
Bimetallic gold nanorods with enhanced biocorona formation for doxorubicin loading and sustained release†
Debolina
Chakraborty‡
a,
Lavanya
Mohan‡
b,
Sruthi Ann
Alex
b,
Natarajan
Chandrasekaran
 b and
Amitava
Mukherjee
*b
b and
Amitava
Mukherjee
*b
aSchool of Advanced Sciences, Vellore Institute of Technology, Vellore, India
bCentre for Nanobiotechnology, Vellore Institute of Technology, Vellore, India. E-mail: amit.mookerjea@gmail.com; amitav@vit.ac.in; Fax: +91-416-224309; Tel: +91 416 2202620
First published on 20th November 2018
Abstract
The biomedical applicability of gold nanorods (AuNRs) arises due to their interesting optical and photothermal properties, which can result in the formation of a protein corona layer when exposed to the physiological system. The current study focuses on the effect of bimetallic coatings of AuNRs (AuNRs@Pd and AuNRs@Cu) on protein corona formation, and the potential application of protein-coronated bimetallic AuNRs was investigated for doxorubicin (dox) loading, release, and in vitro cytotoxicity analysis. Two significant proteins in blood serum, namely, human serum albumin (HSA) and transferrin, were chosen for the protein coronation. The variations in the protein adsorption patterns of monometallic and bimetallic AuNRs were studied based on the protein adsorption, zeta potential, and particle size measurements. A higher adsorption of hard and soft corona was observed for HSA due to their higher abundance and reactivity. The enhanced electropositive nature of these bimetals promoted higher corona formation (AuNR@Pd > AuNR@Cu > AuNRs) when compared with bare AuNRs, which in turn correlated with higher dox loading. The higher corona on bimetallic AuNRs helped to overcome the burst release of dox over a period of 48 h (AuNRs@Pd > AuNR@Cu > AuNRs) when compared to the respective monometallic AuNRs, and the dox release was slightly increased when tested in human plasma. Furthermore, a significant decrease in the cytotoxicity of protein-coronated bimetallic AuNRs as compared to monometallic AuNRs was also observed. Thus, it can be suggested that the use of engineered protein corona on bimetallic nanostructures can open new areas of research for cancer therapeutics.
1. Introduction
Gold nanorods (AuNRs) exhibit distinctive plasmonic properties due to their anisotropic nature and have good colloidal stability, which make them attractive candidates for drug delivery, cancer diagnosis/therapy, biomedical imaging, and biosensing.1–3 In a recent study conducted by Xiao et al. (2012), it was noted that AuNRs conjugated with doxorubicin and a targeting ligand can be used as an efficient multifunctional nanoplatform for both targeting and imaging cancer cells.2 The enhanced uptake by cells due to their rod shape, when aided by several targeting ligands helps them act as carrier vehicles for drug delivery application.4–6 Doxorubicin (dox), an antitumour anthracycline drug, when administered using passive targeting can lead to poor therapeutic efficiency and cause severe side effects, including myelosuppression and cardiotoxicity.7 The therapeutic efficiency of a chemotherapeutic drug can be improved by loading the drugs on the surface of nanostructures, such as metal nanoparticles, nanorods, polymeric nanoparticles, and liposomes, which can improve the bioavailability at a particular site of interest and provide sustained drug release and higher biocompatibility.8,9When these nanorods are used for biological applications, they can encounter a complex mixture of proteins in the physiological fluids,10,11 which can non-specifically and rapidly absorb to form the “protein corona” and thus alter their biological fate.12 Several reports are available on the composition of the corona layer formed by serum proteins or individual proteins on silver and/or gold nanoparticles.13–15 A recent study by García-Álvarez et al. (2018) has exclusively investigated the effect of in vivo formation of protein corona on gold nanoparticles of different sizes and shapes, wherein they found that larger AuNRs could adsorb a higher number of protein types than AuNSs of a similar size.16
The uncontrolled formation of protein corona can alter the performance of the designed nano-therapeutics and can result in unexpected shortcomings.17,18 However, the protein-coronated nanostructures, which are formed by the pre-incubation of specific proteins that can help in targeting or act as sponges to load hydrophilic or hydrophobic molecules of interest, would indicate reduced cellular toxicity and can be exploited for biomedical applications.19 Furthermore, the presence of protein corona on the surface of nanostructures also results in a sustained drug release profile.20 Previous studies conducted by different groups have also highlighted the utility of protein-coronated nanostructures for simultaneous loading of various therapeutic molecules, such as drugs and DNA.19,21 Yeo et al. (2017) demonstrated the use of coronated NRs for loading photosensitizers (chlorin e6) and using these particles for multimodal cancer therapy.22
The preparation of colloidal AuNRs requires a significant amount of CTAB surfactants, which end up forming a bilayer structure on the surface of AuNRs.23 To employ AuNRs for any biomedical application, a series of surface modification steps are carried out to impart biocompatibility since CTAB is known to induce severe cytotoxicity even at lower concentrations.24,25 Our group has previously studied the effect of different AuNR functionalization on the protein corona formation of serum abundant proteins like HSA, IgG, and transferrin.26 One of the possible functionalization methods involve the surface coating of AuNRs with a thin shell of inorganic materials like silica, metal oxides, CNTs, or silver.25 There have been a few recent reports on forming a Cu/Pd shell on Au core nanorods.27 Pd and Cu are known to be more electropositive in nature (i.e. lower electron affinity values) compared to the Au alone.28–30 Therefore, a thin shell of Pd/Cu would possibly induce enhanced cationic surface charge on the AuNR surface. HSA (pI value of 4.3–5.7) and transferrin (pI value 6.14–6.63) both carry predominantly negative surface charge at physiological pH,31 and hence could show enhanced adsorption on these electropositive bimetals compared to monometallic AuNRs. Electrostatic interaction is recognized to be one of the major driving forces for protein corona formation on the AuNRs; however, a complex interplay of various other factors such as covalent interactions, hydrogen bonding, and van der Waals forces cannot be overlooked.32 Additionally, Heinz et al. (2009) suggested that Pd surfaces could indicate stronger adsorption of aromatic and conjugated peptide residues such as Tyr, Phe, His, Arg, Asp, and Asn when compared to Au (111) surfaces, which could be correlated with their higher surface tension. Pd–Au (111) bimetallic surfaces were also observed to indicate additional peptide adsorption.33 The enhanced surface energy of bimetallic coatings may also facilitate the higher adsorption of protein corona.34
In the current work, we hypothesized that the enhancement in electropositivity imparted by the bimetallic coating on the AuNRs could improve the protein corona formation at physiological pH, which as mentioned before, can help in increasing the loading efficiency of the desired drug and also assist in attaining a sustained release profile in the tumor microenvironment. Furthermore, as a consequence of the higher protein corona layer, direct cellular interaction with the AuNR surface will also be minimized, which in turn will reduce the NP-associated cytotoxicity. To the best of our knowledge, this is the first-ever work that explores the effect of bimetallic coating of AuNRs on the enhanced corona formation for two different biologically relevant proteins (HSA and transferrin) and the utilization of these coronated nanostructures as efficient delivery agents for dox. Initially, the bimetallic AuNRs (AuNRs@Pd and AuNRs@Cu) were prepared and characterized, and the effect of the bimetals on the adsorption of HSA and transferrin was studied at different time points. The controllable loading of dox on the corona-conjugated monometallic and bimetallic AuNRs was quantified by the thermal treatment method and was found to be higher for HSA. Bimetallic AuNRs have higher adsorptive properties and could form more protein corona, which helped to provide the sustained release over a period of 48 h (AuNRs@Pd > AuNR@Cu > AuNRs) when compared to their respective monometallic AuNRs, thus, highlighting their potential applicability for developing a drug delivery system. Finally, the lower in vitro cytotoxicity of protein-preincubated bimetallic AuNRs over monometallic AuNRs highlights their comparative advantage for biomedical applications.
2. Materials and methods
2.1. Reagents and solutions
Doxorubicin hydrochloride, human serum albumin (HSA), transferrin, cetlytrimethylammonium bromide (CTAB), sodium borohydride (NaBH4), palladium chloride (PdCl2), and copper oxide (CuO) were procured from Sigma-Aldrich (India). Silver nitrate (AgNO3) was purchased from Merck Specialties Pvt. Ltd (India). Hydrogen tetrachloroaurate hydrate (HAuCl4·2H2O) was purchased from SRL Pvt. Ltd (India). Ascorbic acid and hydrochloric acid (HCl) were purchased from SD Fine Chemicals Ltd (India). Analytical-grade chemicals have been used for the study without further purification. Milli-Q water from Cascada BioWater filtration unit (Pall Corporation, USA) of 18.2 MΩ cm was used for the entire study. The glassware (Borosil) was methodically cleaned using the aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1) ratio, followed by the methodical rinsing and drying in a hot-air oven. Dulbecco's modified Eagle's Medium (DMEM), fetal bovine serum (FBS), antibiotic and antimycotic solution, 10× phosphate buffered saline, trypsin, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), and dimethyl sulphoxide were brought from Himedia Laboratories, India, for performing the in vitro studies.
:HNO3 = 3:1) ratio, followed by the methodical rinsing and drying in a hot-air oven. Dulbecco's modified Eagle's Medium (DMEM), fetal bovine serum (FBS), antibiotic and antimycotic solution, 10× phosphate buffered saline, trypsin, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), and dimethyl sulphoxide were brought from Himedia Laboratories, India, for performing the in vitro studies.
2.2. Synthesis of AuNRs and bimetallic AuNRs
The synthesis of CTAB-capped AuNRs was done according to the protocol reported in our previous studies. In brief, a brown-colored seed solution was prepared by making a mixture solution of HAuCl4 (2.5 mL, 0.5 mM) and CTAB (2.5 mL, 0.2 M), followed by the addition of ice-cold NaBH4 (0.3 mL, 0.01 M). The seed solution was incubated for 3 h. For the growth solution, AgNO3 (12 mL, 0.004 M) and ascorbic acid (2.8 mL, 0.079 M) was added to a mixture solution containing HAuCl4 (200 mL, 0.75 mM) and CTAB (200 mL, 0.2 M). To the above mixture, the 3 h-incubated seed solution (0.48 mL) was added and incubated for 24 h to produce violet-colored CTAB-capped AuNRs. After 24 h of incubation, CTAB-AuNRs were centrifuged at 9000 rpm for 30 min at room temperature (28 °C) to remove the excess CTAB, and the subsequent redispersed solution can be stored for a minimum of 90 days at room temperature (28 °C).35Pd-Coated AuNRs (AuNRs@Pd) and Cu-coated AuNRs (AuNRs@Cu) were prepared from the same initial colloidal solution of AuNRs with an average aspect ratio (η) of 3. In bimetallic nanorods, gold acts as the core and the metal (Pd, Cu) acts as the shell. A mixture of CTAB (5 mL, 30 mM) and AuNRs (5 mL, 0.18 mM) was prepared. To this solution, PdCl2 (1 mM, 1.602 mL) and ascorbic acid (25 mM, 1.602 mL) were added for the preparation of AuNRs@Pd. After 30 min of reaction at 27 °C, the samples were centrifuged at 9000 rpm for 30 min and redispersed in water before characterization. A similar procedure was followed for the preparation of AuNRs@Cu using the precursor CuO.36
2.3. Quantification of protein corona on bimetallic AuNRs
The model protein solutions of HSA and transferrin were prepared in phosphate buffer (10 mM, pH 7.0). 200 μL of the protein with a fixed final concentration of HSA (40 mg mL−1) or transferrin (3 mg mL−1) was added to 200 μL AuNRs (10 mg mL−1). In order to simulate the conditions similar to when AuNRs are utilized for bio-applications, the concentration of AuNRs was lower than the physiological concentration of proteins. The protein nanorod solution was incubated for several time points and centrifuged twice at 7000 rpm for 10 min and redispersed with phosphate buffer. 200 μL of the sample was taken, and 50 μL of 2× Laemmli buffer was added, incubated at 90 °C for 5 min, and centrifuged.37 After each step, the supernatant was collected, and the protein concentration was estimated using the Bradford reagent by reading the absorbance at 595 nm. The protein concentration thus estimated was used for the quantification of hard and soft corona.2.4. Estimation of dox encapsulation efficiency and payload on coronated bimetallic AuNRs by a thermal release method
The dox loaded on the coronated monometallic and bimetallic AuNRs was estimated using a previously established thermal release method. In brief, 100 μL of monometallic or bimetallic AuNRs (5 mg L−1) were taken along with 100 μL of dox with varying final concentrations ranging from 10 to 100 μM and 200 μL of HSA (40 mg mL−1) or transferrin (3 mg mL−1). At concentrations beyond 100 μM of dox, AuNRs were seen to aggregate and lose their stability at the end of 24 h incubation (Table S1, the ESI†). The conjugates were incubated for a period of 24 h under mild stirring at room temperature. The resulting solution contained the dox-loaded protein-coronated nanocarrier. This nanocarrier was heated in a hot water bath at 90 °C for 30 min.19 The bound corona was released from the monometallic and bimetallic AuNRs due to the heat generated. The change in the color of the solution to purple indicated the aggregation of AuNRs. To isolate the payload, the solution was centrifuged at 11000 rpm for 10 min. The supernatant was collected, and the absorbance was read at 485 nm. Using the standard dox curve, the absorbance was equated, and the dox encapsulation efficiency (L.E.) was calculated as the amount of drug present in the NR corona to the amount of the initial loaded drug × 100%.21 Also, the dox payload was calculated as the mass of drug present in the delivery system to the whole mass of the delivery system × 100%. Monometallic and bimetallic AuNRs containing protein corona (either HSA or transferrin) without dox were treated as the respective controls.
2.5. Measurement of dox release
The dox released from the coronated nanocarrier complex was measured by dispersing the sample in buffers of 3 different pH values: acetate buffer (pH 4.6 and 5.3) and phosphate buffer (pH 7.4) under continuous shaking at 37 °C. The coronated dox-loaded monometallic and bimetallic AuNRs were prepared as mentioned in section 2.4. For every 8 h, the pH of the mixture sample was examined and maintained by the addition of HCl as and when required. At a fixed time interval, the mixture samples were centrifuged at 9000 rpm for 10 min. The supernatant was collected, and the absorbance of the released dox was read at 485 nm. The standard curve for dox quantification was previously plotted in the three different buffers that are used in the experiment. The results, thus obtained are expressed as release efficiency, which was calculated as the amount of dox released at a fixed time interval to the amount present in the coronated monometallic and bimetallic AuNRs.Furthermore, the dox release from the coronated dox-conjugated systems was analyzed at different time points (up to 48 h). For obtaining plasma, fresh human blood was obtained from healthy volunteers (aged: 21–25) following the approval from the Institutional Ethical Committee for studies on human subjects (IECH) of VIT University (Ref no: VIT/IECH/014/Jan24.2015). The obtained blood was added to a tube containing EDTA. The resultant mixture was allowed to sit for 15 min and then centrifuged at 5000 rpm for 10 min at 4 °C. The clear supernatant, i.e. the plasma, was then carefully collected and stored at −20 °C until use. The coronated conjugates were dispersed in the plasma and centrifuged at fixed time interval. The absorbance of the released dox in the supernatant was read at 485 nm, using which the release efficiency % was calculated.
2.6. Cytotoxicity assessment of dox-loaded coronated NRs
MCF-7 (human mammary gland carcinoma) cell lines were brought from National Cell Centre Science (NCCS), Pune, India. Confluent cells were obtained by maintaining under sterile conditions in 25 cm2 culture flasks in 5% CO2 incubator at 37 °C. Furthermore, for the cell culture, DMEM medium containing 10% FBS and 1% antibiotic and antimycotic solution was used. Trypsinized cells were mounted on a hemocytometer, after which viable cells in the growth phase were counted using the tryphan blue dye exclusion method.38 The cells were further seeded in 96-well plates at a cell concentration of 1 × 104 cells per well. Following this, the cells were subjected to treatment with phosphate buffer (control), bare, protein-coronated and dox-loaded monometallic and bimetallic AuNRs for two different time points, 24 h and 48 h. The cells were further treated with MTT (5 mg mL−1) and incubated for 4 h in CO2 incubator for the development of purple-colored formazan product. The intensity of the purple color formazan formed was read at 590 nm using an ELISA plate reader after solubilizing it with DMSO (50 μL).262.7. Instrumentation
A high-resolution transmission electron microscope (HR-TEM) (JEOL JEM 2100, Japan) was used to capture the morphological changes of monometallic and bimetallic AuNRs (10 mg L−1) before and after the addition of proteins (HSA, transferrin) at an operating voltage of 200 kV. The grid preparation for the TEM analysis was done by sonicating the colloidal solution for 10 min and followed by the dropwise addition on the carbon-coated copper grids and dried. An ELISA plate reader (BioTek, PowerWave XS2, USA) was used to estimate the proteins using the Bradford assay. UV-2600 UV–visible spectrophotometer (Shimadzu, Tokyo, Japan) in the range, 200–900 nm, was used to analyse the UV–visible spectra of the as-synthesized AuNRs and bimetallic AuNRs. 90 Plus Particle Analyzer (Brookhaven Instruments Corporation, USA) was used for dynamic light scattering (DLS) and zeta potential measurements of the samples.2.8. Statistical significance
All the experiments were done in triplicates (n = 3), and the results are reported as mean ± SD. For statistical significance, two-way ANOVA with Bonferroni's multiple comparison test in GraphPad Prism was used.3. Results and discussion
3.1. Preliminary characterization of monometallic AuNRs and bimetallic AuNRs (AuNRs@Pd and AuNRs@Cu)
The initial characterization of the as-prepared CTAB-AuNRs and bimetallic AuNRs was done using UV–visible spectroscopy, indicating the AuNR peaks: TSPR at 516 nm and LSPR at 761 nm; AuNRs@Pd peaks: TSPR at 502 nm and LSPR at 710 nm; AuNRs@Cu peaks: TSPR at 515 nm and LSPR at 704 nm (Fig. S1, the ESI†). The peaks of AuNRs depicted the distinct surface plasmon resonance of Au about the aspect ratio of nanorods.39 The deposition of Pd and Cu on the AuNRs was found to strongly broaden the LSPR bands.The broadening of the LSPR peak is associated with the modification of dielectric characteristics and the shape of the nanorods upon Pd and Cu deposition. These results can be correlated with previous reports on Au@Pt nanorods.40 The optical absorption in the Pd and Cu layer causes the broadening of the LSPR peak as the imaginary part of the dielectric function of Pd and Cu in this spectral range is greater compared to that of gold.41 The well-dispersed images of AuNRs and bimetallic AuNRs could be noticed under TEM (Fig. 1). The length and breadth of AuNRs, AuNRs@Pd, and AuNRs@Cu were found to be (43.8 ± 5.4 nm, 14.7 ± 1.6 nm), (41.5 ± 5.6 nm, 12.9 ± 1.7 nm), and (42.1 ± 5.1 nm, 13.1 ± 1.6 nm), respectively. Hence, the aspect ratio of bimetallic AuNRs (AuNRs@Pd: 3.2; AuNRs@Cu: 3.1) was only slightly higher than that for bare AuNRs (3.0) based on the measurements taken for the average of 100 nanorods using software ImageJ. Additionally, the smooth-rounded edges of bare NRs changed to sharp edges following coating with Pd/Cu. The percentage composition of the metals is observed using TEM-EDAX. The composition and peaks of monometallic and bimetallic AuNR signals are shown in Fig. 1 (inset), which indicates the presence of additional peaks corresponding to the bimetals (Pd or Cu) upon coating the AuNRs.
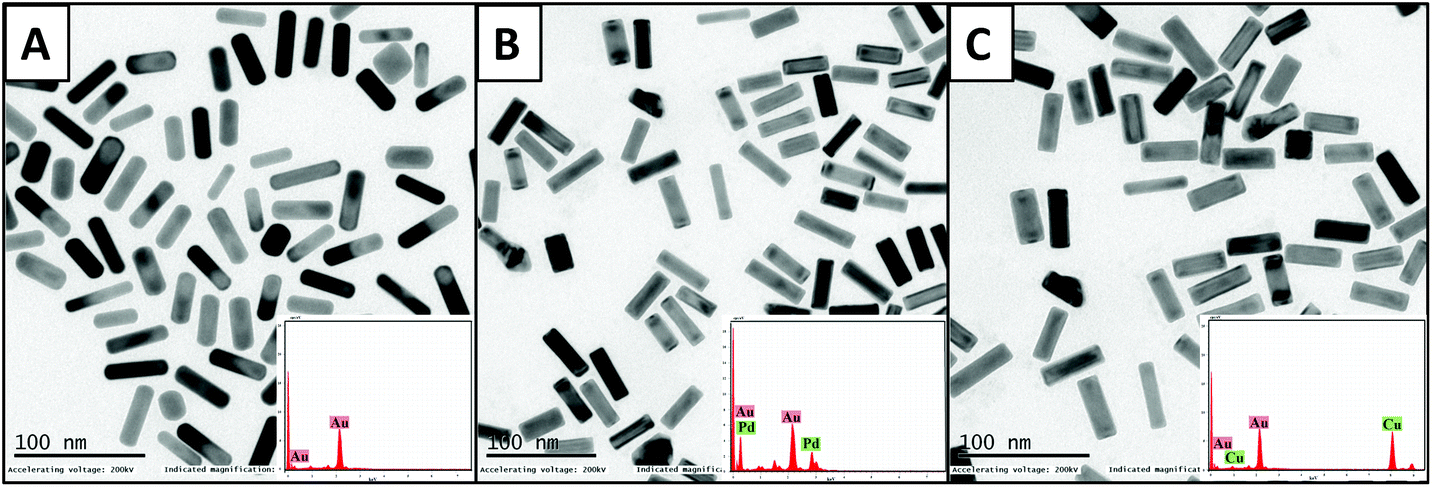 | ||
| Fig. 1 TEM images with their respective EDAX peaks (inset) for (A) monometallic AuNRs, (B) AuNRs@Pd, and (C) AuNRs@Cu. | ||
3.2. Estimation of soft and hard corona based on the individual protein adsorption on monometallic AuNRs and bimetallic AuNRs
The protein adsorption study was done on monometallic AuNRs and bimetallic AuNRs with HSA and transferrin for various time periods, 0, 0.5, 1, 4, 8, 12, and 24 h (Fig. 2). The study helped us to conclude the time point at which maximum adsorption occurred with monometallic AuNRs and bimetallic AuNRs with respect to different proteins. The protein corona formation was maximum at a time point of 1 h, beyond which there was no significance in the variation of protein adsorption. The study was done using the physiological concentrations of these proteins, and hence, varying protein concentrations i.e. HSA (40 mg mL−1) and transferrin (3 mg mL−1) were taken. Thus, rather than the quantified proteins, the percentage of protein bound has been indicated. The time-based study was done by measuring the protein content in the supernatant immediately after the incubation and first centrifugation. Due to the higher availability of HSA, maximum adsorption was seen for HSA rather than transferrin for AuNRs@Pd and AuNRs@Cu when compared to monometallic AuNRs. In addition, among the two proteins used, HSA has a free thiol group on Cys-34 and contributes to ∼80% of the total free thiols in the plasma, which is an important factor contributing to its high binding reactivity.42 HSA (pI value: 4.3–5.7) is negatively charged under physiological conditions (pH 7.4),43 and thus can show higher electrostatic attraction towards bimetallic AuNRs@Pd and AuNRs@Cu when compared to bare AuNRs. Transferrin (pI value: 6.14–6.63)43 has lesser negative charge at pH 7.4 and moderately lower abundance than HSA, hence showing a similar adsorption pattern i.e. higher binding affinity to bimetals when compared to monometallic AuNRs. However, the extent of adsorption was lower for transferrin in comparison with HSA.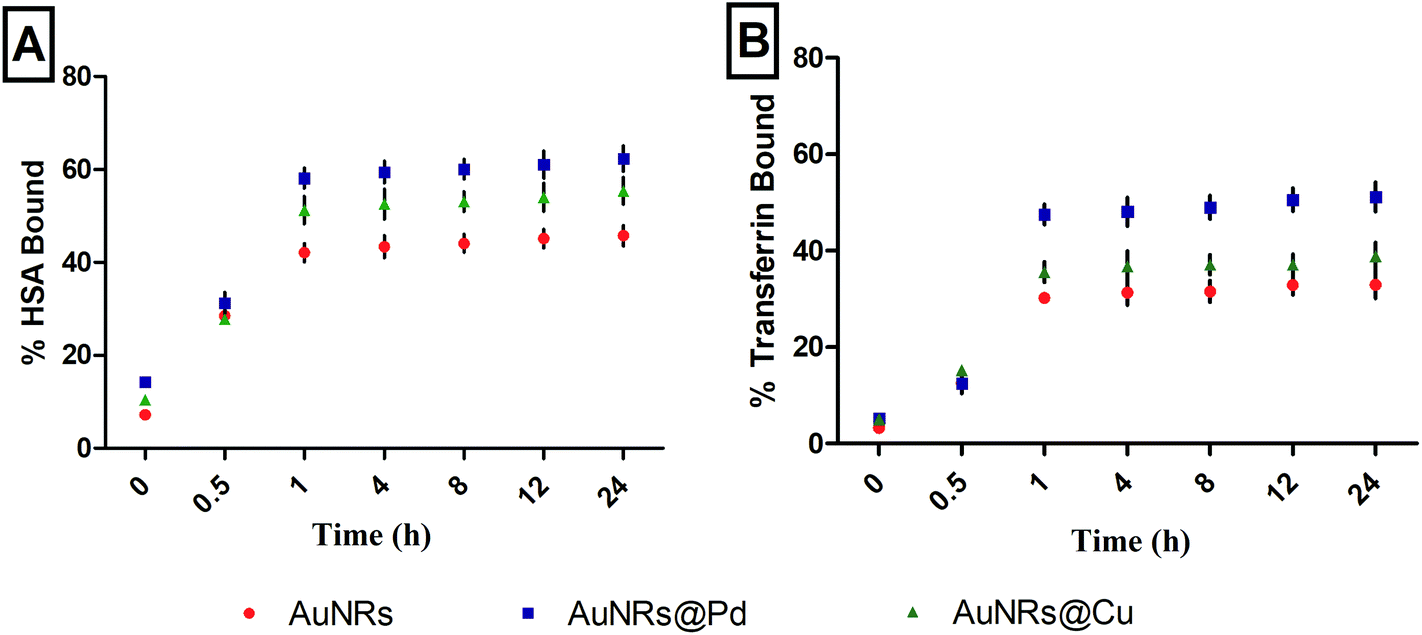 | ||
| Fig. 2 Percentage adsorption of (A) HSA and (B) transferrin over different time points (0, 0.5, 1, 4, 8, 12, and 24 h) on AuNRs and bimetallic AuNRs (AuNRs@Pd, AuNRs@Cu). | ||
3.3. Mean hydrodynamic diameter and zeta potential studies
The particle sizes of monometallic AuNRs and bimetallic AuNRs (AuNRs@Pd and AuNRs@Cu) were observed to be 44.29 ± 0.08 nm, 50.39 ± 2.21 nm, and 48.8 ± 1.92 nm, respectively. The mean hydrodynamic diameters for the bimetallic AuNRs were observed to be slightly higher when compared to monometallic AuNRs due to the adsorptive bimetal coating. Additionally, a higher mean hydrodynamic size compared to TEM-based measurements can be attributed to the hydration layer surrounding AuNRs. Upon interaction with HSA and transferrin, the percentage increase in the mean hydrodynamic size can be seen in Fig. 3. It was found that HSA adsorption caused the highest increase in the size of bimetallic AuNRs when compared to bare AuNRs. HSA-adsorbed AuNRs@Pd (187.31 ± 2.15 nm) and AuNRs@Cu (160.11 ± 1.65 nm) were observed to be larger than AuNRs (128.55 ± 1.36 nm), which can be interrelated with the protein estimation studies in section 3.2. Transferrin showed the least increase in size, which also holds well in the context of the adsorption pattern observed. Despite least adsorption, the hydrodynamic size was significantly higher for AuNRs@Pd (174.56 ± 1.54 nm) and AuNRs@Cu (162.56 ± 2.14 nm) due to their higher electropositivity when compared to bare AuNRs (125.23 ± 3.02 nm).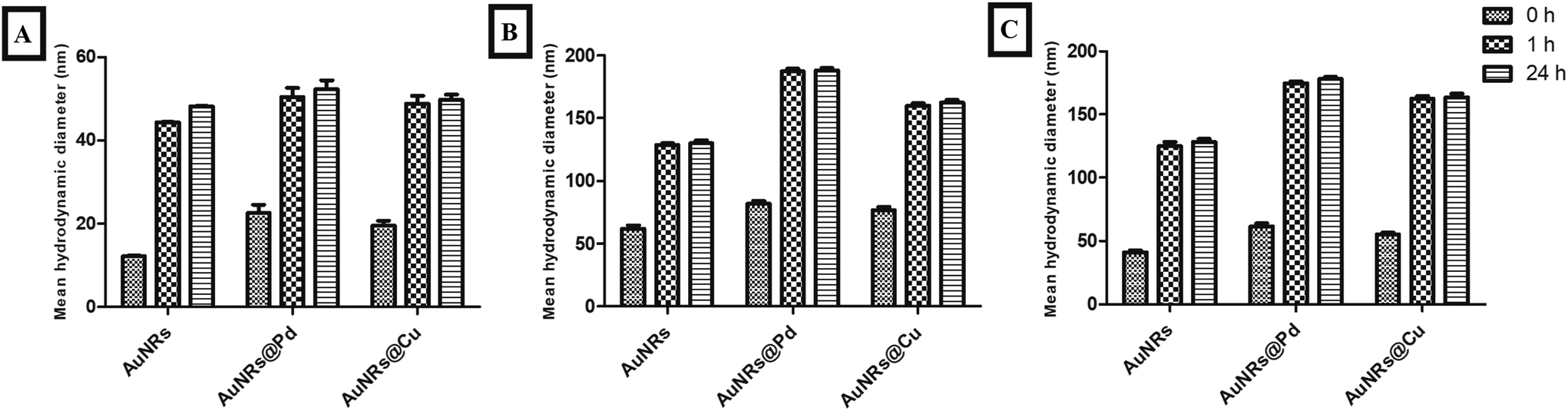 | ||
| Fig. 3 Percentage increase in the size of AuNRs, AuNRs@Pd, and AuNRs@Cu before protein adsorption (A) and after adsorption with HSA (B) and transferrin (C). | ||
The zeta potential measurements for monometallic AuNRs and bimetallic AuNRs (AuNRs@Pd and AuNRs@Cu) were found to be +47.25 ± 3.02, +87 ± 2.65, and +60 ± 2.14 mV, respectively. Since maximum protein adsorption was at 1 h time point, beyond which no significant changes were observed, the zeta potential measurements were done for 0 h, 1 h, and the last time point, 24 h. Table 1 represents the charge distribution on the bare AuNRs and bimetallic AuNRs upon adsorption with proteins, HSA and transferrin, at three different time points. The zeta potential values for HSA and transferrin were found to decrease the positive charge of the monometallic AuNRs and bimetallic AuNRs, but the decrease in the positive charge was relatively higher for HSA due to its higher physiological concentration, better reactivity (due to free thiol groups) and higher negative charge (pI value of 4.3–5.7) at physiological pH.43 Upon interaction with HSA and transferrin, the highly electropositive bimetallic AuNRs became less positive, whereas monometallic AuNRs showing lesser positive charges became negative. The protein adsorption studies and zeta potential measurements showed similar correlation, which was reported earlier by other groups.44,45
| AuNR samples | Zeta potential measurements (mV) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Time | |||||||||
| 0 h | 1 h | 24 h | |||||||
| No protein | HSA | Transferrin | No protein | HSA | Transferrin | No protein | HSA | Transferrin | |
| AuNRs | +43.74 ± 2.64 | +10.57 ± 1.85 | +20.34 ± 2.45 | +47.25 ± 3.02 | −20.34 ± 1.54 | −5.75 ± 2.87 | +46.14 ± 2.42 | −21.47 ± 1.75 | −6.45 ± 2.71 |
| AuNRs@Pd | +74.14 ± 2.13 | +44.12 ± 2.54 | +50.31 ± 2.14 | +87 ± 2.65 | +11 ± 1.84 | +18 ± 3.24 | +85.17 ± 2.95 | +12.2 ± 2.01 | +20.41 ± 2.97 |
| AuNRs@Cu | +58.47 ± 3.14 | +41.28 ± 2.64 | +4.29 ± 2.32 | +60 ± 2.14 | +9 ± 2.36 | +15 ± 1.65 | +60.24 ± 2.65 | +10.47 ± 2.64 | +16.74 ± 2.21 |
3.4. Quantification of bound corona
The protein adsorption was observed to be maximum at 1 h, and hence, the corona formation was quantified for monometallic AuNRs and bimetallic AuNRs at this optimized time point (Table 2). The protein corona formation can be considered to occur in two layers, the soft corona (loosely bound layer) and the hard corona (protein layer tightly bound to the nanosurface). The samples were centrifuged, and the protein was quantified in the supernatant after each step using the Bradford assay.26 The corona layers were estimated to be higher for HSA and transferrin due to higher physiological concentration. The total bound HSA corona on AuNRs@Pd was estimated to be 9.14 ± 0.10 mg mL−1, and that for AuNRs@Cu was estimated to be 7.39 ± 0.07 mg mL−1, which was significantly higher (p < 0.001) than the monometallic AuNRs. In the case of transferrin, the corona on AuNRs@Pd was estimated to be 1.25 ± 0.03 mg mL−1, and that for AuNRs@Cu, was 0.83 ± 0.03 mg mL−1, which was significantly higher (p < 0.001 and p < 0.01, respectively) than the monometallic AuNRs. Among the two bimetallic AuNRs, AuNRs@Pd showed slightly higher protein coronation when compared with AuNRs@Cu due to the higher electropositive nature of palladium. The higher electropositive nature could enhance the electrostatic attraction between bimetallic AuNRs and the proteins, HSA and transferrin, which are negative at physiological pH. The SEP of Au (Au3+ + 3e− → Au) is +1.5 V. Compared with monometallic AuNRs, the adsorptive efficiency improves when AuNRs are coated with a second bimetal like Pd or Cu.46 The higher electropositive nature of Pd can be explained by the higher standard electrode potential (SEP) of Pd (E° = +0.987 V; Pd2+ + 2e− → Pd) when compared to the SEP of Cu (E° = +0.337 V; Cu2+ + 2e− → Cu).47 The forces other than electrostatic interactions that play a significant role in the protein adsorption include, covalent binding via cysteine groups, entropy-driven binding, and hydrophobic interactions. The Vroman effect on the NP–protein complex formation i.e. initial NP adsorption by proteins having higher mobility, followed by their replacement with higher affinity protein can yield NPs having an incomplete surface coverage with either single or multiple protein corona layers surrounding it.48,49| AuNRs | Measured protein concentration (mg mL−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Initial | Unbound | Soft corona | Hard corona | Total corona | ||||||
| HSA | Transferrin | HSA | Transferrin | HSA | Transferrin | HSA | Transferrin | HSA | Transferrin | |
| ***, **, and * are the statistical significance values for protein concentrations measured for bimetallic AuNRs in comparison with the respective monometallic form (i.e. p = 0.001, 0.01, 0.05, respectively). | ||||||||||
| AuNRs | 39.6 ± 1.07 | 2.98 ± 0.10 | 33.2 ± 0.73 | 2.26 ± 0.01 | 3.25 ± 0.06 | 0.45 ± 0.01 | 2.23 ± 0.05 | 0.12 ± 0.02 | 5.48 ± 0.05 | 0.57 ± 0.01 |
| AuNRs@Pd | 39.58 ± 0.61 | 2.9 ± 0.20 | 30.22 ± 0.19*** | 1.61 ± 0.10 | 6.01 ± 0.11*** | 1.0 ± 0.02*** | 3.13 ± 0.09*** | 0.25 ± 0.04 | 9.14 ± 0.10*** | 1.25 ± 0.03*** |
| AuNRs@Cu | 39.98 ± 0.39 | 2.94 ± 0.10 | 32.49 ± 0.12 | 2.08 ± 0.05 | 5.15 ± 0.12*** | 0.63 ± 0.01* | 2.24 ± 0.02 | 0.2 ± 0.04 | 7.39 ± 0.07*** | 0.83 ± 0.03** |
The HSA- and transferrin-adsorbed monometallic AuNRs and bimetallic AuNRs indicated aggregation in the TEM images (Fig. 4). The corona layer formed on the AuNRs can act as a sponge to load different drugs, and hence, taking into consideration the variation in corona formation between the bare AuNRs and bimetallic AuNRs, further studies on the drug encapsulation efficiency and drug payload estimation were continued.
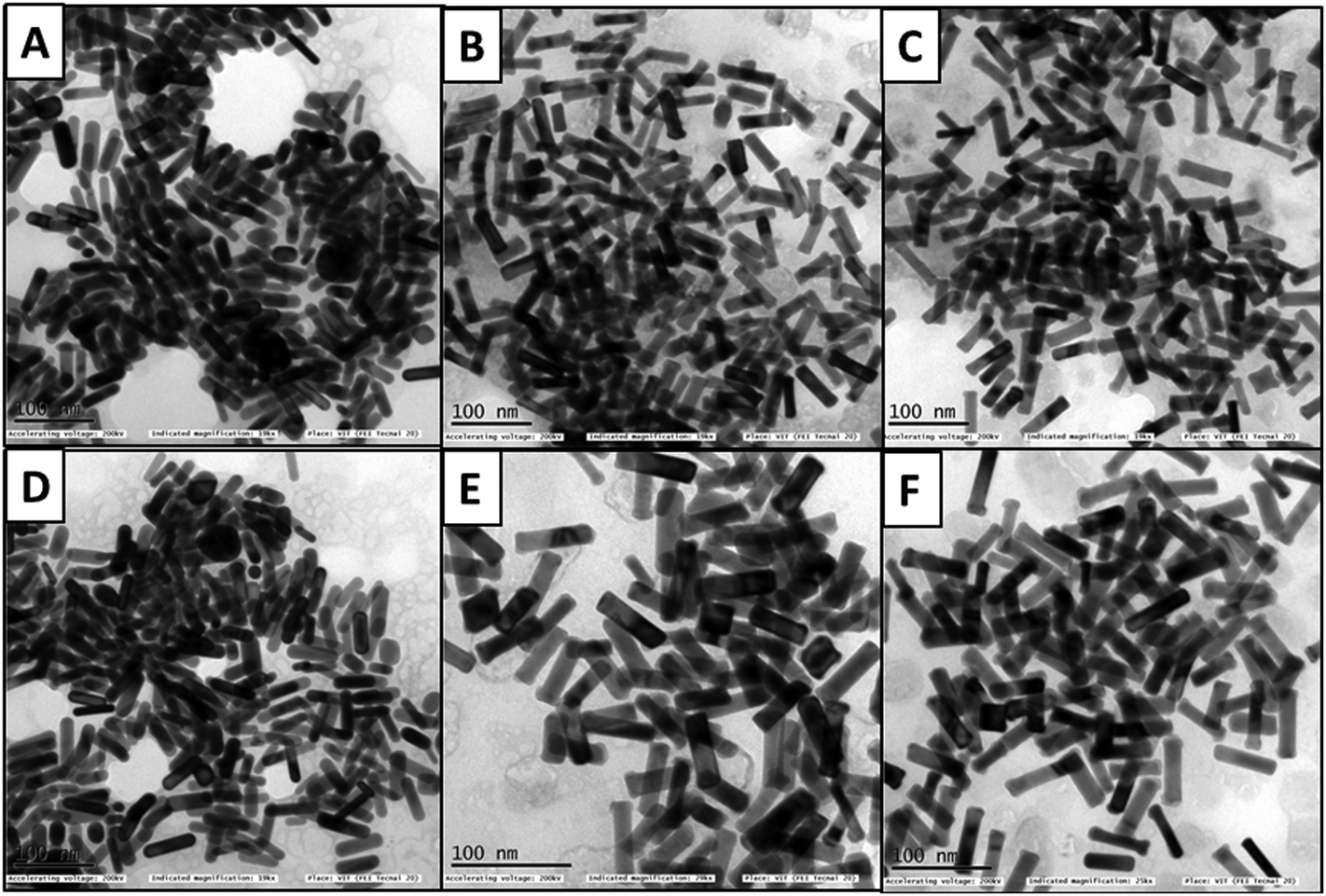 | ||
| Fig. 4 TEM images of AuNRs, AuNRs@Pd, and AuNRs@Cu after adsorption with HSA (A–C), and transferrin (D–F), respectively, after 1 h incubation. | ||
3.5. Analyzing the effect of corona on dox encapsulation
The different metal coating on monometallic AuNRs could result in significant differences in the corona formation. As the dox molecules are more bound to the corona rather than the AuNR surface, it is important to evaluate the role of the adsorbed corona in holding the payload. Initially, the dox binds to the soft corona because of its small size, and later, the dox can get rearranged to be tightly bound to the hard corona.19 Thus, a thermal heat treatment was done to release the dox molecule bound to the corona and to quantify the payload, based on which the encapsulation efficiency and drug payload were calculated. The dox encapsulation efficiency (Fig. 5) observed for different concentrations of dox (i.e. 10, 20, 50, and 100 μM) was evaluated to estimate dox concentration at which maximum encapsulation efficiency could be achieved. The release of the payload from the dox-loaded coronated samples was seen when the samples were heated at 90 °C for 30 min. This could lead to the aggregation of AuNRs, which was indicated by the formation of the purple color, resulting in the disassembly of the bound corona with the subsequent release of the payload from AuNRs. An increase in further heating time did not enhance the release of dox significantly (results not shown). For different loading concentrations of dox (10–100 μM), the L.E. was seen to increase, and the highest encapsulation efficiency was obtained for 100 μM (Fig. 5).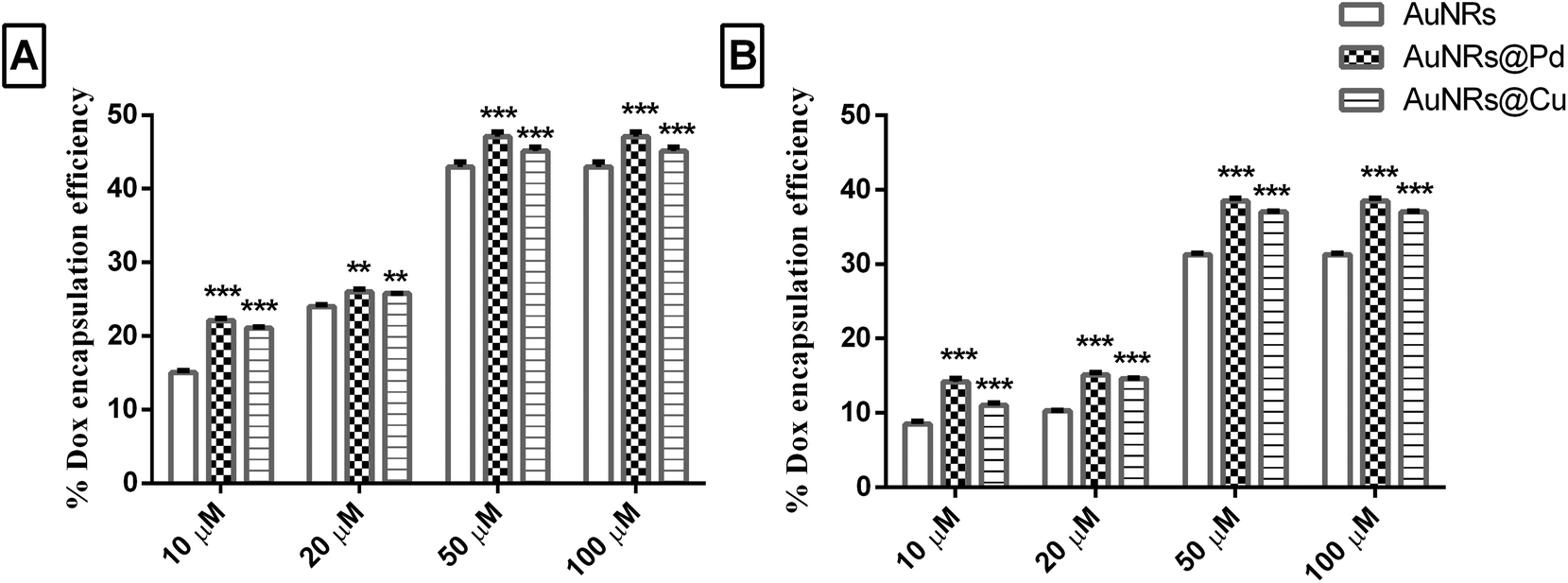 | ||
| Fig. 5 Doxorubicin encapsulation efficiency % of (A) HSA and (B) Transferrin on coronated nanoparticles for different initial dox concentrations: 10, 20, 50, and 100 μM. ***, **, and * are the statistical significance for encapsulation efficiency % assessed for bimetallic AuNRs in comparison with the respective monometallic form (i.e. p = 0.001, 0.01, 0.05, respectively). | ||
The highest encapsulation efficiency was obtained for bimetallic AuNRs for all the loading concentrations of dox tested. At 100 μM dox loading concentration, a significant increase (p < 0.001) in the dox encapsulation efficiency (47.08 ± 0.65% and 45.10 ± 0.5%) was seen for HSA-coronated AuNRs@Pd and AuNRs@Cu, respectively, as compared to HSA-coronated monometallic AuNRs (42.97 ± 0.66%). In the case of transferrin, a significant increase (p < 0.001) in the dox encapsulation efficiency was seen for transferrin-coronated AuNRs@Pd (38.48 ± 0.39%) and AuNRs@Cu (37.01 ± 0.14%) when compared to transferrin-coronated monometallic AuNRs (31.26 ± 0.17%). Higher dox encapsulation efficiency for the highly electropositive bimetallic AuNRs could be attributed to the more protein corona on the AuNRs coated with metals (higher protein corona and thereby dox encapsulation efficiency on AuNRs@Pd > AuNRs@Cu due to the more electropositive nature of Pd)47 when compared to monometallic AuNRs having lesser protein coronation.
A significantly lower L.E. (p < 0.001) was seen for transferrin when compared to HSA due to their lesser abundance and reduced reactivity (Fig. 5). The calculated dox payload has also been indicated in Fig. S2 (the ESI†), which suggested a significant increase in payload for transferrin-coronated conjugates compared to HSA-coronated counterparts. Taking into consideration that large amounts of therapeutic molecules can be held by the protein corona, the possible interpretation for the varying amounts of payload on the monometallic AuNRs and bimetal AuNRs could be due to the difference in the amount of total bound corona.50
3.6. Dox release from protein-coronated monometallic and bimetallic AuNRs under pH triggers and in blood plasma
When the drug-loaded nanocarrier is administered into the body, it can encounter different physiological fluids that can alter the drug release from the nanocarrier. The release behavior of the drug for HSA- and transferrin-coronated AuNRs (monometallic and bimetallic) was studied over a period of 48 h under the effect of different pH triggers (Fig. 6). Neutral pH (pH 7.4) was selected to mimic the normal physiological plasma, whereas different acidic pH conditions were selected to mimic the endosomal (pH 4.9–6.0) and lysosomal (pH 4.5–5.0) environment.51 At the end of 48 h, a substantial amount of drug (more than 50%) was released for all the coronated particles. An inverse relation pattern could be observed between the drug release % and pH trigger, wherein a decrease in the pH of the medium boosted the drug release. The least amount of drug was released by the coronated particles at pH 7.4. This reduced drug release at higher pH could be explained by the reduced solubility of dox i.e. acidic pH of 4.6 could maximize dox solubility and promote a stronger and faster release profile. More abundance of HSA when compared to transferrin caused an increase in the encapsulation profile of dox on the former. Hence, at the end of 48 h, the dox release observed for the HSA-coronated bimetallic AuNRs (AuNRs@Pd: 61.98 ± 2.12% and AuNRs@Cu: 72.76 ± 3.12%) was lesser and more gradual when compared with transferrin-coronated bimetallic AuNRs (AuNRs@Pd: 64.03 ± 2.14% and AuNRs@Cu: 73.93 ± 2.84%). This decrease in the release profile for HSA-coronated nanostructures could be due to the higher amount of protein corona on it when compared to transferrin-coronated counterparts. A similar observation, wherein the presence of the soft corona protein like HSA impeded the passive release of the payload from the nanoparticle surface, was reported by Cifuentes-Rius et al. (2013).50 Additionally, AuNRs@Pd indicated a more gradual dox release when compared with the lesser electropositive AuNRs@Cu due to the slightly higher protein coronation, which can act as a reservoir for the drug molecules. A burst release effect can be witnessed for monometallic AuNRs due to the relatively lesser amount of protein corona. Here, around 50% of the dox was released by 1 h, attaining a maximum value of 76.37 ± 3.45% and 78.81 ± 3.1% for HSA- and transferrin-coronated monometallic AuNRs at the end of 48 h. A similar study also reported that the presence of corona on the drug-loaded nanocarrier could reduce the burst effect.20 This indicates that the drug release is not dependent on the amount of drug loaded on the corona, but rather was influenced by the amount of protein bound.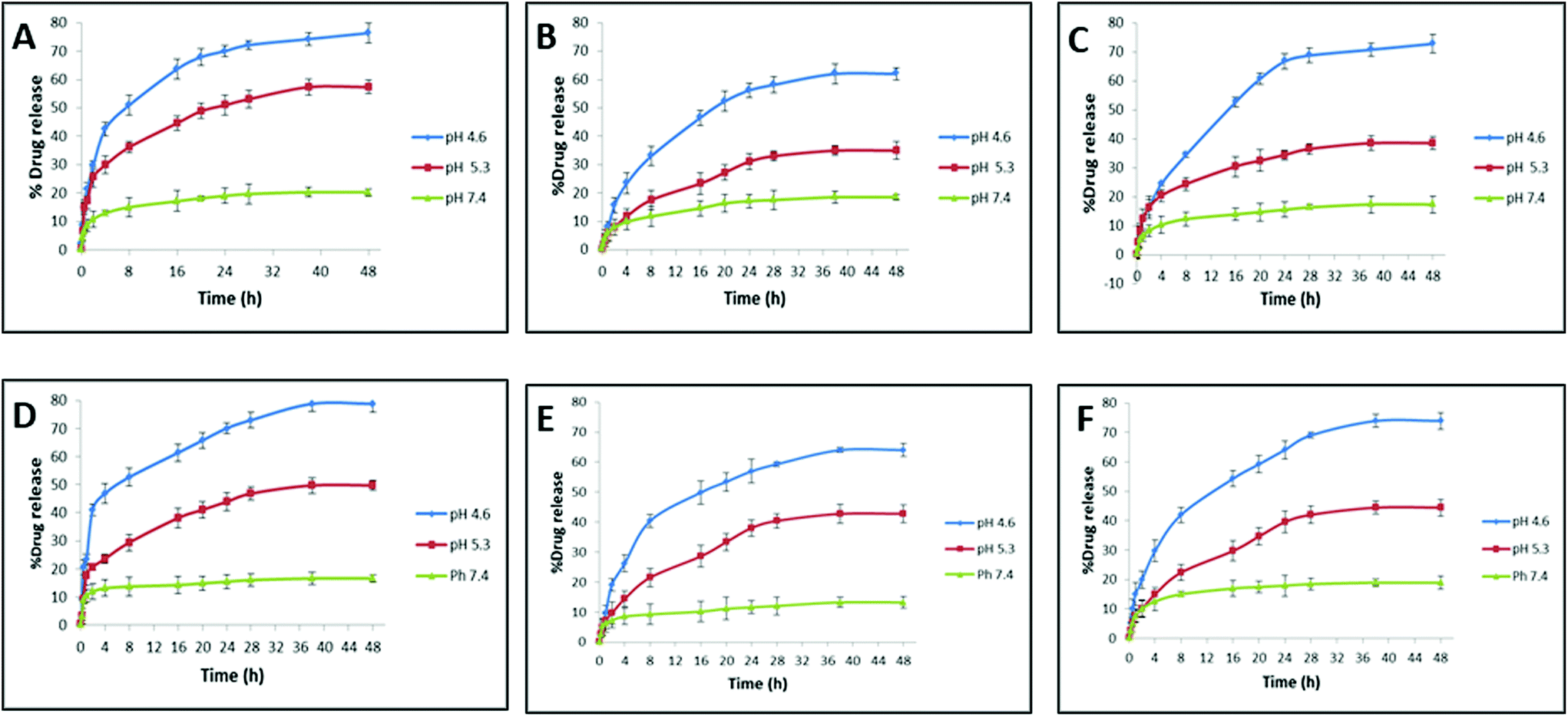 | ||
| Fig. 6 Doxorubicin release % under the effect of different pH triggers from AuNRs, AuNRs@Pd, and AuNRs@Cu coronated with HSA (A–C), and transferrin (D–F), respectively. | ||
The dox release behavior from the coronated NPs was also studied in the presence of multiple proteins in the plasma (Fig. 7). It was seen that at the end of 48 h, the dox release % for HSA- and transferrin-coronated monometallic AuNRs were 26.08 ± 1.37% and 29.87 ± 1.42%, respectively. For HSA- and transferrin-coronated bimetallic AuNRs, the dox release was observed to decrease (Pd: 15.86 ± 1.74% and 19.24 ± 1.53%; Cu: 21.62 ± 1.04% and 25.15 ± 1.34%). Since the physiological pH of blood plasma is ∼7.4, the results were compared with dox release % noted for the phosphate buffer at pH 7.4. It could be suggested that due to the inherent Vroman effect, there was a dynamic exchange of bound corona proteins from the surface of the nanorods, and this in turn led to a higher release of the associated dox molecules in plasma when compared to incubation in phosphate buffer (pH 7.4).
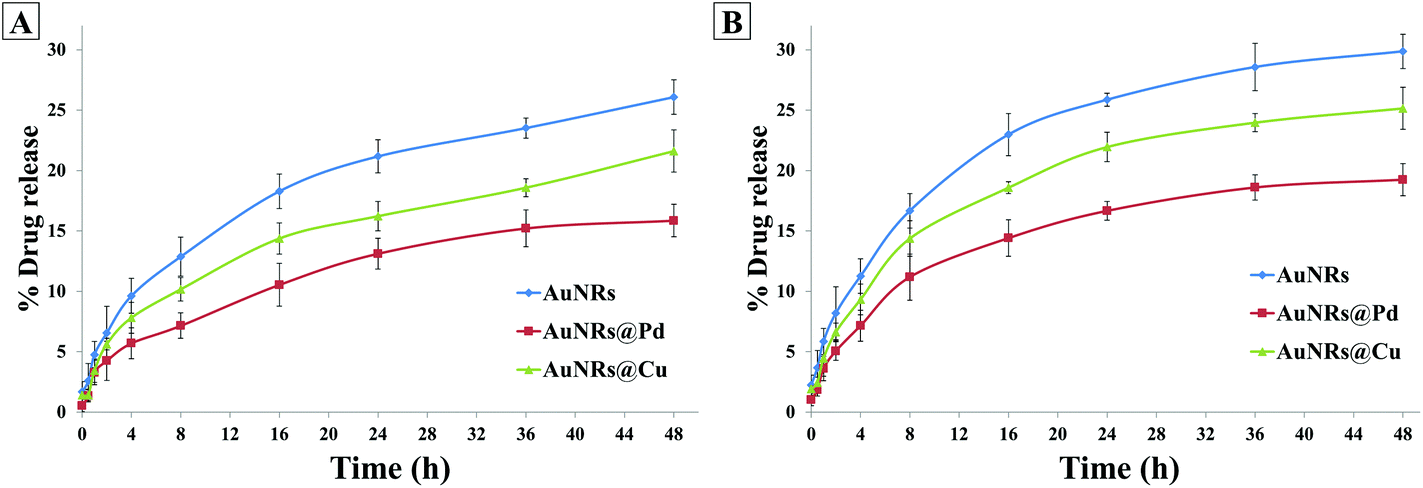 | ||
| Fig. 7 Doxorubicin release % for AuNRs, AuNRs@Pd, and AuNRs@Cu coronated with (A) HSA and (B) transferrin over different time points. | ||
3.7. Influence of bimetallic coating on cellular toxicity
To further use the coronated dox-loaded AuNRs for the nanocarrier system, the in vitro cellular toxicity of the prepared AuNRs and conjugates was analyzed against the MCF-7 cell line. Earlier reports suggested that the presence of protein corona lowers cellular toxicity, possibly due to the reduction in the direct interaction of the NPs with the cells and also due to a possible increase in the native size of the NPs, thereby minimizing their uptake.52 Bare monometallic and bimetallic AuNRs were seen to have nearly the same toxicity. The higher toxicity observed could be due to the concentration of AuNRs used and the presence of the surfactant (CTAB) on their surface which causes membrane disruption by inducing pore formation in them (Fig. 8).26 The formation of HSA corona around the nanoparticles significantly reduced the toxicity (p < 0.001). Following 24 h incubation, bare AuNRs that had a comparatively lesser amount of HSA corona showed considerably higher toxicity as compared to AuNRs@Pd (p < 0.001) and AuNRs@Cu (p < 0.05). For transferrin-coronated AuNRs, a slight decrease (p < 0.001) in cell viability was seen when compared to HSA-coronated AuNRs, which could be attributed to the fact that the presence of transferrin possibly enhances the cellular uptake of respective NRs by transferrin receptors present on the cell surface.53 Further loading of dox on both HSA- and transferrin-coronated AuNRs increased their toxicity. The cell viability % values for different concentrations of free doxorubicin (0.25, 0.5, 1, 1.5, 2, and 2.5 μM) for 24 and 48 h have been provided in Fig. S3 (the ESI†). AuNRs@Pd, which had the highest dox-loading efficiency, significantly lowered (p < 0.001) the cellular viability from 92.59 ± 1.84% and 82.82 ± 0.31% for HSA-coronated AuNRs@Pd and transferrin-coronated AuNRs@Pd, to 60.87 ± 1.83% and 54.68 ± 1.06% for the respective dox-loaded conjugates. Furthermore, it can be seen that both bare and coronated AuNRs did not show a significant increase in cellular toxicity upon incubation for 48 h; however, a significant decrease in cell viability (p < 0.001) for dox-loaded conjugates could be seen following 48 h incubation, suggesting that there was a sustained release of the bound drug over the given incubation period. Fig. 9 provides the graphical representation of the preparation of bimetallic AuNRs, their higher dox loading capability compared to the monometallic form due to the higher protein coronation, and finally, their impact on dox release and in vitro toxicity, thereby emphasizing their potential drug delivery applications.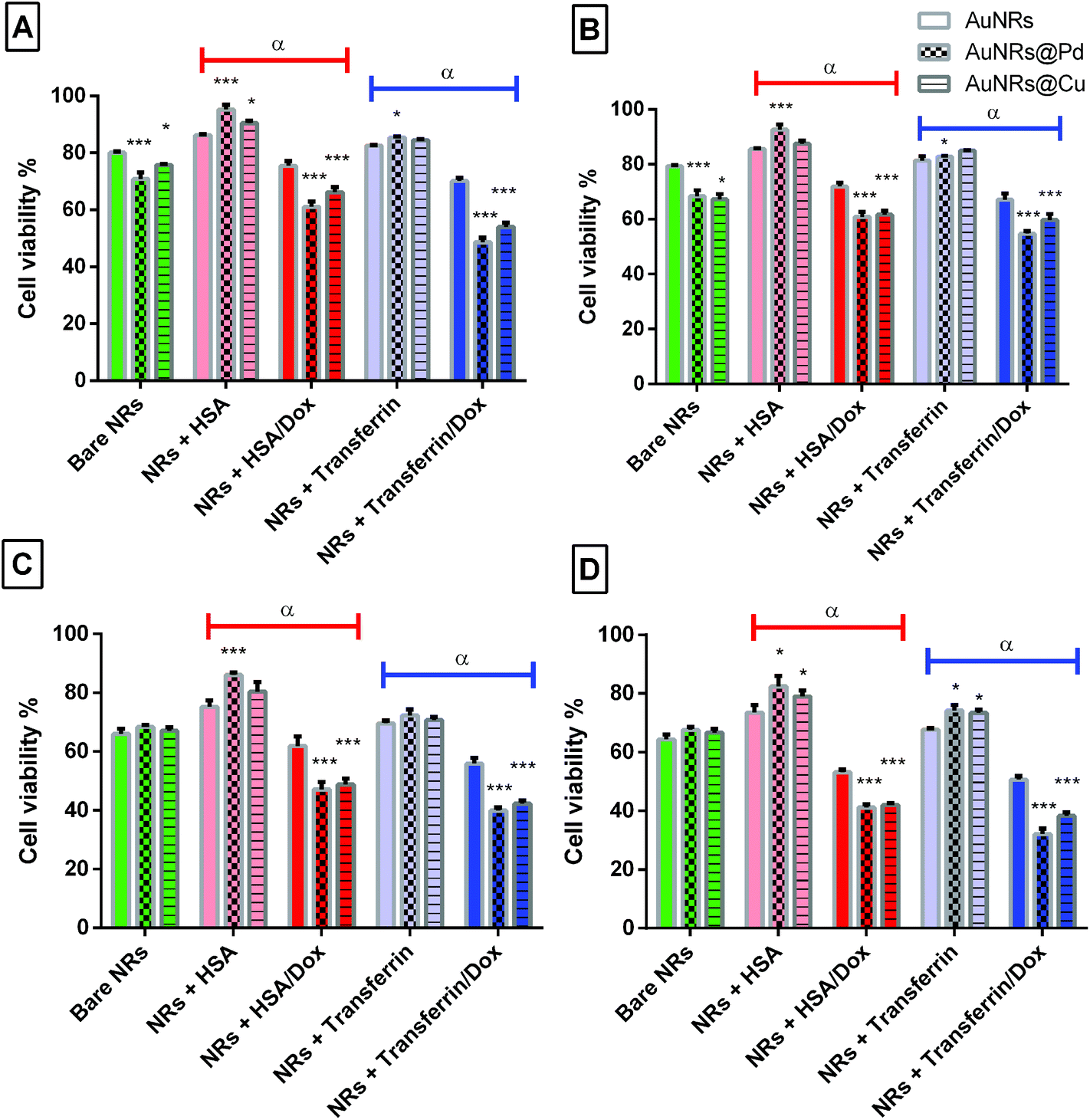 | ||
| Fig. 8 Cell viability % for monometallic and bimetallic AuNRs upon protein coronation and dox loading for (A–B) 24 h and (C–D) 48 h for two different loaded dox concentrations (0.5 and 1 μM), respectively. ***, **, and * are the statistical significance for the bimetallic cell viability assessed in comparison with the respective monometallic form (i.e. p = 0.001, 0.01, 0.05, respectively), while α (p = 0.001) is the statistical significance for the cell viability assessed for dox-loaded AuNRs in comparison with the respective protein-coronated form. | ||
 | ||
| Fig. 9 Schematic representation of the preparation of bimetallic AuNRs and their impact on protein coronation, dox loading and release, and in vitro toxicity compared to monometallic AuNRs. | ||
4. Future perspectives
Considering the multiple molecule binding ability of proteins, the corona on the surface of such bimetallic nanostructures can be exploited for the simultaneous loading of multiple therapeutic molecules and their release.21 Furthermore, a follow-up study can be carried out, wherein the thickness of bimetallic coating (Pd/Cu) can be varied, and the influence of shell thickness on protein corona formation and drug loading can be evaluated. Additionally, the comparative effect of other electropositive bimetal coatings on protein coronations can also be explored for drug delivery applications. Though the thermal property of AuNRs could be reduced following bimetallic coating due to their lower plasmonic resonance, their higher dox loading efficiency upon protein coronation can compensate for the loss of the opto-thermal property when used as a combinatorial therapeutic agent. Yang et al. (2018) have explored dox-loaded bimetallic Au@Pt nanospheres for chemo-photothermal combinational therapy.545. Conclusions
The preparation of bimetallic AuNRs (AuNRs@Pd, AuNRs@Cu) and the formation of individual protein corona, HSA and transferrin, could help in the development of a versatile nanocarrier for the encapsulation of the drug. Using a time-based study, the maximum adsorption of protein was found to occur within 1 h. Based on the bimetal coating, charge and the abundance of protein tested, the corona layer quantification was observed to vary. The protein quantification data for HSA and transferrin was seen to follow a pattern similar to the percentage increase in the mean hydrodynamic size i.e. AuNRs@Pd > AuNRs@Cu > AuNRs. This higher adsorption of protein corona could have resulted due to the electropositive nature of the bimetals with Pd showing higher electropositivity due to its higher standard electrode potential and also due to their higher peptide binding. Apart from electrostatic interactions, other forces may also play a role in the protein adsorption on AuNRs, such as covalent binding via cysteine groups, entropy-driven binding, and hydrophobic interactions. The HSA and transferrin protein corona layer formed on AuNRs can be used to carry small drug molecules. In particular, the efficiency of the bound corona for the encapsulation of dox on monometallic and bimetallic AuNRs was tested. Distinct dox binding/release efficiency of the bound corona was seen in the case of bimetallic AuNRs, i.e. their higher protein corona layer was seen to cause a proportional enhancement in the drug encapsulation efficiency. The substantial amount of bound corona on bimetallic AuNRs reduced the overall burst effect of dox release, and thereby, could induce a gradual dox release. From the study, it can be concluded that the coating of a bimetal over AuNRs can enhance the adsorption of protein, which can in turn improve the drug payload and the sustained release of the drug. The improved dox release for lower pH suggested that these systems could be triggered to release dox only after reaching the endosome or lysosome of the target cells. When tested in human plasma, the dox release was slightly higher due to the dynamic exchange of the bound proteins containing dox on the AuNR surface. Thus, the improved dox loading of bimetallic AuNRs provided an added advantage when coronated with transferrin as they would exhibit enhanced cell uptake specifically for cancerous cells due to the higher number of transferrin receptors on the cancerous cell surface. In summary, this study creates a basis for the future development of a better and enhanced drug delivery system, which can be used in future studies to understand the in vivo behavior of the nanotherapeutic system.Conflicts of interest
There are no conflicts to declare.References
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, V. Z. Matson, A. Javadi, W. Xu, Y. Yang, Y. Zhang, J. W. Engle, R. J. Nickles and W. Cai, Theranostics, 2012, 2, 757 CrossRef CAS PubMed.
- C. Wang, Z. Ma, T. Wang and Z. Su, Adv. Funct. Mater., 2006, 16, 1673–1678 CrossRef CAS.
- A. Malugin and H. Ghandehari, J. Appl. Toxicol., 2010, 30, 212–217 Search PubMed.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387 CrossRef CAS.
- N. Zhang, D. Zhu, F. Li, H. Hua, X. Tian and Y. Zhao, J. Biomed. Nanotechnol., 2017, 13, 134–143 CrossRef CAS.
- P. Changenet-Barret, T. Gustavsson, D. Markovitsi, I. Manet and S. Monti, Phys. Chem. Chem. Phys., 2013, 15, 2937–2944 RSC.
- C. M. Alexander, M. M. Maye and J. C. Dabrowiak, Chem. Commun., 2011, 47, 3418–3420 RSC.
- R. Venkatesan, A. Pichaimani, K. Hari, P. K. Balasubramanian, J. Kulandaivel and K. Premkumar, J. Mater. Chem. B, 2013, 1, 1010–1018 RSC.
- M. Mahmoudi, S. Laurent, M. A. Shokrgozar and M. Hosseinkhani, ACS Nano, 2011, 5, 7263–7276 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- A. Sasidharan, J. E. Riviere and N. A. Monteiro-Riviere, J. Mater. Chem. B, 2015, 3, 2075–2082 RSC.
- N. A. Monteiro-Riviere, M. E. Samberg, S. J. Oldenburg and J. E. Riviere, Toxicol. Lett., 2013, 220, 286–293 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, B. W. Neun, S. Man, X. Ye, M. Hansen, A. K. Patri, R. M. Crist and S. E. McNeil, Nanomedicine, 2014, 10, 1453–1463 CrossRef CAS PubMed.
- R. García-Álvarez, M. Hadjidemetriou, A. Sánchez-Iglesias, L. M. Liz-Marzán and K. Kostarelos, Nanoscale, 2018, 10, 1256–1264 RSC.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- E. Mahon, A. Salvati, F. B. Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS PubMed.
- J. C. Y. Kah, J. Chen, A. Zubieta and K. Hamad-Schifferli, ACS Nano, 2012, 6, 6730–6740 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, R. Sakhtianchi, B. Müller, K. Landfester, D. Crespy and M. Mahmoudi, Colloids Surf., B, 2014, 123, 143–149 CrossRef CAS PubMed.
- S. Shahabi, L. Treccani, R. Dringen and K. Rezwan, Nanoscale, 2015, 7, 16251–16265 RSC.
- E. L. L. Yeo, P. S. P. Thong, K. C. Soo and J. C. Y. Kah, Nanoscale, 2018, 10, 2461–2472 RSC.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS PubMed.
- J. Zhou, Z. Cao, N. Panwar, R. Hu, X. Wang, J. Qu, S. C. Tjin, G. Xu and K.-T. Yong, Coord. Chem. Rev., 2017, 352, 15–66 CrossRef CAS.
- S. A. Alex, N. Chandrasekaran and A. Mukherjee, J. Mol. Liq., 2017, 248, 703–712 CrossRef CAS.
- N. Toshima and Y. Wang, Chem. Lett., 1993, 22, 1611–1614 CrossRef.
- J. F. Sanchez-Ramirez, G. Díaz, A. Vázquez and U. Pal, J. New Mater. Electrochem. Syst., 2005, 8, 127–131 CAS.
- J. G. Speight, Lange's handbook of chemistry, McGraw-Hill, New York, 2005 Search PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- J. A. Kellum and P. W. G. Elbers, Stewart's Textbook of Acid-Base, 2009 Search PubMed.
- D. Chakraborty, P. Chauhan, S. A. Alex, S. Chaudhary, K. Ethiraj, N. Chandrasekaran and A. Mukherjee, J. Mol. Liq., 2018, 268, 335–342 CrossRef CAS.
- H. Heinz, B. L. Farmer, R. B. Pandey, J. M. Slocik, S. S. Patnaik, R. Pachter and R. R. Naik, J. Am. Chem. Soc., 2009, 131, 9704–9714 CrossRef CAS PubMed.
- Y. Li, P. Wang, C. Hu, K. Wang, Q. Chang, L. Liu, Z. Han, Y. Shao, Y. Zhai and Z. Zuo, Sci. Rep., 2018, 8, 1939 CrossRef PubMed.
- S. A. Alex, S. Rajiv, S. Chakravarty, N. Chandrasekaran and A. Mukherjee, Mater. Sci. Eng., C, 2017, 71, 744–754 CrossRef CAS.
- M. F. Cardinal, D. Mongin, A. l. Crut, P. Maioli, B. Rodríguez-González, J. Pérez-Juste, L. M. Liz-Marzán, N. Del Fatti and F. Vallée, J. Phys. Chem. Lett., 2012, 3, 613–619 CrossRef CAS PubMed.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Biomaterials, 2016, 75, 295–304 CrossRef CAS PubMed.
- J. Conde, J. Rosa, M. Jesús and P. V. Baptista, Biomaterials, 2013, 34, 2516–2523 CrossRef CAS PubMed.
- L. Vigderman, B. P. Khanal and E. R. Zubarev, Adv. Mater., 2012, 24, 4811–4841 CrossRef CAS PubMed.
- M. Grzelczak, J. Perez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2007, 111, 6183–6188 CrossRef CAS.
- P. Johnson and R. Christy, Phys. Rev. B: Solid State, 1974, 9, 5056–5070 CrossRef CAS.
- M. J. Torres, L. Turell, H. Botti, L. Antmann, S. Carballal, G. Ferrer-Sueta, R. Radi and B. Alvarez, Arch. Biochem. Biophys., 2012, 521, 102–110 CrossRef CAS PubMed.
- G.-C. Funk, Wien. Klin. Wochenschr., 2007, 119, 390–403 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanomedicine, 2009, 5, 106–117 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS.
- G. Sharma, A. Kumar, S. Sharma, M. Naushad, R. P. Dwivedi, Z. A. ALOthman and G. T. Mola, J. King Saud Univ., Sci., 2017 DOI:10.1016/j.jksus.2017.06.012.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- M. Raoufi, M. J. Hajipour, S. M. K. Shahri, I. Schoen, U. Linn and M. Mahmoudi, Nanoscale, 2018, 10, 1228–1233 RSC.
- J. Piella, N. G. Bastús and V. Puntes, Bioconjugate Chem., 2016, 28, 88–97 CrossRef PubMed.
- A. Cifuentes-Rius, H. de Puig, J. C. Y. Kah, S. Borros and K. Hamad-Schifferli, ACS Nano, 2013, 7, 10066–10074 CrossRef CAS PubMed.
- Y. Yan, J. Fu, X. Liu, T. Wang and X. Lu, RSC Adv., 2015, 5, 30640–30646 RSC.
- K. Saha, D. F. Moyano and V. M. Rotello, Mater. Horiz., 2014, 1, 102–105 RSC.
- N. A. Monteiro-Riviere, M. E. Samberg, S. J. Oldenburg and J. E. Riviere, Toxicol. Lett., 2013, 220, 286–293 CrossRef CAS PubMed.
- Q. Yang, J. Peng, Y. Xiao, W. Li, L. Tan, X. Xu and Z. Qian, ACS Appl. Mater. Interfaces, 2017, 10, 150–164 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01127c |
| ‡ Both the authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |