rhBMP-2, CDC42 was not activated significantly. After treatment for 30, 60, and 120 min with rhBMP-2, the activation of CDC42 was significantly higher compared with the control group. We concluded that the expression of GTP-CDC42 became enhanced over time following rhBMP-2 treatment and peaked at 60 min (Fig. 8B and C).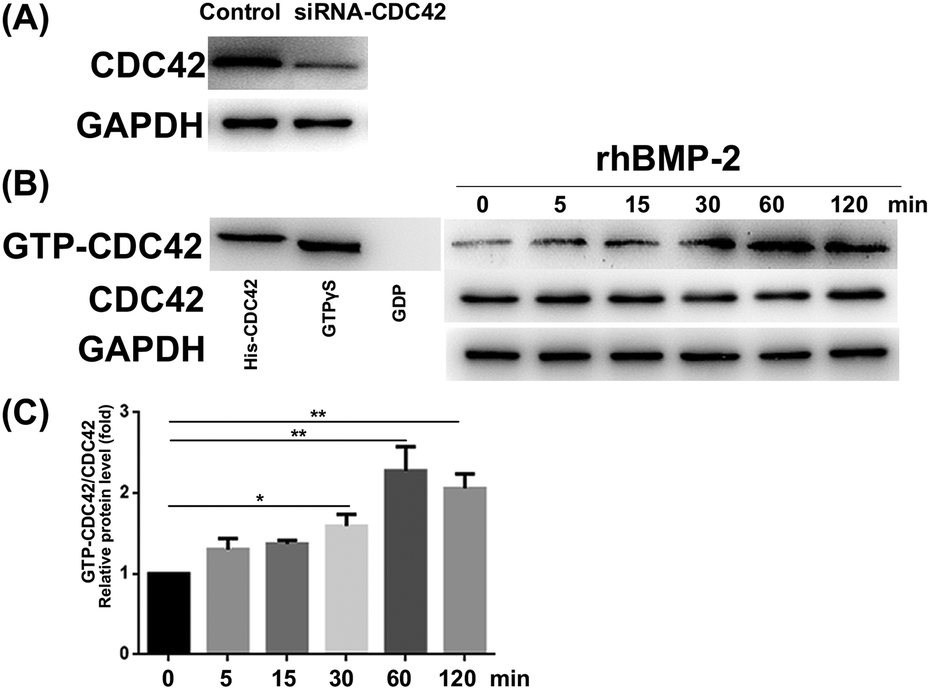 | ||
| Fig. 8 rhBMP-2 induced CDC42 activation. (a) The knockdown efficiency of siRNA. (b) Representative bands visible after gst-pull-down assays. His-CDC42 protein and GTPγs-treated protein sample were used as a positive control, while a gdp-treated protein sample was used as a negative control. (c) The degree of CDC42 activation at different time points after rhBMP-2 treatment. *P < 0.05, **P < 0.01, n = 5, compared to the group of BMSCs without rhBMP-2 treatment. | ||
RhBMP-2 induced migration via the CDC42/PAK1/LIMK1 pathway
To analyze whether rhBMP-2 facilitates BMSC migration via the CDC42/PAK1/LIMK1 pathway, we examined the expression of related proteins at various times after rhBMP-2 treatment. Remarkably, rhBMP-2 increased the activation of PAK1 and LIMK1, particularly in the 60 min group and the 90 min group. As a downstream factor of LIMK, cofilin protein phosphorylation was increased by treatment with rhBMP-2 in a time-dependent manner (Fig. 9A). In an inactive state, p-cofilin inhibits actin depolymerization to stabilize actin filaments.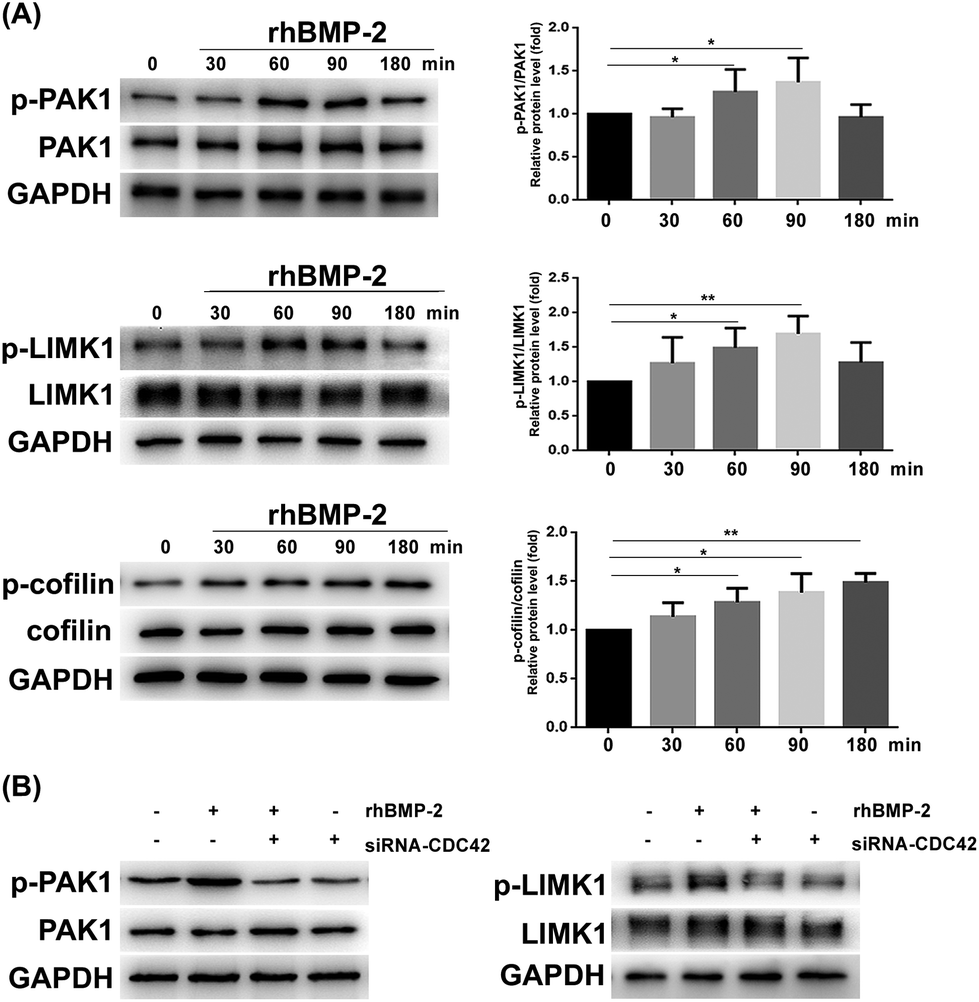 | ||
| Fig. 9 rhBMP-2 induces bmsc migration via the CDC42/PAK1/LIMK1 pathway. (a) Phosphorylation of PAK1, LIMK1, and cofilin at various time points after rhBMP-2 treatment. *P < 0.05, **P < 0.01, n = 5. (b) Phosphorylation of PAK1 and LIMK1 decreased in BMSCs depleted of CDC42, and this decrease was not reversed by rhBMP-2. | ||
Phosphorylation of both PAK1 and LIMK1 was enhanced by rhBMP-2 stimulation at 60 min, whereas silencing of CDC42 reduced levels of phosphorylation (Fig. 9B). In contrast to normal BMSCs, inhibition using CDC42 siRNA led to a significant reduction of PAK1 and LIMK1 phosphorylation at 60 min. Furthermore, PAK1 and LIMK1 from CDC42-siRNA-treated BMSCs could not be activated by rhBMP-2. These data confirm that rhBMP-2 activates PAK1 and LIMK1 via the activation of CDC42.
Discussion
The recruitment of stem cells forms the basis of bone repair and reconstruction. For improved function, BMSCs must promptly migrate to the appropriate site. BMSCs are recruited to sites requiring bone repair and migrate to them in response to various cytokines and chemokines at the site of injury.5 Numerous studies have demonstrated that the migration ability of BMSCs affects their function;22,23 therefore, a better understanding of the mechanisms underlying BMSC migration could promote the application of these cells in tissue engineering. As it is a potent osteogenic growth factor, the roles of BMP-2 in osteogenesis and bone synthesis have been widely investigated,1,24,25 while little research has been conducted into the process of cell migration during bone repair. In this study, we investigated changes in BMSC migration in vitro in response to rhBMP-2, and determined that the regulatory mechanism underlying the process of stem cell migration and recruitment involves the CDC42/PAK1/LIMK1 signaling pathway. Meanwhile, we implanted collagen sponge with rhBMP-2 incorporated within its microstructure, to investigate regulation of the homing and enrichment of BMSCs in vivo.The early features of cell migration include stress fiber polarization and the formation of cortical protrusions resulting from the accumulation of cortical actin at the cell periphery. Our results showed that rhBMP-2 treatment significantly expanded the proportion of BMSCs with actin filament accumulation and increased the recruitment of BMSCs. Our results agree with previous data obtained in C2C12 cells. Gamell et al. showed that BMP-2 treatment leads to rapid actin cytoskeleton rearrangement and increases the rate of C2C12 cell migration, and that these effects require the co-activation of CDC42 and PI3Kα.12 In addition, the authors also found that the p38/MK2/Hsp25 pathway is involved in actin rearrangement and cell migration in response to BMP-2 treatment.17
When we silenced CDC42, the cytoskeletal reorganization and migration could be blocked, which indicated that CDC42 has significant influence on cytoskeletal re-modelling. As a small GTPase of the Rho family, CDC42 is involved in developmental processes and tumor growth. Woodham et al. described the important role of CDC42 in cell proliferation and migration in mouse embryonic melanoblasts. Loss of CDC42 led to severe pigmentation defects and revealed that CDC42 acts as a coordinator for multiple cell migration-related mechanisms in the melanocyte lineage, including actin motility, myosin activity, and integrin-based adhesion.26 In ovarian cancer cells, phosphorylated p70 (S6K) can increase the expression of CDC42, downstream of PI3K/AKT, resulting in activation of PAK1 and cytoskeletal changes.14
To verify whether BMP-2 has an effect on CDC42, we conducted CDC42 pull-down activation assays and found that CDC42 was swiftly activated to the GTP-CDC42 form by the action of rhBMP-2. This indicated that the CDC42-related pathway was involved in the rhBMP-2-induced BMSC migration. Although few studies have proven the relationship between CDC42 and BMP-2-induced BMSC migration, several studies have indicated the influence of CDC42 in cytoskeletal reorganization and the migration of stem cells. Park et al. found that ceramide-induced embryonic stem cell migration was regulated by PKC and FAK/Paxillin-dependent N-WASP/CDC42/ARP2/3 complex formation,27 while Xu et al. showed that the Ang II-angiotensin II type 2 receptor (AT2R) caused focal contacts through the activation of FAK and CDC42, causing cytoskeletal reorganization and thereby, BMSC migration.28 Studies have shown that CDC42 signaling not only mediates the migration process of BMSCs, but is also closely related to its osteogenic differentiation process. Li et al. demonstrated that CDC42 plays a key role in the morphological changes and remodeling of BMSCs on the surface of biomaterials, while both CDC42 and Wnt/β-catenin signaling regulate the osteogenic differentiation of BMSCs.29 Ichida et al. found that the migration, morphology, and adherence of mesenchymal stem cells altered dramatically during osteogenic differentiation. Cell migration was up-regulated in the early stages, while in later stages, migration decreased and adhesion increased. This process was accompanied by activation of CDC42 and RAC1, during the early stages of differentiation, and a decrease of FAK phosphorylation as differentiation progressed.19 Many studies have confirmed that the SDF-1/CXCR4 pathway is important for mesenchymal stem cell migration.2,16,21,30 During wound healing, SDF-1 can improve the ability of BMSCs to migrate and differentiate by promoting the expression of BMP-2.31 Meanwhile, SDF-1 regulates actin reorganization via CDC42.15,32 In addition, PI3K/Akt signaling is important for the migration of BMSCs during tissue regeneration as a pathway upstream of CDC42 and PAK1.33
Research has shown that BMP-2 can increase cell migration by activating PAK1 and its downstream effectors LIMK1 in C2C12 cells in vitro.12 The PAK family takes part in several cytoskeletal changes, usually in response to small GTPase.12 LIMK1, as the substrate of PAK, is involved in the regulation of the cytoskeleton via the phosphorylation of cofilin.13,34 Studies have shown that binding of BMP to BMP II receptors increases the activity of LIMK1, thus playing a role in maintaining synaptic growth and stability.35 In our study, for BMSCs, rhBMP-2 was shown to promote the phosphorylation of PAK1, LIMK1, and cofilin. Furthermore, we also demonstrated that the activation of PAK1 and LIMK1 by rhBMP-2 was abolished by silencing CDC42 in BMSCs. This suggested that CDC42 plays a central role in the process of rhBMP-2-mediated BMSC migration.
To further understand the role of rhBMP-2 in promoting the migration of BMSC in vivo and the function of CDC42 during this process, we used CM-Dil for direct cell viability staining, which causes minimal cell damage, and observed BMSC growth in collagen sponge materials implanted in SD rats and nude mice. As a carrier, the collagen sponge exhibits good biocompatibility, and can be degraded into terminal physiological products. Its structure also has the benefit of being permeable to infiltration by cells and various cytokines, thus increasing their mutual contact.11,36,37 Collagen sponges loaded with high doses of rhBMP-2 have been in use for years, and several drawbacks have become evident, such as edema, ectopic bone growth, and the risk of tumor recurrence.38–40 Some recent studies have made changes to the rhBMP-2/Absorbable Collagen Sponge (ACS), which have been approved for clinical use, such as changing the dose of BMP-2,40,41 or adding other factors or material components.42,43 Efforts have been made to reduce the dose of rhBMP-2. RhBMP-2/ACS containing 1.25–2.5 μg of rhBMP-2 was shown to promote local bone formation in a model of skull defects in rats, and increasing the dose did not further enhance osteogenesis.44 With protamine-based polyelectrolyte complex carrier, 0.5 μg of rhBMP-2 is sufficient to achieve solid posterolateral spinal fusion in the rat model.41 In this experiment, we focused on the role of low-dose rhBMP-2 in migration, and the mechanisms involved, to explore the efficacy and value of low-dose rhBMP-2-loaded collagen sponges. Furthermore, we tried to provide a theoretical basis for the construction of materials conducive to osteogenesis-related cell migration and enrichment.
To identify BMP-2-induced migration in vivo, Kimura et al. established an ectopic bone formation model by transplanting male mouse-derived EGFP-positive myeloid cells into female mice and placing gelatin gel implants incorporating BMP-2 into the backs of animals. The number of bone marrow-derived osteoprogenitors undergoing migration was consistent with the release profile of BMP-2, and BMP-2 recruited osteoprogenitors to ectopic bone formation sites through the circulatory system.45 Similarly, Otsuru et al. reported that BMP-2 accelerated the recruitment of osteoprogenitors from the bone marrow to the circulation, which contributed to the formation of ectopic bone.46 This recruitment is regulated by the CXCR4/SDF-1 signaling pathway.16 Through in vivo imaging and observation by fluorescence microscopy, we determined that BMSCs in the circulatory system accumulated in response to induction by rhBMP-2 in collagen sponge material in vivo. With CDC42 siRNA treatment, BMSCs decreased the homing ability. Our research was the first to show that collagen sponge material loaded with rhBMP-2 could recruit BMSCs injected into the circulatory system, while with CDC42 siRNA inhibition, recruitment decreased significantly. Along with these studies, our present findings demonstrate that CDC42 and its related pathways are important for cell migration. Further studies are now needed to identify the mechanisms involved in the BMP-2-mediated migration of other osteogenesis-related cell types and to find ways to activate the CDC42/PAK1/LIMK1 pathway to improve the effect of rhBMP-2.
The repair and reconstruction of bone tissue is a longstanding and continuing challenge for modern medicine.47 The ability of bioactive materials to recruit osteogenesis-related cells is a criterion used for their evaluation. It is logical to attempt to modify existing established materials, such as the rhBMP-2/collagen sponge, to improve cell recruitment, which potentially has wide applications in various clinical settings.
There are several limitations to the present study. First, our investigations of material release were not completely accurate, owing to experimental limitations. Second, we did not identify ways to activate the CDC42/PAK1/LIMK1 pathway to improve the effect of rhBMP-2 in cell migration. Furthermore, we did not study the ability of osteogenesis to be induced by rhBMP-2. These issues will be the focus of our future research.
Conclusions
In this investigation, we verified that rhBMP-2 treatment activated CDC42 and downstream factors, thus promoting the migration of BMSCs and enhancing cytoskeletal reorganization in vitro, and confirmed that an in vivo collagen sponge incorporating rhBMP-2 can significantly promote BMSC recruitment from the circulatory system. Silencing of the CDC42 gene inhibited its downstream pathways and thereby led to a significant reduction in the number of BMSCs within the material. In conclusion, our data demonstrate that rhBMP-2 accelerates BMSC migration via the CDC42/PAK1/LIMK1 pathway both in vivo and in vitro. This research will facilitate a deeper understanding of the mechanisms involved in the BMP-2-mediated migration of BMSCs. CDC42 might also act as a target to increase the recruitment of sufficient numbers of BMSCs to defect sites for bone repair with rhBMP-2-loaded materials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was supported by the National Natural Science Foundation of China (31330028 and 81702177).Notes and references
- F. Yang, J. Wang, J. Hou, H. Guo and C. Liu, Biomaterials, 2013, 34, 1514–1528 CrossRef CAS PubMed.
- X. Wang, Y. Wang, W. Gou, Q. Lu, J. Peng and S. Lu, Int. Orthop., 2013, 37, 2491–2498 CrossRef PubMed.
- L. P. Cramer, Nat. Cell Biol., 2010, 12, 628–632 CrossRef CAS PubMed.
- T. D. Pollard and J. A. Cooper, Science, 2009, 326, 1208–1212 CrossRef CAS PubMed.
- H. Ito, Mod. Rheumatol., 2011, 21, 113–121 CrossRef CAS PubMed.
- S. Sinha and W. Yang, Cell. Signalling, 2008, 20, 1927–1934 CrossRef CAS PubMed.
- S. Etienne-Manneville and A. Hall, Nature, 2002, 420, 629–635 CrossRef CAS PubMed.
- D. C. Edwards, L. C. Sanders, G. M. Bokoch and G. N. Gill, Nat. Cell Biol., 1999, 1, 253–259 CrossRef CAS PubMed.
- J. Cau and A. Hall, J. Cell Sci., 2005, 118, 2579–2587 CrossRef CAS PubMed.
- H. R. Dawe, L. S. Minamide, J. R. Bamburg and L. P. Cramer, Curr. Biol., 2003, 13, 252–257 CrossRef CAS PubMed.
- M. Geiger, Adv. Drug Delivery Rev., 2003, 55, 1613–1629 CrossRef CAS PubMed.
- C. Gamell, N. Osses, R. Bartrons, T. Rückle, M. Camps, J. L. Rosa and F. Ventura, J. Cell Sci., 2008, 121, 3960–3970 CrossRef CAS PubMed.
- A. A. John, R. Prakash, J. Kureel and D. Singh, J. Mol. Med., 2018, 96, 427–444 CrossRef CAS PubMed.
- C. K. Ip, A. N. Cheung, H. Y. Ngan and A. S. Wong, Oncogene, 2011, 30, 2420–2432 CrossRef CAS PubMed.
- K. Kowalski, A. Kolodziejczyk, M. Sikorska, J. Placzkiewicz, P. Cichosz, M. Kowalewska, W. Streminska, K. Janczyk-Ilach, M. Koblowska, A. Fogtman, R. Iwanicka-Nowicka, M. A. Ciemerych and E. Brzoska, Cell Adhes. Migr., 2017, 11, 384–398 CrossRef CAS PubMed.
- S. Otsuru, K. Tamai, T. Yamazaki, H. Yoshikawa and Y. Kaneda, Stem Cells, 2008, 26, 223–234 CrossRef CAS PubMed.
- C. Gamell, A. G. Susperregui, O. Bernard, J. L. Rosa and F. Ventura, PLoS One, 2011, 6, e16477 CrossRef CAS PubMed.
- L. Fourel, A. Valat, E. Faurobert, R. Guillot, I. Bourrin-Reynard, K. Ren, L. Lafanechere, E. Planus, C. Picart and C. Albiges-Rizo, J. Cell Biol., 2016, 212, 693–706 CrossRef CAS PubMed.
- M. Ichida, Y. Yui, K. Yoshioka, T. Tanaka, T. Wakamatsu, H. Yoshikawa and K. Itoh, FEBS Lett., 2011, 585, 4018–4024 CrossRef CAS PubMed.
- W. Friess .
- S. Liu, H. Liang, S. M. Lee, Z. Li, J. Zhang and Q. Fei, Acta Biochim. Biophys. Sin., 2017, 49, 101–109 CAS.
- Y. C. Huang and T. J. Liu, Acta Biomater., 2012, 8, 1048–1056 CrossRef CAS PubMed.
- S. Kumar and S. Ponnazhagan, Bone, 2012, 50, 1012–1018 CrossRef CAS PubMed.
- C. Dai, H. Guo, J. Lu, J. Shi, J. Wei and C. Liu, Biomaterials, 2011, 32, 8506–8517 CrossRef CAS PubMed.
- R. X. Shao, R. F. Quan, T. Wang, W. B. Du, G. Y. Jia, D. Wang, L. B. Lv, C. Y. Xu, X. C. Wei, J. F. Wang and D. S. Yang, J. Tissue Eng. Regener. Med., 2018, 12, e1813–e1825 CrossRef CAS PubMed.
- E. F. Woodham, N. R. Paul, B. Tyrrell, H. J. Spence, K. Swaminathan, M. R. Scribner, E. Giampazolias, A. Hedley, W. Clark, F. Kage, D. J. Marston, K. M. Hahn, S. W. Tait, L. Larue, C. H. Brakebusch, R. H. Insall and L. M. Machesky, Curr. Biol., 2017, 27, 624–637 CrossRef CAS PubMed.
- S. S. Park, M. O. Kim, S. P. Yun, J. M. Ryu, J. H. Park, B. N. Seo, J. H. Jeon and H. J. Han, Biochim. Biophys. Acta, 2013, 1831, 350–360 CrossRef CAS PubMed.
- X. P. Xu, H. L. He, S. L. Hu, J. B. Han, L. L. Huang, J. Y. Xu, J. F. Xie, A. R. Liu, Y. Yang and H. B. Qiu, Stem Cell Res. Ther., 2017, 8, 164 CrossRef PubMed.
- G. Li, Y. Song, M. Shi, Y. Du, W. Wang and Y. Zhang, Acta Biomater., 2017, 49, 235–246 CrossRef CAS PubMed.
- A. E. Rapp, R. Bindl, A. Heilmann, A. Erbacher, I. Müller, R. E. Brenner and A. Ignatius, Eur. Cells Mater., 2015, 29, 22–34 CrossRef CAS PubMed.
- F. Granero-Molto, J. A. Weis, M. I. Miga, B. Landis, T. J. Myers, L. O'Rear, L. Longobardi, E. D. Jansen, D. P. Mortlock and A. Spagnoli, Stem Cells, 2009, 27, 1887–1898 CrossRef CAS PubMed.
- J. L. Z. Elie Haddad, F. Louache, N. Debili, C. Crouin, K. Schwarz, A. Fischer, W. Vainchenker and J. Bertoglio, Blood, 2001, 97, 33–38 CrossRef.
- J. Chen, R. Crawford, C. Chen and Y. Xiao, Tissue Eng., Part B, 2013, 19, 516–528 CrossRef CAS PubMed.
- R. W. Scott and M. F. Olson, J. Mol. Med., 2007, 85, 555–568 CrossRef CAS PubMed.
- B. A. Eaton and G. W. Davis, Neuron, 2005, 47, 695–708 CrossRef CAS PubMed.
- C. Domene, C. Jorgensen and S. W. Abbasi, Phys. Chem. Chem. Phys., 2016, 18, 24802–24811 RSC.
- L. Mahesh, G. M. Kurtzman and S. Shukla, Compend. Contin. Educ. Dent., 2015, 36, 358–363 Search PubMed.
- C. J. Kowalczewski and J. M. Saul, Front. Pharmacol., 2018, 9, 513 CrossRef PubMed.
- D. S. Geller, M. Y. Singh, W. Zhang, J. Gill, M. E. Roth, M. Y. Kim, X. Xie, C. K. Singh, H. D. Dorfman, E. Villanueva-Siles, A. Park, S. Piperdi and R. Gorlick, Clin. Cancer Res., 2015, 21, 3003–3012 CrossRef CAS PubMed.
- L. Krishnan, L. B. Priddy, C. Esancy, B. S. Klosterhoff, H. Y. Stevens, L. Tran and R. E. Guldberg, Acta Biomater., 2017, 49, 101–112 CrossRef CAS PubMed.
- T. Hu, S. A. Abbah, M. Wang, S. Y. Toh, R. W. Lam, M. Naidu, G. Bhakta, S. M. Cool, K. Bhakoo, J. Li, J. C. Goh and H. K. Wong, Spine, 2015, 40, 613–621 CrossRef PubMed.
- C. Susin, J. Lee, T. Fiorini, R. M. de Freitas, H. C. Chiu, H. S. Prasad, A. N. Buxton and U. M. Wikesjo, J. Clin. Periodontol., 2018 DOI:10.1111/jcpe.12921.
- C. R. Kinsella Jr., M. R. Bykowski, A. Y. Lin, J. J. Cray, E. L. Durham, D. M. Smith, G. E. DeCesare, M. P. Mooney, G. M. Cooper and J. E. Losee, Plast. Reconstr. Surg., 2011, 127, 1865–1873 CrossRef PubMed.
- M. Pelaez, C. Susin, J. Lee, T. Fiorini, F. C. Bisch, D. R. Dixon, J. C. McPherson 3rd, A. N. Buxton and U. M. Wikesjo, J. Clin. Periodontol., 2014, 41, 827–836 CrossRef CAS PubMed.
- Y. Kimura, N. Miyazaki, N. Hayashi, S. Otsuru, K. Tamai, Y. Kaneda and Y. Tabata, Tissue Eng., Part A, 2010, 16, 1263–1270 CrossRef CAS PubMed.
- S. Otsuru, K. Tamai, T. Yamazaki, H. Yoshikawa and Y. Kaneda, Biochem. Biophys. Res. Commun., 2007, 354, 453–458 CrossRef CAS PubMed.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |