
Self-assembling peptides cross-linked with genipin: resilient hydrogels and self-standing electrospun scaffolds for tissue engineering applications†
Raffaele
Pugliese
 ab,
Mahboubeh
Maleki
a,
Ronald N.
Zuckermann
c and
Fabrizio
Gelain
*ab
ab,
Mahboubeh
Maleki
a,
Ronald N.
Zuckermann
c and
Fabrizio
Gelain
*ab
aIRCSS Casa Sollievo della Sofferenza, Unità di Ingegneria Tissutale, Viale Cappuccini 1, San Giovanni Rotondo, FG 71013, Italy. E-mail: f.gelain@css-mendel.it
bCenter for Nanomedicine and Tissue Engineering (CNTE), ASST Grande Ospedale Metropolitano Niguarda, Piazza dell'Ospedale Maggiore 3, Milan 20162, Italy
cThe Molecular Foundry, Lawrence Berkeley National Laboratory, 1 Cyclotron road, Berkeley, California 94720, USA
First published on 19th November 2018
Abstract
Self-assembling peptides (SAPs) are synthetic bioinspired biomaterials that can be feasibly multi-functionalized for applications in surgery, drug delivery, optics and tissue engineering (TE). Despite their promising biocompatibility and biomimetic properties, they have never been considered real competitors of polymers and/or cross-linked extracellular matrix (ECM) natural proteins. Indeed, synthetic SAP-made hydrogels usually feature modest mechanical properties, limiting their potential applications, due to the transient non-covalent interactions involved in the self-assembling phenomenon. Cross-linked SAP-hydrogels have been recently introduced to bridge this gap, but several questions remain open. New strategies leading to stiffer gels of SAPs may allow for a full exploitation of the SAP technology in TE and beyond. We have developed and characterized a genipin cross-linking strategy significantly increasing the stiffness and resiliency of FAQ(LDLK)3, a functionalized SAP already used for nervous cell cultures. We characterized different protocols of cross-linking, analyzing their dose and time-dependent efficiency, influencing stiffness, bioabsorption time and molecular arrangements. We choose the best developed protocol to electrospin into nanofibers, for the first time, self-standing, water-stable and flexible fibrous mats and micro-channels entirely made of SAPs. This work may open the door to the development and tailoring of bioprostheses entirely made of SAPs for different TE applications.
Introduction
The field of self-assembling peptides (SAPs) has undergone an outstanding growth since the early 1990s, when Shuguang Zhang serendipitously discovered a segment of a yeast protein capable of self-assembling.1 Since then, self-assembling peptides have been used as hemostat solutions,2–4 nanocarriers of drugs,5–8 bone fillers9–11 and wound healers,12–14 but also as injectable scaffolds for the regeneration of injured heart,15–17 cartilage and18,19 nucleus pulposus.20,21 The field of peptide scaffolds is still expanding at an accelerating pace, and a few clinical trials are currently assessing their potential for remineralization in dental repair22,23 and their sealing properties in post-operative lymphorrhea following pelvic surgery.24SAPs are made of amino acids and self-assemble into various nanostructures (nanofibers, nanotubes and nanovesicles) upon exposure to shifts of pH, temperature and osmolarity. These structures can mimic the natural peptide-based extracellular matrix (ECM), and can also display multiple, specific functional motifs capable of interacting with cells25,26 and proteins27,28 (if designed to do so). Furthermore, SAPs are synthetic, pathogen-free, and biodegradable and mainly used at low concentrations in water (less than 8%, w/v), all desirable properties for translational therapies in the future.
Despite their extensive use in different areas of materials science and regenerative medicine, several applications of SAPs are still precluded because of their poor mechanical stability, mainly arising from the non-covalent interactions within supramolecular assemblies.29 As a matter of fact, most of the SAP-based therapies focused their efforts on the biochemical composition of the target tissue to be regenerated, while the biomechanical properties were still out-of-reach. Indeed, scaffold biomechanical properties should be reliably tuned in order to presumably match those of the target tissue and to expand their potential range of applications. In addition, the improvement of both architectural (e.g. nano-topography) and mechanical (e.g. elasticity and stiffness) features could make these materials tailorable via electrospinning (ES), a versatile nano- and micro-fabrication technique widely used in TE,30–32 but rarely used with SAPs because of their intrinsic limited molecular weight and viscoelastic properties. So far, electrospun SAP fibers have been produced in biopolymer blends or alone as coatings.33,34
We recently demonstrated how cross-linking could be a viable strategy to fill some of these goals;35 however, the biocompatibility of the cross-linking agent we used (i.e. sulfo-SMCC) as well as the specific SAP sequence requirements (comprising both cysteine and lysine residues) may pose some limitations in the future.
Genipin, extracted from Gardenia jasminoides Ellis fruits, is a naturally occurring cross-linking agent used in Chinese medicine36,37 that specifically reacts with primary amines of peptides and proteins. To date, it has been employed for the cross-linking of gelatin,38–40 chitosan41,42 and Fmoc-triphenyalanine hydrogel43 with far less cytotoxicity (5000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) than glutaraldehyde. It was demonstrated to reverse the clinical symptoms of diabetes44 and to exert anti-inflammatory,45–48 anti-oxidative,49 neuroprotective (in Alzheimer's diseases),50,51 and anti-cancer effects in colorectal,52,53 breast,54 and prostate55 cancers, as well as pancreatic adeno-,56 hepato-57,58 and gastric carcinomas.59–61
000 times) than glutaraldehyde. It was demonstrated to reverse the clinical symptoms of diabetes44 and to exert anti-inflammatory,45–48 anti-oxidative,49 neuroprotective (in Alzheimer's diseases),50,51 and anti-cancer effects in colorectal,52,53 breast,54 and prostate55 cancers, as well as pancreatic adeno-,56 hepato-57,58 and gastric carcinomas.59–61
We here introduced and optimized a genipin (gp) cross-linking reaction to stabilize biomimetic hydrogels made of an FAQ(LDLK)3 (i.e. NH2-FAQRVPPGGGLDLKLDLKLDLK-CONH2) SAP. By using genipin, we significantly increased the stiffness and failure stress of FAQ(LDLK)3, both increments dependent on the dose and exposure-time to the cross-linking agent. While still preserving the SAP secondary structure arrangements, the stress-relaxation properties of the hydrogels were modulated by using different genipin concentrations and delivery strategies. These enhancements allowed us to electrospin cross-linked SAPs to obtain, for the first time, water-stable (but bioabsorbable), self-standing and flexible nano- and micro-fibrous mats and micro-channels entirely made of functionalized SAPs. Literally, this work will open the door of SAPs to the field of electrospun scaffolds, yielding biomimetic synthetic bioprostheses with tunable resilience, bioabsorption times and/or bioactivities, for several TE applications like heart patches, skin dressing, blood vessel implants and so on.
Experiment
Peptide synthesis and purification
Peptides were synthesized using fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis with a Liberty-Discovery (CEM) microwave automated synthesizer and were purified as previously described.26 Briefly, the synthesis was carried out with 0.5 M HBTU in DMF and 2 M DIEA in NMP as the activator and activator base solutions respectively. The peptide was synthesized by using a rink-amide resin (0.5 mmol g−1). Fmoc-protected amino acids were dissolved at 0.2 M in DMF, the deprotection solution for Fmoc-group removal was 20% (v/v) 4-methylpiperidin in DMF. The removal of side-chain protecting groups and cleavage was obtained with a TFA:TIS:H2O (95:2.5:2.5) cocktail. A three-glycine spacer, interposed between the FAQRVPP functional motif26 and the (LDLK)3 self-assembling backbone, constitutes the FAQ(LDLK)3 peptide. The raw peptide was precipitated using cold ethyl ether and then lyophilized (Labconco). The resulting raw peptide was purified using a Waters binary high-performance liquid chromatography (HPLC) apparatus (>95%). The molecular weight of the purified peptide was determined via single quadrupole mass detection (Waters LC-MS Alliance-3100). The purified peptide powder was subsequently dissolved in 0.1 M HCl solution in order to remove possible TFA salts.
Cross-linked SAP hydrogel preparation
:5 v/v; pH = 7.4) and filtered (0.22 μm pore size). Genipin cross-linked hydrogels were prepared by adding 86 mM and 170 mM of genipin to 50 μl of peptide (5% w/v) and incubating at 37 °C for 72 h. At the end of the reaction, the unreacted genipin in the supernatant was removed by aspiration with a vacuum pipette, and the resulting cross-linked hydrogel was washed and suspended in 1.5 ml of PBS for 1 h. Washes were repeated 5 times before use.
:PBS:EtOH (47.5:47.5:5 v/v) to the purified peptide powder to achieve a final 5% (w/v) concentration. The mixed solution was sonicated for 30 min and incubated at 37 °C for 72 h.
:EtOH (95:5 v/v) to the purified peptide powder to achieve a final 1% (w/v) concentration. The mixed solutions were then sonicated for 30 min and incubated at 37 °C for 24 h. See Table S1 in the ESI† for all abbreviations used in the manuscript.
All experiments were performed over a wide range of concentrations and treatment times to assess genipin-dosing regimens influencing the mechanical properties of SAP hydrogels.
Electrospinning of cross-linked SAP scaffolds
Electrospun nanofibers (es-FAQ(LDLK)3/gp) were fabricated using in situ partial cross-linked FAQ(LDLK)3/gp (14.6 mM, 72 h) with a single-jet customized electrospinning system. The spinning solvent to dissolve FAQ(LDLK)3/gp (14.6 mM, 72 h) was 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Sigma Aldrich). The ES solution was sonicated for 30 min, and after 1 h of rest it was employed for ES. All sample preparations were carried out under ambient conditions. The peptide solution at 35% (w/v) in 100% HFIP was placed in a syringe with a 29-gauge needle (1 ml BD™ U-100 insulin syringe) mounted on a syringe pump (NewEra NE-1000). The syringe pump was used to provide a constant stream of solution at the tip of the needle. An electrical potential of 12–15 kV was applied to the needle by using a power supply (Spellman High Voltage, 0–30 kV). A 33G micro-needle (Hamilton needle, stainless steel, outer diameter = 210 μm, length 51 mm) as the collector was fixed to a rotating arm of a non-conductive mandrel (length 12 cm, ≈40 rpm) and the grounding electrode was indirectly (using a copper ring) placed at the end of the micro-needle in order to provide a constant speed for a uniform fiber collection around the micro-needle. Spinning was performed under ambient conditions (23–25 °C) with a humidity range of 35–40%. A seamless micro-conduit of es-FAQ(LDLK)3/gp was obtained when the selected micro-channel was post-treated (PT). The optimized procedure involved exposure to the vapor phase of 2.5 mM NaOH in PBS (PT-I) for 3 days to slightly activate genipin while stabilizing the morphological stability of the fibers, followed by 1 day of immersion of 170 mM genipin in H2O:EtOH (90:10 v/v) (PT-II). Subsequently, the cross-linking process was terminated by soaking the micro-channel in PBS for 1 day (PT-III) to remove any trace amounts of used ethanol. All the PT phases were performed at 37 °C under a 95% O2/5% CO2 atmosphere (Caron's Oasis™ CO2/O2 Incubator, Marietta, OH, USA).
Rheological tests
The rheological properties of the assembled nanostructures were investigated with a controlled stress AR-2000ex Rheometer (TA Instruments). A truncated cone–plate geometry (acrylic truncated diameter, 20 mm; angle, 1°; truncation gap, 34 μm) was used. All measurements were performed at 25 °C (Peltier cell) and all samples were tested one day after dissolution at 1% and 5% (w/v) concentrations. To monitor the sol–gel transition and to evaluate the storage (G′) and loss (G′′) moduli increase as a function of time, a time-sweep test at a constant angular frequency (ω = 1 Hz) was carried out for 15 h. For FAQ(LDLK)3 (without genipin) the assembly was triggered by adding PBS laterally to the peptide solution positioned in the 34 μm cone–plate truncation gap, while for the genipin cross-linked peptides (FAQ(LDLK)3/gp) both assembly and cross-linking were triggered by adding genipin in PBS:EtOH (95:5 v/v). Afterwards, in the linear viscoelastic region, a frequency sweep test (0.1–1000 Hz, 1% strain) was performed to measure G′ and G′′ of the scaffolds. Stress/strain sweeps were performed (0.01%–1000%) to identify the limits of the linear viscoelastic region and the maximum strain and stress to which the sample can be subjected. On the assembled genipin cross-linked hydrogels (0.5 cm diameter, 2 mm thickness, equilibrated in PBS for 24 h), stress-relaxation tests were performed at 10% strain that was held constant and with a deformation rate of 1 mm min−1. The load was recorded as a function of time. Stress-relaxation data and τ½ (time for the initial stress of the material to halve throughout the test) were evaluated using a two-element Maxwell–Weichert linear viscoelastic model.62 Lastly, a temperature ramp was recorded as a function of G′ (Trate = 5 °C min−1, 1% strain, ω = 1 Hz). Each experiment was performed in triplicate.
2,4,6-Trinitrobenzene sulfonic acid (TNBSA) assay
The cross-linking degree of FAQ(LDLK)3/gp was assessed using TNBSA (Thermo Scientific), which specifically reacts with primary amine groups in peptides/proteins, yielding a yellow-colored product that can be monitored at 335 nm. Primary amine groups are quantified based on molar absorptivity using the extinction coefficient of TNBSA (10000 M−1 cm−1).63 100 μl of peptides were treated with TNBSA (50 μl, 0.01% w/v in 0.1 M NaHCO3, pH = 8.5) and incubated for 2 h at 37 °C. To stop the reaction, 25 μl of 1 N HCl was added to the solution. The measurements were performed via a 1 cm spectrophotometric cuvette using an Infinite M200 Pro plate reader (Tecan). For each sample, the primary amine groups were estimated at 0, 2, 4, 6, 8, 24, 32, 48, 72 and 120 hours. 0.1 M NaHCO3 was used as the blank.64 All measurements were performed with the Origin™8 software using Logistic fitting.
Fluorescence measurements
The effect of the formation of blue pigmentation that qualitatively indicates genipin cross-linking65 was measured through fluorescence intensity as a function of cross-linking time. The FAQ(LDLK)3 peptide was used as a control. Genipin fluorescence intensity was recorded using an Infinite M200 PRO plate reader (Tecan) with λex = 590 nm and λem = 630 nm, from 0 h to 120 h at 25 °C. All experimental runs were repeated three times per each different timepoint. Background fluorescence was subtracted from each spectrum. Lastly, the spectra were averaged and processed with the Origin™8 software using Boltzmann fitting.Fourier transform infrared spectroscopy (FTIR) analysis
The FTIR analysis of the assembled nanostructures was performed on FAQ(LDLK)3 dissolved at a concentration of 1% and 5% (w/v) in distilled water, and on the DCR, ISCR and ISPCR peptides after a 24 h incubation at 4 °C. All spectra were recorded in attenuated total reflection (ATR) using a PerkinElmer Spectrum 100 spectrometer. A 2 μl aliquot of the FAQ(LDLK)3 peptide solution and of the films obtained via DCR, ISCR and ISPCR were deposited on the reflection diamond element and allowed to evaporate. Twenty acquisitions were recorded for each spectrum, using the following conditions: 4 cm−1 spectrum resolution, 25 kHz scan speed, 1000 scan co-addition and triangular apodization. All the obtained spectra were reported after ATR correction, smoothing and automatic baseline correction using the Origin™8 software. Each sample preparation was repeated three times.X-ray diffraction (XRD) analysis
XRD patterns were obtained using a multiple-wavelength anomalous diffraction and monochromatic macromolecular crystallography beamline (8.3.1) located at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory, as previously described.35,66 All samples were prepared 24 h before the analysis and stored at 4 °C. On the day of the analysis the FAQ(LDLK)3 peptide solution at a concentration of 1% and 5% (w/v) was centrifuged at 12000 rpm for 10 min and the resulting concentrated pellet was placed on a MicroRT™ X-ray Capillary (Mitegen) and allowed to evaporate. Genipin cross-linked peptides were placed directly on the MicroRT™ X-ray Capillary and allowed to evaporate. The data were analyzed with FIT2D67 and processed with the Origin™8 software.
Atomic force microscopy (AFM) analysis
AFM images were captured in tapping mode by using a Multimode Nanoscope V system (Digital Instrument, Veeco), using single-beam silicon cantilever probes (Veeco RFESP MPP-21100-10, cantilever f0, resonance frequency 59–69 kHz, constant force 3 N m−1). The FAQ(LDLK)3 peptide was dissolved in distilled water a day prior to imaging, whereas the DCR and ISCR peptides were measured at 32 h of cross-linking reaction time. Right before the analysis, peptide solutions were diluted to a final concentration of 0.001% (w/v) and deposited on a freshly cleaved mica surface. 2 μl of each solution was kept on the mica for 4 min at RT. Samples were subsequently rinsed with distilled water to remove loosely bound peptides, then dried at RT for 30 min. 100 different nanofibers of approximately 10 independent fields per sample were measured and characterized as previously described.68Scanning electron microscopy (SEM) analysis
Electrospun fibers from mats and microchannels were analyzed with the aid of scanning electron microscopy (SEM; Tescan Vega) after sputter coating with ≈12 nm of Au (Quorum Q150R S), as previously described.34The average and standard deviation of the electrospun fiber diameters were measured by choosing 100 fibers and analyzing them using the ImageJ software (v.1.45s, National Institutes of Health, USA).
Trypsin degradation in vitro
The degradation of genipin cross-linked peptides was investigated by monitoring the scaffold weight loss upon trypsin exposure (10 mg ml−1 PBS, pH = 7.4, Sigma Aldrich) at 37 °C.69 In brief, DCR (100 μl, 5% w/v) and ISPCR (100 μl, 1% w/v) peptides were immersed in trypsin solution (2 ml, pH 7.4) and incubated at 37 °C. The enzyme solution was changed every week. At given time points, the cross-linked peptides were taken out, washed and lyophilized. The degradation ratio was calculated by comparing the weight loss of the scaffolds with the initial dry weight using the following equation:| Degradation (%) = [(W0 − Wt)/W0] × 100 |
Cell morphology, viability and differentiation assays
Human neural stem cells (hNSCs) were obtained according to good manufacturing practice protocols (GMP) in agreement with the guidelines of the European Medicines Agency (EMA) and the Agenzia Italiana del Farmaco (AIFA).70 hNSCs were expanded as previously described.71 Briefly, hNSCs were cultured to a neurosphere state and mechanically dissociated. The day after, the cells were seeded on the surface of FAQ(LDLK)3 hydrogel (3 × 104 cells per cm2), exposed to different concentrations of genipin and cultured for 1 day in vitro (1DIV). The Cultrex-BME substrate (R&D systems; 150 μg mL−1 in basal medium) was used as a positive control substrate. Cell viability was assessed using a commercially available LIVE/DEAD cell viability assay kit (Thermo Fisher Scientific). hNSCs were differentiated in standard differentiation medium on top of es-FAQ(LDLK)3/gp mats and immunostained at 7DIV. Further details are provided in the ESI.†Statistical analysis
The data were processed using the GraphPad Prism 7 software. Rheological tests were assessed by one-way ANOVA followed by Tukey's multi-comparison test. p < 0.05 was considered statistically significant.Results and discussion
Design and fabrication of cross-linked functionalized-SAP hydrogels
LDLK12 peptides containing alternating charged hydrophilic and hydrophobic amino acid residues have a strong propensity for cross-β-sheet formation under physiological conditions of pH and temperature; also, hydrophobic forces and electrostatic and VDW interactions drive their assembly into nanofibers featuring charged hydrophilic and hydrophobic residues respectively exposed to the aqueous environment and packed in a hydrophobic inner pocket.29,72 These designer SAPs can be feasibly functionalized with functional motifs at the C- or -N terminus to obtain biomimetic scaffolds customized for specific applications.26,73,74 Usually, functional motifs are short peptides (2–16 amino acids) linked to the self-assembling backbone through a spacer (comprising a few glycines) to ensure flexibility and exposure to target binding.75 Also, multiple different functionalizations can be included with ease within the same scaffold in order to merge different TE strategies within the same implant (e.g. cell transplants, drug delivery and in vivo tracking).We previously demonstrated that upon exposure to neutral-pH solutions, the designer FAQRVPP-LDLK12 functionalized sequence self-assembles into nanofibers flanked by the added motifs,26 providing functionalized microenvironments with specific biological cues capable of stimulating adult neural stem cell (NSC) adhesion and differentiation. Indeed, the phage display-derived FAQRVPP motif enhanced both neuronal and oligodendroglial differentiations of mNSCs and hNSCs in in vitro 2D cultures and favored nervous regeneration in acute spinal cord injuries (SCI) in rats without altering the physiological inflammatory response following the initial insult.26 However, FAQRVPP-LDLK12 spontaneously forms weak and fragile hydrogel scaffolds with limited flexibility and processing potential, i.e. suitable mainly for the regeneration of soft tissues or as fillers. Conversely, by applying a genipin cross-linking reaction to stabilize the primary amines of FAQRVPP-LDLK12 (described in “Crosslinked SAP hydrogel preparation” paragraph), we obtained scaffolds made of cross-linked functionalized SAPs displaying remarkable improvements in resilience, stress-relaxation and processability. Thus, by using in situ cross-linking reaction (ISCR), we managed to cross-link functionalized hydrogels with tunable mechanical properties (Fig. 1a) suitable for TE applications precluded so far. Vice versa, by using diffusive cross-linking reaction (DCR), we obtained hollow microchannels (Fig. 1b) and flexible membranes resilient to compression (Fig. 1c and Video S1 in the ESI†). Furthermore, as genipin mainly interacts with the lysine-rich backbone sequence, different functionalizations68,76 can be added to the same self-assembling backbone and mixed prior to self-assembly in order to have multi-functionalized scaffolds for multi-target therapies.
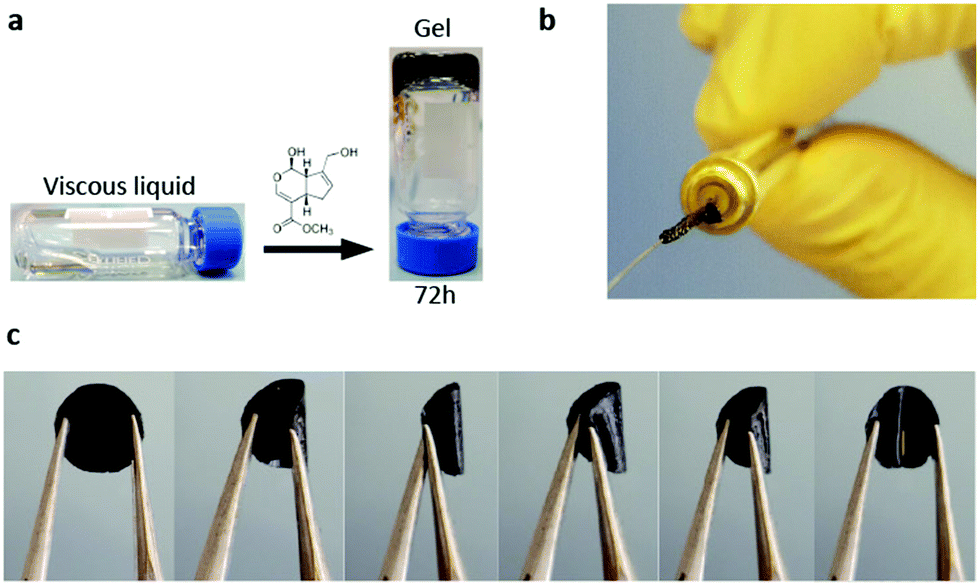 | ||
| Fig. 1 Examples of SAP cross-linked scaffolds: (a) ISCR-FAQ(LDLK)3/gp functionalized hydrogel with tunable mechanical properties after 72 h of reaction; (b) hollow flexible microchannel obtained via DCR; (c) flexible DCR-FAQ(LDLK)3/gp membrane resilient to compression. | ||
Biomechanical characterization
SAP-hydrogels are materials of choice in diverse applications (i.e. scaffolds for regenerative medicine,2,29,71,77–79 carriers for drug delivery,80–82 actuators for optics and fluidics,83 and ECM models for biological studies84), but their usage is often limited by their poor mechanical properties. Indeed, most SAPs are brittle and do not exhibit enough stretchability. Accordingly, it was crucial to assess how the different genipin cross-linking strategies influenced their mechanical strength, thus enlarging their range of potential applications. In the case of hydrogels the most relevant biomechanical features to be characterized are the storage (G′) and loss (G′′) moduli. The former reflects the stiffness trend of the biomaterial, and its increase as a function of time can be indicative of over-structural kinetic and networking processes of the sample, while the latter represents the energy dissipated during the test and correlates with the liquid-like response of the hydrogel. The ratio between G′ and G′′ provides insights into the viscoelastic profile of the tested material, i.e. whether it behaves as a viscous liquid (G′ < G′′) or as an elastic solid (G′ > G′′).85 We investigated two different protocols for the cross-linking reaction, DCR and ISCR (see Experiment for details), in order to elucidate the differences, if any, arising from two ways of administration of genipin to the functionalized SAP. Indeed, DCR could be seen to be interesting for post-assembling treatments of a preformed scaffold while ISCR may look similar to an injectable self-polymerizing agent.By monitoring the temporal evolution of G′ and G′′, the increasing hydrogel stiffness was observed for the FAQ(LDLK)3/gp peptides using DCR (Fig. 2a) and ISCR (Fig. 2b) and for the un-cross-linked FAQ(LDLK)3 peptide (Fig. 2a). The latter reached a plateau after 5 h, displaying an average G′ value of 0.7 kPa, which is typical of soft SAP hydrogels and in agreement with our previously published results.25 By contrast, the storage modulus of genipin cross-linked peptides increased linearly as a function of the reaction time, reaching a plateau after ≈11 h. Close to the equilibrium, the average G′ profile of DCR-FAQ(LDLK)3/gp (86 mM) and DCR-FAQ(LDLK)3/gp (170 mM) was 72 kPa and 110 kPa respectively, while for ISCR-FAQ(LDLK)3/gp (86 mM) and ISCR-FAQ(LDLK)3/gp (170 mM), the average G′ value is shifted respectively to 39.5 kPa and 70 kPa. This increase in the storage modulus was likely due to the formation of covalent cross-links concomitant with self-assembly, both activated by the shift of pH and temperature. Self-assembly is usually a fast process (the macroscopic hydrogelation of SAPs can be a matter of a few seconds), while genipin cross-linking could be much slower (from 24 h to 72 h). Hence, the difference of stiffness between the FAQ(LDLK)3/gp peptides with DCR and ISCR (at the same genipin concentration) can be attributed to the fact that in DCR, where the samples feature pre-assembled β-rich structures, the interactions among genipin and the already self-assembled fibers lead to the formation of an efficiently entangled nanofibrous network with increased values of G′. We hypothesize that in the case of ISCR, genipin is presumably uniformly distributed throughout the sample before self-assembly and the two phenomena are concomitant (even if for a short period of time) after the pH shift (see Experiment for details); this may have influenced the stabilization of the forming β-sheet domains and consequently the mechanical strength of the hydrogel. This consideration will be better investigated in the next sections, but, in our opinion, will need a mandatory confirmation through hybrid atomistic/coarse-grained MD simulations in the near future. In all cases, the trends of G′ and G′′ showed the elastic solid-like behavior (G′) of the samples to be predominant as compared to the viscous component (G′′) (Fig S1a and b in the ESI†). In subsequent frequency-sweep tests, G′ profiles were almost unchanged along the tested frequency range (0.1–100 Hz) for both DCR-FAQ(LDLK)3/gp (Fig. 2aI) and ISCR-FAQ(LDLK)3/gp (Fig. 2bI) peptides, maintaining higher average G′ values for FAQ(LDLK)3/gp (170 mM) as depicted in Fig. 2aII and bII (p < 0.05).
 | ||
| Fig. 2 Rheological characterization of the DCR- and ISCR-FAQ(LDLK)3/gp hydrogels. Peptide solutions were monitored via a 15 h time-sweep test (a–b) immediately after the initiation of both self-assembly of FAQ(LDLK)3 and cross-linking reaction (for DCR and ISCR only), followed by a frequency sweep test (0.1–1000 Hz). (aI) Close to the equilibrium, the FAQ(LDLK)3 peptide had a G′ value of 0.7 kPa typical of soft SAP hydrogels, whereas the average G′ values of DCR-FAQ(LDLK)3/gp (86 mM) and DCR-FAQ(LDLK)3/gp (170 mM) were 72 kPa and 110 kPa respectively. (bI) For ISCR-FAQ(LDLK)3/gp (86 mM) and ISCR-FAQ(LDLK)3/gp (170 mM), the average G′ values were respectively 39.5 kPa and 70 kPa. In both cases cross-linking led to a significant stiffness increase compared with the uncross-linked peptide (aII–bII) (n = 3; p < 0.05). In a strain-failure test (aIII–bIII), the DCR- and ISCR-peptides were less prone to deformation than the soft self-assembled FAQ(LDLK)3 hydrogel, due to their “solid-like” structure. In stress-failure tests (aIV–bIV), DCR- and ISCR-FAQ(LDLK)3/gp showed a substantial failure stress increase compared with FAQ(LDLK)3 (n = 3) (aV–bV), likely thanks to the additional covalent interactions holding up the FAQ(LDLK)3/gp self-assembled nanostructures. | ||
Failure strain and stress tests were also performed within the linear viscoelastic region to assess the hydrogel failure when subjected to a linear strain/stress progression. As expected, FAQ(LDLK)3, yielding a soft hydrogel, showed a strain-to-failure of 8% (Fig. 2aIII), while the FAQ(LDLK)3/gp peptides exhibited less deformation before failure than the soft self-assembled hydrogel, thus leading us to believe that we had a more fragile “solid-like” structure. Indeed, DCR-FAQ(LDLK)3/gp exhibited a strain-to-failure of 2.9% and 3.5% respectively for 86 mM and 170 mM of genipin (Fig. 2aIII), while the ISCR hydrogels with 86 mM and 170 mM of genipin showed a strain-to-failure of 2.7% and 3%, respectively (Fig. 2bIII). In contrast, the stiffening of the peptide chains, due to the cross-links, led to an increment of failure stress compared to the standard soft hydrogel. While failure occurred for a stress of 39 Pa for FAQ(LDLK)3, this was not the case for the DCR-FAQ(LDLK)3/gp peptides, which failed at 1738 Pa and 2576 Pa respectively for 86 mM and 170 mM of genipin (Fig. 2aIV). An increase in failure stress was also observed in ISCR-FAQ(LDLK)3/gp, with failure occurring at stresses of 779 Pa (86 mM) and 1997 Pa (170 mM) (Fig. 2bIV). This manifold difference of stress failure (p < 0.05), between cross-linked and un-cross-linked peptides (Fig. 2aV and bV) can be attributed to the efficient formation of covalent cross-links added to the standard weak intermolecular interactions already present in soft self-assembled hydrogels.
In the same way, we tested the possibility of lowering the concentrations of genipin (ISPCR) to improve the biomechanics of FAQ(LDLK)3 (1% w/v) partially (Fig. 3). For both ISPCR-FAQ(LDLK)3/gp peptides (14.6 mM and 29.6 mM), we found the increments of G′ values to be proportional to genipin concentrations (Fig. 3a) and always higher than that of G′′ (Fig. S1c in the ESI†), confirming a prevailing scaffold elastic component over the viscous one. The average G′ values of ISPCR-FAQ(LDLK)3/gp (14.6 mM) and ISPCR-FAQ(LDLK)3/gp (29.6 mM) were 3.3 kPa and 4.8 kPa respectively (Fig. 3aI). Both ISPCR-FAQ(LDLK)3/gp peptides exhibited a greater G′ value (p < 0.05) compared to FAQ(LDLK)3 (Fig. 3aII). In failure strain and stress tests, both ISPCR-FAQ(LDLK)3/gp peptides exhibited a modest decrease of strain-to-failure (Fig. 3aIII) and an increment of their failure stresses (Fig. 3aIV–aV), likely due to the stiffening of the cross-linked nanofibers.
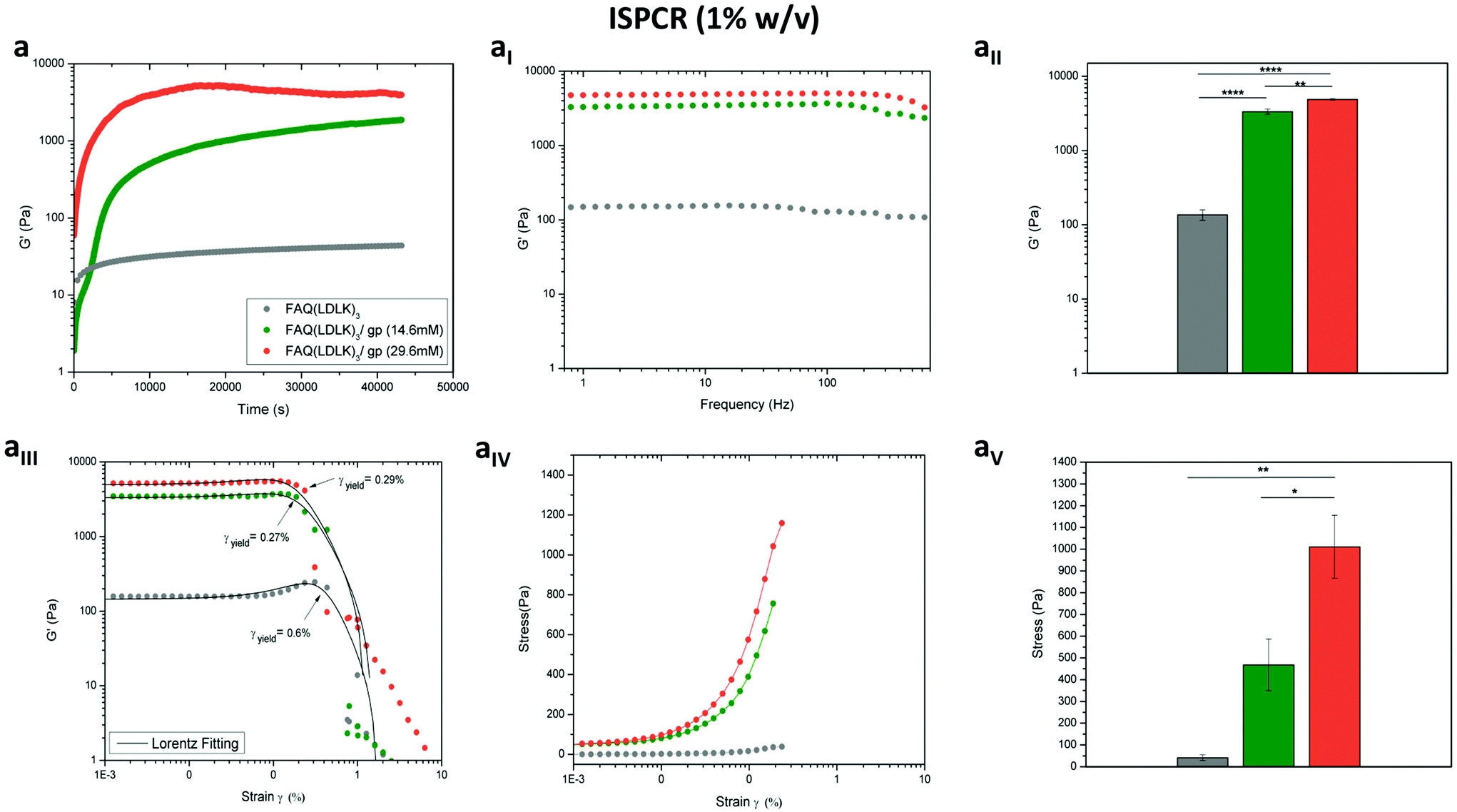 | ||
| Fig. 3 Rheological characterization of the ISPCR-FAQ(LDLK)3/gp hydrogels. The improved biomechanics of ISPCR-FAQ(LDLK)3 (1% w/v) at lower concentrations of genipin were monitored via time-sweep tests (a) followed by frequency sweep tests (0.1–1000 Hz). The average G′ of ISPCR-FAQ(LDLK)3/gp (14.6 mM) and ISPCR-FAQ(LDLK)3/gp (29.6 mM) was 3.3 kPa and 4.8 kPa respectively (aI). Both ISPCR-FAQ(LDLK)3/gp hydrogels exhibited greater G′ values (n = 3; p < 0.05) compared to FAQ(LDLK)3 (aII). Failure strain and stress tests confirmed the previous findings at higher concentrations of genipin, i.e. both ISPCR-FAQ(LDLK)3/gp hydrogels exhibited decreased strain-to-failure (aIII) but increased failure stresses (aIV–aV) compared soft FAQ(LDLK)3 hydrogels (n = 3; p < 0.05). | ||
Next, we looked at the stress-relaxation profile of the SAP-hydrogels. The rate of stress-relaxation under constant strain was quantified as the time for the initially measured stress to relax to half of its original value (τ½). We hypothesized that the stress-relaxation properties of the hydrogels could be modulated by using different cross-linker densities (170 mM, 86 mM and 14.6 mM) on DCR and, as a consequence, different degrees of entanglement of the assembled peptides. Indeed, a low concentration of genipin gave a rate of stress-relaxation around 3.5 s, whereas a higher cross-linking density led to a significantly longer timescale of τ½ ranging from 112 to 390 s (Fig. S2a in the ESI†). Interestingly, the stress relaxation behavior of these materials closely fitted with a two-element Maxwell–Weichert linear viscoelastic model (Fig. S2aI in the ESI†). Lastly, when temperature ramps were applied at physiological pH (Fig. S2b in the ESI†), the solid-like DCR-FAQ(LDLK)3/gp (170 mM) gel did not convert into a viscous-liquid state and G′ remained constant, suggesting that the genipin-mediated cross-linking is not sensitive to temperature shifts within the tested temperature range. These results show that genipin could be a precious tool to modulate the mechanical properties of SAP-scaffolds, allowing one to meet the needs of so far precluded different applications. Furthermore, the tunability of their stress-relaxation profiles could be relevant for cell behavior studies since cells usually respond to oscillation forces in a few seconds, exert traction forces on timescales of minutes, and undergo proliferation/spreading on minutes-to-hours timescales.62
TNBSA assessment of cross-linking on SAP hydrogels
TNBSA was used to quantitatively measure the free primary amino groups during/after the cross-linking reaction to evaluate the reaction kinetics and the degree of cross-linking in genipin-treated SAP-hydrogels. TNBSA is a rapid and sensitive compound: it forms a highly chromogenic derivative, which can be measured at 335 nm. As expected, the control FAQ(LDLK)3 did not show changes over time, while both DCR- and ISCR-FAQ(LDLK)3/gp displayed a decrease of free-amine groups during the cross-linking reaction (Fig. 4). In DCR, free amine values of 43% and 28% were achieved after 72 h with 86 mM and 170 mM of genipin respectively (Fig. 4a). In contrast, in ISCR, 51.7% and 35% free amine-groups were detected after 72 h, respectively, for 86 mM and 170 mM (Fig. 4b). These different degrees of cross-linking corroborate the hypothesis that stiffness differences between DCR- and ISCR-FAQ(LDLK)3/gp can correlate to the different ways of interaction of genipin with, respectively, the already-assembled and the still-forming peptide nanostructures. At low concentrations of genipin, the reaction kinetics of ISPCR (Fig. 4c) was similar to those obtained with DCR and ISCR, with free amine values of 46.2% and 34.8% respectively for FAQ(LDLK)3/gp (14.6 mM) and FAQ(LDLK)3/gp (29.6 mM).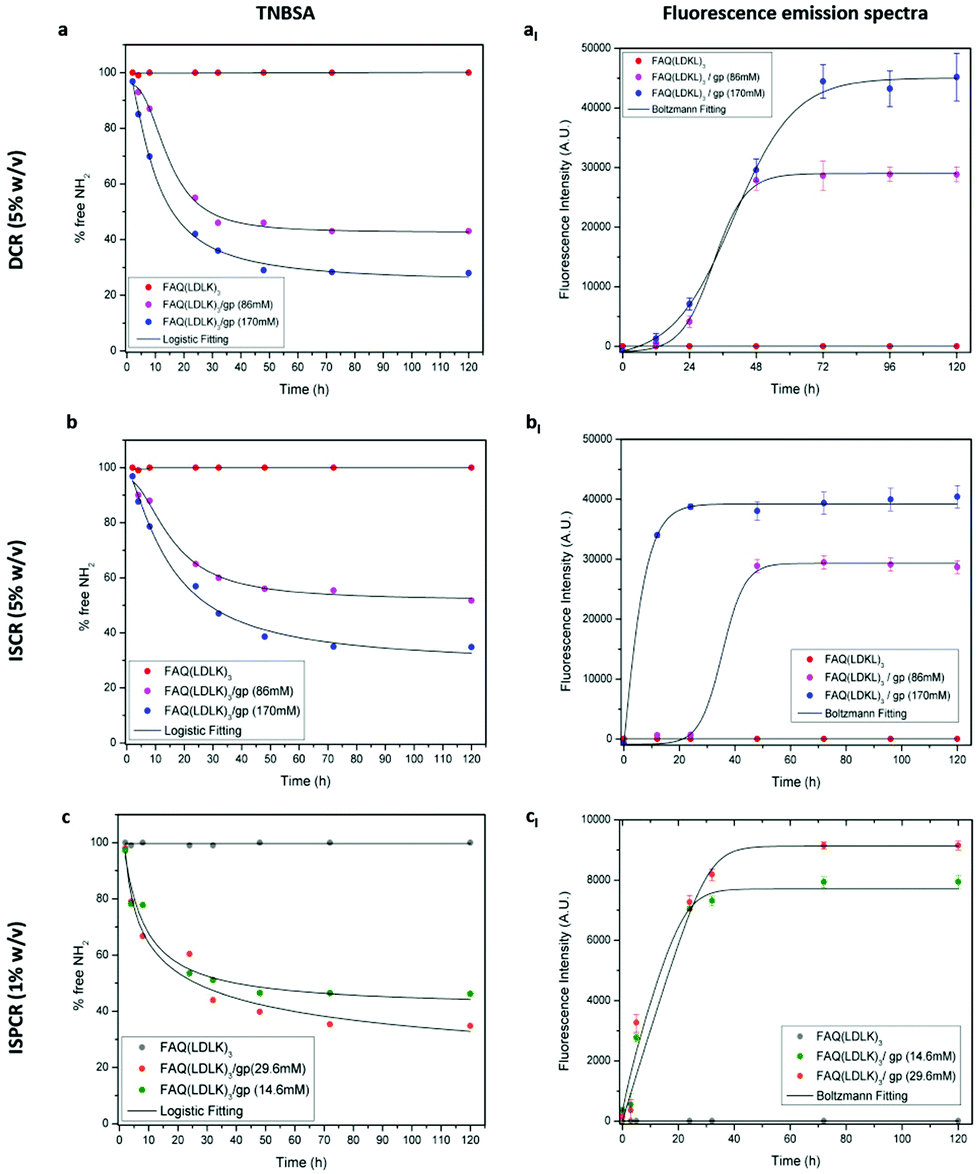 | ||
| Fig. 4 Cross-linking reaction kinetics: tracking the percentages of free primary amines of FAQ(LDLK)3 and DCR-, ISCR- and ISPCR-FAQ(LDLK)3/gp peptides over time. All peptide solutions were monitored by the spectrophotometric TNBSA amine assay for 120 h. While the control FAQ(LDLK)3 (in red) did not display changes in free NH2-groups over time, the FAQ(LDLK)3/gp peptides showed a decrease in free NH2-groups during the time course of the reaction, related to genipin concentration and manner of administration. (a) At 72 h, DCR-FAQ(LDLK)3/gp reached free amine values of 43% and 28% for 86 mM and 170 mM of genipin respectively. (b) In ISCR, 51.7% and 35% free amine-groups were detected respectively for 86 mM and 170 mM. (c) By contrast, ISPCR-FAQ(LDLK)3/gp at 14.6 mM and 29.6 mM of genipin reached free-NH2 values of 46.2% and 34.8%, respectively. The extent of the cross-linking reaction was also monitored via fluorescence measurements (aI–bI–cI). The reaction of the genipin compound with primary amines produces a blue pigmentation that correlates with the degree of cross-linking and can be tracked via fluorescence intensity measurements. Fluorescence intensity was recorded for FAQ(LDLK)3, DCR-, ISCR- and ISPCR-FAQ(LDLK)3/gp peptides from 0 h to 120 h. No fluorescence emission was detected from the uncross-linked FAQ(LDLK)3 hydrogels (in red). In contrast, the DCR-, ISCR- and ISPCR-FAQ(LDLK)3/gp peptides led to an increase of fluorescence intensity during the time course of the reaction. At 72 h, a stable blue pigmentation was completely formed in all cross-linked SAP-hydrogels. | ||
The extent of the cross-linking reaction was also assessed via fluorescence measurements. In brief, the reaction of the genipin compound with primary amines in peptides or proteins produces a blue pigmentation65 that qualitatively correlates with the degree of cross-linking and can be tracked by analyzing its fluorescence (see the fluorescence measurement section in Experiment for further details). When exposed to a clear solution of genipin, the initially translucent DCR (Fig. 4aI) and ISCR (Fig. 4bI) hydrogels gradually became light blue within a few hours, leading to a concomitant increase of fluorescence intensity. At 72 h of cross-linking reaction fluorescence intensity got to the highest values and plateaued. At this time-point a stable blue pigmentation was completely formed in all SAP-hydrogels, with the highest fluorescence values in the samples with 170 mM genipin. A similar trend was observed in ISPCR as well (Fig. 4cI). As expected, the fluorescence emission was not detected at 630 nm (λex = 590 nm) from the un-cross-linked FAQ(LDLK)3 hydrogels. In addition, to assess whether the functional motif FAQRVPP could interact with genipin, thereby interfering with the cross-linking reaction, we monitored the fluorescence intensity of the functional motif FAQRVPP in solutions of genipin. No fluorescence emission was observed during the time course of the reaction (data not shown), suggesting a poor, or even absent, interaction of genipin with the NH2 group of arginine (R) and/or the not-acetylated N-terminus of the chosen functional motif. Therefore we may assume a negligible interference of the cross-linking reaction with the biomimetic properties of the original FAQ(LDLK)3 peptide, already proven to be a promising candidate for nervous tissue regeneration.25 Nonetheless, the effect of genipin cross-linking on functional motifs comprising lysine (K) should be carefully weighed as they indeed may take part in the cross-linking reaction.
Influence of cross-linking on the assembled secondary structures
The structural characterization of the assembled SAPs was performed by ATR-FTIR. β-Sheet structures can be tracked by analyzing the Amide I region (1600–1700 cm−1), which is mainly associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and related to the SAP-backbone conformation. Conventionally, in parallel β-sheet structures, the Amide I region exhibits a peak around 1630 cm−1, while in anti-parallel β-sheet structures, the Amide I region displays both a major component at 1630 cm−1 and a minor component at 1695 cm−1. The ratio of the peak intensities at 1695 cm−1 and 1630 cm−1, named β-sheet organizational index, is proportional to the ratio of antiparallel/parallel β-sheet structures.86 The FTIR spectra of FAQ(LDLK)3 showed anti-parallel β-sheet features (β-sheet organizational index = 21.6%). A predominantly anti-parallel β-sheet structure was also seen in DCR-FAQ(LDLK)3/gp, with a slight variation of the β-sheet organizational index in FAQ(LDLK)3/gp (86 mM) and (170 mM), i.e. 16.32% and 19.65% respectively (Fig. 5a). In contrast, β-sheet bands in ISCR-FAQ(LDLK)3/gp were still present but the Amide I peak decreased and broadened, likely due to NH2-group deformation (Fig. 5b). In both ISCR-FAQ(LDLK)3/gp peptides, the β-sheet organizational index significantly decreased compared to FAQ(LDLK)3, showing values of 13.68% and 15.63% respectively for 86 mM and 170 mM of genipin. This decrease in β-structuring highlights that in ISCR, since the cross-linking reaction is concomitant with the self-assembly phenomenon, genipin could affect the stabilization of the forming β-sheet domains, thus influencing not only the mechanical strength of the resulting SAP-hydrogels, as previously observed in biomechanical characterization, but also their macromolecular organization. In the Amide II region (1480–1575 cm−1), β-sheet aggregation for all tested SAPs was confirmed by the presence of a peak at ≈1543 cm−1 directly related to CN stretching and NH bending.
O stretching vibration and related to the SAP-backbone conformation. Conventionally, in parallel β-sheet structures, the Amide I region exhibits a peak around 1630 cm−1, while in anti-parallel β-sheet structures, the Amide I region displays both a major component at 1630 cm−1 and a minor component at 1695 cm−1. The ratio of the peak intensities at 1695 cm−1 and 1630 cm−1, named β-sheet organizational index, is proportional to the ratio of antiparallel/parallel β-sheet structures.86 The FTIR spectra of FAQ(LDLK)3 showed anti-parallel β-sheet features (β-sheet organizational index = 21.6%). A predominantly anti-parallel β-sheet structure was also seen in DCR-FAQ(LDLK)3/gp, with a slight variation of the β-sheet organizational index in FAQ(LDLK)3/gp (86 mM) and (170 mM), i.e. 16.32% and 19.65% respectively (Fig. 5a). In contrast, β-sheet bands in ISCR-FAQ(LDLK)3/gp were still present but the Amide I peak decreased and broadened, likely due to NH2-group deformation (Fig. 5b). In both ISCR-FAQ(LDLK)3/gp peptides, the β-sheet organizational index significantly decreased compared to FAQ(LDLK)3, showing values of 13.68% and 15.63% respectively for 86 mM and 170 mM of genipin. This decrease in β-structuring highlights that in ISCR, since the cross-linking reaction is concomitant with the self-assembly phenomenon, genipin could affect the stabilization of the forming β-sheet domains, thus influencing not only the mechanical strength of the resulting SAP-hydrogels, as previously observed in biomechanical characterization, but also their macromolecular organization. In the Amide II region (1480–1575 cm−1), β-sheet aggregation for all tested SAPs was confirmed by the presence of a peak at ≈1543 cm−1 directly related to CN stretching and NH bending.
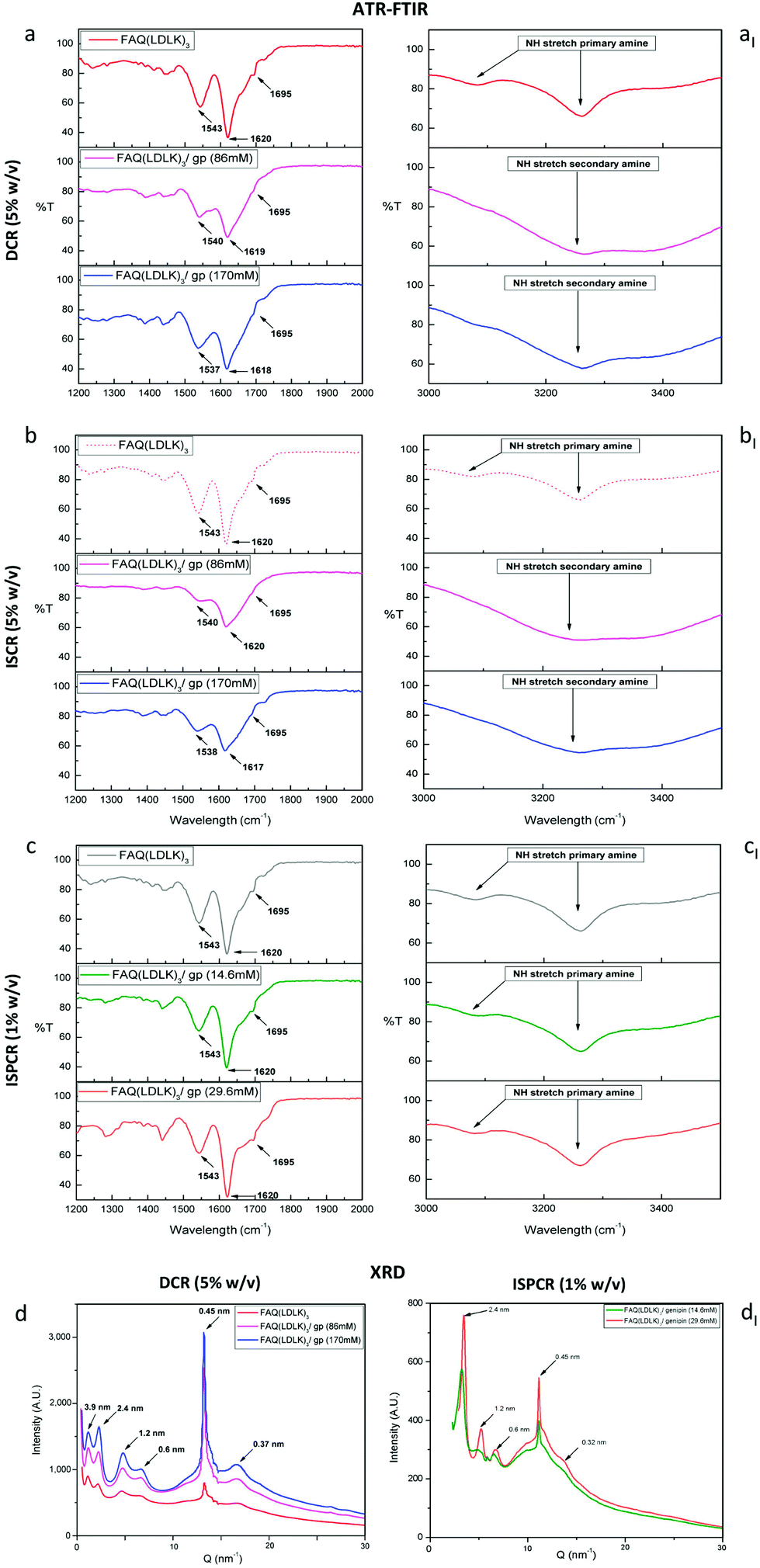 | ||
| Fig. 5 Structural characterization of the assembled scaffolds. ATR-FTIR spectra in the Amide I and Amide II absorption regions of DCR-FAQ(LDLK)3/gp (a), ISCR-FAQ(LDLK)3/gp (b) and ISPCR-FAQ(LDLK)3/gp (c). All peptide spectra displayed a broad band near 1540 cm−1 (Amide II region, β-sheet aggregation) and peaks at 1620 and 1695 cm−1 (Amide I region, antiparallel β-sheet structures). In addition, NH stretching in the 3100–3300 cm−1 band has been assessed. The FAQ(LDLK)3 peptide displayed two peaks ascribable to primary amine stretching, while DCR- and ISCR-FAQ(LDLK)3/gp showed only one peak (aI–bI), suggesting that the carboxymethyl group of genipin reacted with the majority of the primary amino-groups of SAPs, yielding secondary amines. The ISPCR-FAQ(LDLK)3/gp (cI) peaks assigned to primary amines are still detectable, likely due to the partial linking of the free amine groups after a crosslinking reaction run for just 24 hours. (d–dI) X-ray diffraction (XRD) data of FAQ(LDLK)3 (red), DCR-FAQ(LDLK)3/gp 86 mM (magenta), DCR-FAQ(LDLK)3/gp 170 mM (blue), ISPCR-FAQ(LDLK)3/gp 14.6 mM (green), and ISPCR-FAQ(LDLK)3/gp 29.6 mM (orange); all of these SAPs show similar XRD peaks, suggesting the presence of similar structures. | ||
In addition, we assessed NH stretching in the 3100–3300 cm−1 region, where primary amines (R-NH2) feature two bands, due to the asymmetrical and symmetrical NH stretching respectively. In contrast, secondary amines (R2-NH) show only a single weak and broad band given by their single NH bond. The FAQ(LDLK)3 peptide displayed two peaks ascribable to primary amine stretching, while DCR- and ISCR-FAQ(LDLK)3/gp showed only one NH stretching (Fig. 5aI and bI). Hence, these features suggest that the carboxymethyl groups of genipin reacted with the primary amino groups of SAPs yielding secondary amines.
The FTIR spectra of ISPCR-FAQ(LDLK)3/gp showed β-sheet features in the Amide I and II regions, characterized by the presence of peaks at 1620, 1695 and 1543 cm−1 (Fig. 5c). However, it has to be noted that the β-sheet signature appeared more evident and similar to those of FAQ(LDLK)3, suggesting that low concentrations of genipin and a shorter reaction time moderately influence the β-sheet formation propensity of the tested SAP. Lastly, primary amine peaks are still present in the 3100–3300 cm−1 band (Fig. 5cI), likely because of a partial linking of the free amine groups in a cross-linking reaction of just 24 hours.
X-ray diffraction characterization
In X-ray diffraction analyses (see Experiment for details), the first low Q peaks (d = 3.9 nm in Fig. 5d–dI) matched the height of SAP fibers obtained with the AFM tests (see the next paragraph and Fig. S3a in the ESI†). The peaks at 2.4 nm and 1.2 nm likely pointed at, respectively, the total thickness and the intra-layer distance of typical bilayered β-sheet structures found in similar SAPs.87 The peak at 0.6 nm could be assigned to the length of the lysine side-chain, while the strong peak at 0.45 nm was ascribable to the peptide backbone distance in the β-sheets.88 The FAQ(LDLK)3/gp peptides displayed a significant increment of the latter peak, correlated to the concentration of genipin: this was probably due to stronger backbone packing after cross-linking. Lastly, the 0.327 nm spacing could be ascribed to the distance among residues along the peptide chains. Based on these results, we hypothesize that cross-linking presumably strengthened the peptide backbone packing, and significantly contributed to the stabilization of the assembled structures without affecting the standard SAP assembly into cross-β structures.35Morphological characterization
Atomic force microscopy (AFM) morphological analysis was carried out to monitor the effects of cross-linking on the self-aggregated nanostructures of the FAQ(LDLK)3 peptides. All tested peptides self-assembled into nanofibers but with slightly different morphologies. FAQ(LDLK)3 yielded short and single fibers with ≈13 nm width, consistent with those previously obtained (Fig. 6a).26 The FAQ(LDLK)3 height ranged from 2.4 to 3.9 nm (Fig. S3a in the ESI†), in agreement with the data obtained from XRD analysis. By contrast, the nanofiber morphology of DCR-FAQ(LDLK)3/gp featured a tight and clustered bundle network of presumably cross-linked nanofibers; average values of 14.63 nm and 3.16 nm for the width and height respectively. In contrast, ISCR-FAQ(LDLK)3/gp (Fig. 6b) showed less tangled fibers more similar to those of FAQ(LDLK)3 (Fig. 6c). | ||
| Fig. 6 Atomic force microscopy (AFM) morphological analysis. (a) DCR-FAQ(LDLK)3/gp self-organized into a tight and clustered network of 14.63 nm wide and 3.16 nm long nanofibers. In contrast, ISCR-FAQ(LDLK)3/gp (b) showed a less tangled fiber morphology. (c) FAQ(LDLK)3 self-assembled into single short nanofibers. | ||
The AFM results confirmed the assembly propensity of all peptides into nanofibers and suggested that genipin cross-linking has fostered the formation of clusters of nanofibers.
In vitro degradation of cross-linked hydrogels
The weight loss of hydrogels upon exposure to trypsin digestion (at 37 °C) was used to test the degradation of the assembled SAPs. All samples showed significant weight loss over time (Fig. S4a in the ESI†). The weight of the FAQ(LDLK)3 peptide decreased faster than those of the cross-linked samples, showing 86% degradation after 4 weeks. Both FAQ(LDLK)3/gp peptides at 14.6 mM and 170 mM of genipin, needed 8–10 weeks respectively to be degraded to that value. Indeed, the weight loss rate of these samples was relatively constant throughout the degradation test; however, a total degradation was not observed in either of the cross-linked samples. It is interesting to report the morphological differences that arose during the degradation tests: in FAQ(LDLK)3/gp (170 mM), trypsin-mediated degradation was evenly distributed on the sample surface, leading to an increase in surface roughness and a significant decrease in hydrogel thickness from 2 mm to 0.5 mm after 6 weeks. In contrast, the digestion of FAQ(LDLK)3/gp (14.6 mM) was more localized in the central part of the hydrogel.Since particulate debris was present in the supernatant, this was analyzed to evaluate the presence of both degraded peptides and genipin released during degradation. In absorbance measurements, all tested SAPs exhibited the typical 230 nm and 270 nm peaks of the peptide bond and aromatic amino acids respectively. In contrast, in fluorescence measurements (see Experiment for details) we detected the presence of genipin released during the trypsin degradation experiment (data not shown).
We also evaluated the behavior of the genipin cross-linked SAPs in the presence of organic solvents (such as dichloromethane, dimethylformamide, chloroform and methanol) that usually lead to the dissolution of PCL, PLGA and PLA polymers. When genipin cross-linked SAPs were dipped in these solutions for 48 h, they did not show changes in terms of degradation, dissolution or shrinkage: an interesting quality of cross-linked SAPs (not present in standard soft SAPs) which can be taken into account to design composite materials made of polymers/functionalized peptides or to develop novel SAP casting protocols.
Fabrication and characterization of electrospun cross-linked nanofibrous scaffolds
In the last two decades, the electrospinning technique has emerged as a promising approach for the large-scale production of nanofibers. Among other qualities, electrospinning allows for the fabrication of well-defined 3D porous nano- and micro-fibrous scaffolds, an asset useful for many TE applications,33,89–93 but currently missing in all SAP-made scaffolds. So far, electrospinning has been mainly used for natural and synthetic polymers but, as such, electrospun fibrous scaffolds do not feature an easiness of functionalization with bioactive peptides comparable to SAPs. Indeed, biomimetic scaffolds coaxing specific cell behaviors like, for example, migration, differentiation and proliferation have become a hot topic in TE.94In order to overcome this issue, modification of electrospun fibrous scaffolds with the incorporation of bioactive molecules (i.e. RGD, YIGSR, IKVAV or FAQRVPP) using covalent binding,95–97 noncovalent host–guest interactions,98 physical adsorption (plasma treatment)99 or blended electrospinning procedures33 became interesting strategies to mimic the biochemical properties of the ECM, thus creating biomimetic scaffolds fostering tissue regeneration at the site of implant. However, to take full advantage of the several benefits coming from SAP technology (e.g. multi-functionalization, high biocompatibility, pathogen-free technology, ease of production and tailoring, etc.) researchers moved their efforts toward the ES of pure SAPs,34,100 unfortunately obtaining just soft fibrous-based SAP coatings because of the poor mechanical properties of the peptides. Driven by the idea that scaffolds should be designed both at the biochemical and biomechanical levels we successfully applied the cross-linking technique to nanofibrous electrospun scaffolds to obtain, for the first time, flexible self-standing 3D scaffolds made of pure SAPs (Fig. 7a).
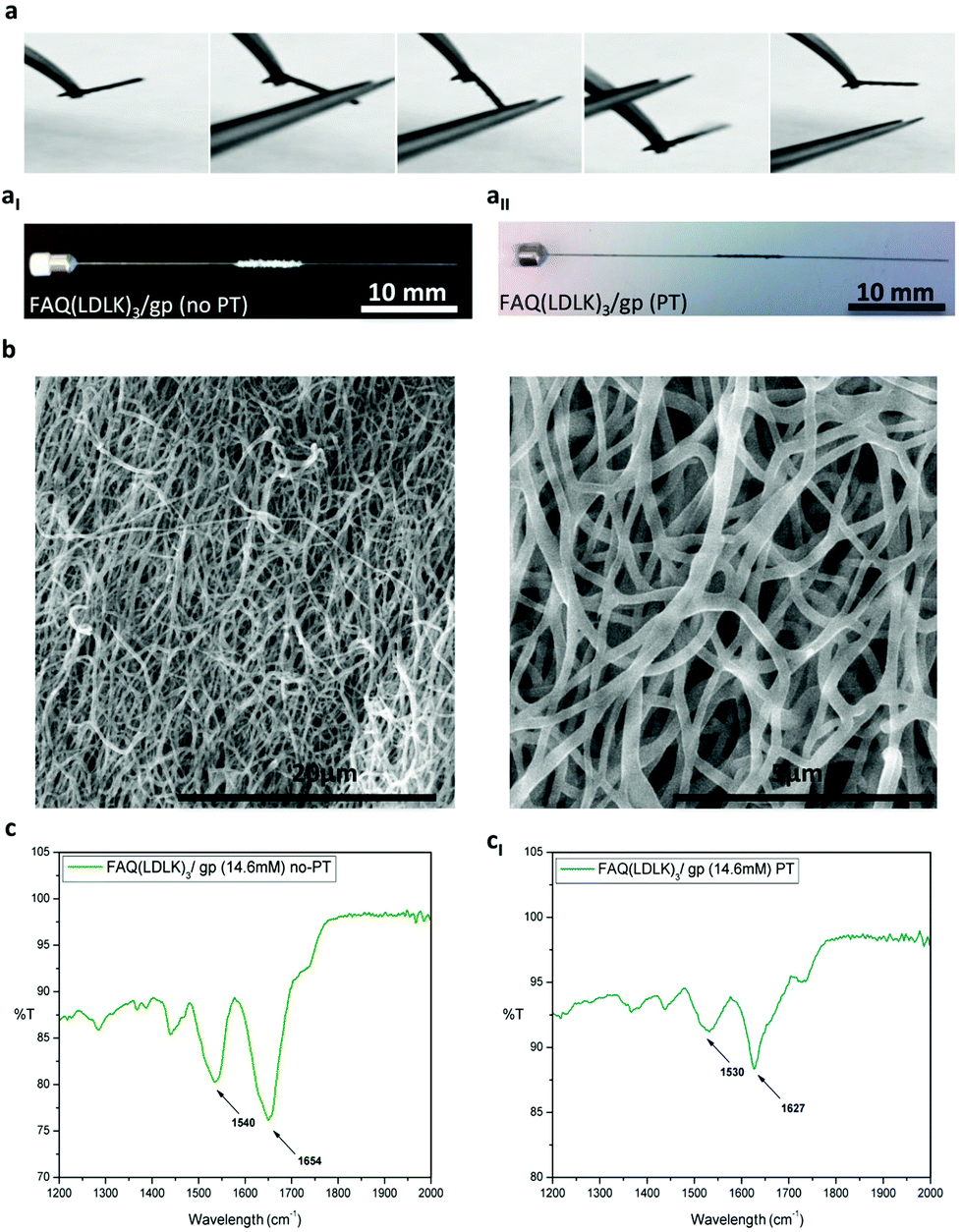 | ||
| Fig. 7 (a) Self-standing electrospun microchannels made of the ISPCR-FAQ(LDLK)3/gp (14.6 mM, 72 h) peptide. (aI) After electrospinning, ISPCR-FAQ(LDLK)3/gp fibers were white in color (no-PT), whereas after PT, the ISPCR-FAQ(LDLK)3/gp fibers appeared dark-blue due to the increase of the cross-linking degree (aII). Even if the PT-steps led to a latitude shrinkage of the ISPCR-FAQ(LDLK)3/gp microchannel, they preserved its interconnected-porous 3D fibrous network. (b) SEM images of the ISPCR-FAQ(LDLK)3/gp fibers after PT at low and high magnifications. The ISPCR-FAQ(LDLK)3/gp fibers were randomly oriented, giving a very porous construct with average fiber diameters of 294 nm. ATR-FTIR spectra in the Amide I and Amide II absorption regions of electrospun ISPCR-FAQ(LDLK)3/gp before and after PT. (c) Electrospun ISPCR-FAQ(LDLK)3/gp before PT showed a peak at 1654 cm−1 related to random coil conformations, whereas in the Amide II region, it exhibited a broad peak at 1540 cm−1 ascribable to β-sheet aggregation. (cI) Electrospun ISPCR-FAQ(LDLK)3/gp after PT showed peaks at 1627 cm−1 (Amide I) and 1530 cm−1 (Amide II) indicative of a stronger β-sheet formation, highlighting that the PT-steps influenced the cross-linking reaction and also the macromolecular organization of the resulting electrospun SAP-fibers. | ||
Our ES studies suggested that DCR was not feasible with the electrospinning of FAQ(LDLK)3 as electrospun nanofibers dissolved in aqueous post-treatment solutions before cross-linking was consolidated (data not shown). On the other hand, only a low concentration of genipin could be added to the starting ES solution in order to prevent gelling and clogging of the ES setup. The best method developed for FAQ(LDLK)3/gp fiber fabrication was based on ISPCR-FAQ(LDLK)3 with 14.6 mM genipin.
We focused our efforts on the fabrication of fibrous mats and micro-channels as described in the Experiment section. After electrospinning, the ISPCR-FAQ(LDLK)3/gp fibers were white in color with both round and flat shapes (Fig. 7aI). As previously described, ISPCR kinetics told us that some genipin inside the fibers could be still activated in the subsequent post-treatment (PT) steps. Firstly, the white electrospun fibers were exposed to the vapor phase of 2.5 mM NaOH in PBS (PT-I) at 37 °C for 3 days in order to slightly activate the cross-linking reaction and stabilize the fiber morphology. As this step was not enough to provide stable scaffolds, a post-treatment through immersion in a high-concentrated genipin solution was added to achieve a higher degree of cross-linking. Hence, the partially cross-linked fibers were immersed in 170 mM genipin in H2O:EtOH (90:10 v/v) for 1 day at 37 °C (PT-II), and finally soaked in PBS for 1 day (PT-III) to remove any remnants of ethanol. After each PT-step, the increase of the cross-linking degree was testified by the appearance and increase of a darker blue color throughout the procedures (Fig. 7aII). Overall, the PT-steps lead to a latitude shrinkage of the FAQ(LDLK)3/gp scaffolds, but they preserved its interconnected-porous 3D fibrous network (Fig. S5 in the ESI†). As shown in the SEM images of ISPCR-FAQ(LDLK)3/gp after PT (Fig. 7b), the fibers were randomly oriented giving a very porous construct with average fiber diameters of 294 nm. Interestingly, a significant number of linked fibers in the fiber-to-fiber interconnections was observed in the junctions of adjacent fibers for electrospun FAQ(LDLK)3/gp after PT. This feature made a stable 3D-interconnected network structure with preserved fibrous morphology. Notably, the fibers were not merged before PT (Fig. S6 in the ESI†).
The obtained fibrous scaffolds were characterized through ATR-FTIR tests. Before PT, electrospun FAQ(LDLK)3/gp showed a 1654 cm−1 peak in the Amide I region (1600–1700 cm−1) mainly related to random coil conformations, whereas in the Amide II region (1480–1575 cm−1), it exhibited a broad peak at 1540 cm−1 ascribable to β-sheet aggregation (Fig. 7c). In contrast, the narrow characteristic peaks at 1627 cm−1 (Amide I) and 1530 cm−1 (Amide II) showed stronger β-sheet formation of FAQ(LDLK)3/gp after PT (Fig. 7cI), highlighting that the PT-steps influence not only the cross-linking reaction but also the macromolecular organization of the resulting electrospun SAP-fibers. Un-cross-linked electrospun FAQ(LDLK)3 revealed broader bands (with relatively less intensity) centered near 1654 cm−1 and 1540 cm−1 which were characteristic of mainly random coil and β-sheet conformations, respectively (data not shown). Furthermore, mechanical characterization was performed on cross-linked electrospun FAQ(LDLK)3/gp nanofibrous mats having two different thicknesses, after the PT-steps. Electrospun mats were cut into a rectangular shape (50 mm × 30 mm; thickness = 0.185 mm or 0.36 mm) and placed on the rheometer plate. Storage (G′) and loss modulus (G′′) were measured by varying the frequencies of the applied oscillatory stress. The average G′ and G′′ values were respectively 2.095 kPa and 50 Pa for the electrospun mats with 0.185 mm thickness, whereas they were 7.365 kPa and 394.7 Pa for the electrospun mats with 0.36 mm thickness (Fig. S7a in the ESI†). As expected, the mechanical properties well-correlate with the overall scaffold thickness due to the increased concentration of the entangled nanofibers and covalent binding. For both the FAQ(LDLK)3/gp nanofibrous mats, G‘/G′′ remained relatively constant along the tested frequency range (0.1–1000 Hz) suggesting that the electrospun scaffolds are resistant to deformations applied at different oscillatory frequencies. Stress/strain failure test was also performed (0.1%–1000% strain) to identify the maximum strain/stress to which the sample can be subjected. The electrospun FAQ(LDLK)3/gp nanofibrous mats (thickness = 0.185 mm) showed a strain-to-failure of 57.82% with failure stress occurring at 509 Pa; in contrast, the electrospun FAQ(LDLK)3/gp nanofibrous mats (thickness = 0.36 mm) exhibited a strain-to-failure of 108.2% with failure stress at 1130 Pa (Fig. S7b in the ESI†). These values of strain-to-failure are particularly interesting as, to the best of our knowledge, they have never been achieved with SAP-based scaffolds so far and are desirable features for deformable biomaterials to be used in tissue engineering. Nonetheless, this mechanical behavior of electrospun cross-linked constructs is comparable with previous reports of polymeric electrospun nanofibrous mats.33,101 Lastly, temperature ramps were recorded as a function of G′ (see Experiment for further details). When temperature ramps were applied to the electrospun FAQ(LDLK)3/gp nanofibrous mats, a phase transition was not observed and the G′ remained constant through the tested temperature range (Fig. S7c in the ESI†) similarly to the DCR-FAQ(LDLK)3/gp membrane, highlighting their stability when subjected to temperature increments.
Moreover, as the ES solution came from ISPCR-SAPs, the cross-linker was likely well-distributed within the electrospun FAQ(LDLK)3/gp fibers; this led to the manufacture of a single unified, self-standing tubular channel with spring-like properties (e.g. recoverable deformation under load) (Video S2 in the ESI†). Furthermore, in contrast to uncross-linked FAQ(LDLK)3, the electrospun fibers of FAQ(LDLK)3/gp were stable in the wet state (upon exposure to PBS at 37 °C) over time (Fig. S8 in the ESI†), allowing for their long-term use. Also, after cross-linking, electrospun SAP scaffolds showed an interesting stability (data not shown) in different organic solvents, unusual of standard electrospun SAPs.
Taken together, these results make electrospun scaffolds from FAQ(LDLK)3/gp particularly promising candidates for tissue engineering applications thanks to their stable 3D fibrous-based structure capable of retaining the fiber morphology when immersed in aqueous solutions and/or when subjected to external deformations.
Cell morphology and viability assays
Since the system investigated here has structural features that are biomimetic of ECMs, and can also provide a functionalized microenvironment with specific biological cues capable of stimulating adult neural stem cell adhesion and differentiation,26 we chose to investigate if genipin can have cytotoxic effects on hNSCs and how the organization of the cross-linked electrospun fibers affects cells in culture. We selected GMP-grade hNSCs for these experiments, since they are well known and characterized on SAP scaffolds,25,35,68,71,102,103 and also because of their demonstrated safety and efficacy in a phase I clinical trial of amyotrophic lateral sclerosis (ALS).70To evaluate whether genipin can affect the hNSC viability, different concentrations of genipin (21 mM, 42 mM and 64 mM) were added to the cell culture medium and incubated for one day at 37 °C (see Experiment for further details). Cell viability was found to be high in each of the tested conditions (Fig. S9a in the ESI†), suggesting a negligible cytotoxicity ascribable to genipin. We next investigated whether cross-linked electrospun scaffolds made of functionalized FAQ(LDLK)3/gp could be still considered biomimetic scaffolds for their future use in neural tissue engineering applications. After 7DIV, we assessed the phenotype of the differentiated hNSC progenies cultured on the top surface of es-FAQ(LDLK)3/gp scaffolds (Fig. S9b in the ESI†). The differentiated hNSC progeny was immunostained with markers for astrocytes (GFAP), post-mitotic neurons (βIII-tubulin) and oligodendrocytes (GalC and O4). hNSCs cultured on cross-linked electrospun scaffolds exhibited a spread and branching neural morphology similar to that found on previously reported FAQ(LDLK)3 hydrogel.26
Conclusions
Over the last two decades, self-assembling peptides came under the spotlight because of their ease of design, functionalization and synthesis. They showed promising potential in regenerative therapies as soft hydrogels, but so far lacked the biomechanical properties required for their engraftment into medium-hard tissues and for the production of complex 3D scaffolds typical of polymers and natural cross-linked proteins.In this work we developed a feasible cross-linking strategy to modulate the mechanical properties of biomimetic SAPs and introduced different genipin cross-linking protocols with profound effects on stiffness, bioabsorption time and molecular arrangements of scaffolds. Biomechanical enhancements were dependent on the dose and time of exposure to genipin, thus allowing their tuning to suit the specific needs of different applications. We showed that the cross-linking reaction did not involve the residues of the chosen functional motifs, thus leaving the door open to the crosslinking of multi-functionalized SAPs. In addition, thanks to the improved SAP chain length and biomechanics, we managed, for the first time, to electrospin the cross-linked SAPs into single unified and self-standing mats and microchannels having interconnected fibers. Indeed, our electrospun highly cross-linked SAP constructs retained their fibrous morphology when immersed in aqueous solution, showing flexibility, spring-like behaviour and better stability to degradation. Lastly, the evidence of preserved functional motif bioactivity was established by detecting the three main phenotypes and respective morphologies of differentiated hNSC progenies on the surface of electrospun cross-linked nanofibrous scaffolds (Fig. S9†).
This work may open the door to the development and tailoring of functionalized synthetic bioprostheses entirely made of SAPs with tunable resilience, bioabsorption times and/or bioactivities for different tissue engineering applications and beyond.
Author contributions
R.P. and F.G. conceived the project. R.P. synthesized the peptides and carried out the hydrogel cross-linking experiments while M.M. took care of the electrospinning ones. F.G. supervised the project. R.Z. co-supervised the XRD experiments. R.P. and F.G. wrote the manuscript. All authors have approved the final article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described and performed by R. P., M. M. and F. G. was funded by the “Ricerca Corrente” funding granted by the Italian Ministry of Health and by the “5 × 1000” voluntary contributions. Financial support also came from Revert and Vertical Onlus donations. The XRD experiments were conducted at the Advanced Light Source and at the Molecular Foundry (the Lawrence Berkeley National Laboratory) both of which are supported by the Office of Science, under Contract No. DE-AC02-05CH11231. We thank Amanda Marchini for running all the in vitro experiments assessing the cytotoxicity and differentiation of hNSC on cross-linked substrates. We thank Prof. Luca Beverina for allowing our FTIR experiments to be performed at his facility at the Material Science department of the University of Milan-Bicocca.References
- S. Zhang, Interface Focus, 2017, 7, 20170028 CrossRef PubMed.
- B. B. Hsu, W. Conway, C. M. Tschabrunn, M. Mehta, M. B. Perez-Cuevas, S. Zhang and P. T. Hammond, ACS Nano, 2015, 9, 9394–9406 CrossRef CAS PubMed.
- S. Yang, S. Wei, Y. Mao, H. Zheng, J. Feng, J. Cui, X. Xie, F. Chen and H. Li, BMC Biotechnol., 2018, 18, 12 CrossRef PubMed.
- C. E. Morgan, A. W. Dombrowski, C. M. Rubert Perez, E. S. Bahnson, N. D. Tsihlis, W. Jiang, Q. Jiang, J. M. Vercammen, V. S. Prakash, T. A. Pritts, S. I. Stupp and M. R. Kibbe, ACS Nano, 2016, 10, 899–909 CrossRef CAS PubMed.
- M. Zhao, Y. Zhou, S. Liu, L. Li, Y. Chen, J. Cheng, Y. Lu and J. Liu, Drug Delivery, 2018, 25, 546–554 CrossRef CAS PubMed.
- Z. Song, X. Chen, X. You, K. Huang, A. Dhinakar, Z. Gu and J. Wu, Biomater. Sci., 2017, 5, 2369–2380 RSC.
- A. Zhou, S. Chen, B. He, W. Zhao, X. Chen and D. Jiang, Drug Des., Dev. Ther., 2016, 10, 3043–3051 CrossRef CAS PubMed.
- C. Karavasili, M. Spanakis, D. Papagiannopoulou, I. S. Vizirianakis, D. G. Fatouros and S. Koutsopoulos, J. Pharm. Sci., 2015, 104, 2304–2311 CrossRef CAS PubMed.
- M. Ozeki, S. Kuroda, K. Kon and S. Kasugai, J. Biomater. Appl., 2011, 25, 663–684 CrossRef CAS PubMed.
- H. Nakahara, H. Misawa, A. Yoshida, T. Hayashi, M. Tanaka, T. Furumatsu, N. Tanaka, N. Kobayashi and T. Ozaki, Cell Transplant., 2010, 19, 791–797 Search PubMed.
- B. He, Y. Ou, S. Chen, W. Zhao, A. Zhou, J. Zhao, H. Li, D. Jiang and Y. Zhu, Mater. Sci. Eng., C, 2017, 74, 451–458 CrossRef CAS PubMed.
- A. Schneider, J. A. Garlick and C. Egles, PLoS One, 2008, 3, e1410 CrossRef PubMed.
- F. Paladini, S. T. Meikle, I. R. Cooper, J. Lacey, V. Perugini and M. Santin, J. Mater. Sci.: Mater. Med., 2013, 24, 2461–2472 CrossRef CAS PubMed.
- Y. Loo, Y. C. Wong, E. Z. Cai, C. H. Ang, A. Raju, A. Lakshmanan, A. G. Koh, H. J. Zhou, T. C. Lim, S. M. Moochhala and C. A. Hauser, Biomaterials, 2014, 35, 4805–4814 CrossRef CAS PubMed.
- Y. Ichihara, M. Kaneko, K. Yamahara, M. Koulouroudias, N. Sato, R. Uppal, K. Yamazaki, S. Saito and K. Suzuki, Biomaterials, 2018, 154, 12–23 CrossRef CAS PubMed.
- M. Zhang, W. W. Ai, Z. L. Mei, Y. H. Hu and Z. L. Zhang, Exp. Ther. Med., 2017, 14, 3441–3446 CrossRef CAS PubMed.
- M. E. Davis, J. P. Motion, D. A. Narmoneva, T. Takahashi, D. Hakuno, R. D. Kamm, S. Zhang and R. T. Lee, Circulation, 2005, 111, 442–450 CrossRef CAS PubMed.
- B. He, X. Yuan, A. Zhou, H. Zhang and D. Jiang, Expert Rev. Mol. Med., 2014, 16, e12 CrossRef PubMed.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS PubMed.
- X. C. Li, Y. H. Wu, X. D. Bai, W. Ji, Z. M. Guo, C. F. Wang, Q. He and D. K. Ruan, Tissue Eng., Part A, 2016, 22, 1218–1228 CrossRef CAS PubMed.
- Y. Wu, Z. Jia, L. Liu, Y. Zhao, H. Li, C. Wang, H. Tao, Y. Tang, Q. He and D. Ruan, Artif. Organs, 2016, 40, E112–E119 CrossRef CAS PubMed.
- P. A. Brunton, R. P. Davies, J. L. Burke, A. Smith, A. Aggeli, S. J. Brookes and J. Kirkham, Br. Dent. J., 2013, 215, E6 CrossRef CAS PubMed.
- M. Schlee, F. Rathe, C. Bommer, F. Broseler and L. Kind, J. Periodontol., 2018, 89(6), 653–660 CrossRef CAS PubMed.
- Y. Kondo, T. Nagasaka, S. Kobayashi, N. Kobayashi and T. Fujiwara, Hepato-Gastroenterology, 2014, 61, 349–353 Search PubMed.
- A. Caprini, D. Silva, I. Zanoni, C. Cunha, C. Volonte, A. Vescovi and F. Gelain, New Biotechnol., 2013, 30, 552–562 CrossRef CAS PubMed.
- F. Gelain, D. Cigognini, A. Caprini, D. Silva, B. Colleoni, M. Donega, S. Antonini, B. E. Cohen and A. Vescovi, Nanoscale, 2012, 4, 2946–2957 RSC.
- S. Koutsopoulos, L. D. Unsworth, Y. Nagai and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef CAS PubMed.
- F. Gelain, L. D. Unsworth and S. Zhang, J. Controlled Release, 2010, 145, 231–239 CrossRef CAS PubMed.
- R. Pugliese and F. Gelain, Trends Biotechnol., 2017, 35, 145–158 CrossRef CAS PubMed.
- S. Panseri, C. Cunha, J. Lowery, U. Del Carro, F. Taraballi, S. Amadio, A. Vescovi and F. Gelain, BMC Biotechnol., 2008, 8, 39 CrossRef PubMed.
- T. T. Yuan, A. M. DiGeorge Foushee, M. C. Johnson, A. R. Jockheck-Clark and J. M. Stahl, Nanoscale Res. Lett., 2018, 13, 88 CrossRef PubMed.
- S. Hong and G. Kim, J. Biomed. Mater. Res., Part B, 2010, 94, 421–428 Search PubMed.
- A. Raspa, A. Marchini, R. Pugliese, M. Mauri, M. Maleki, R. Vasita and F. Gelain, Nanoscale, 2016, 8, 253–265 RSC.
- M. Maleki, A. Natalello, R. Pugliese and F. Gelain, Acta Biomater., 2017, 51, 268–278 CrossRef CAS PubMed.
- R. Pugliese, A. Marchini, G. A. A. Saracino, R. N. Zuckermann and F. Gelain, Nano Res., 2018, 11, 586–602 CrossRef CAS.
- S. Habtemariam and G. Lentini, Biomedicines, 2018, 6(2), 70 CrossRef PubMed.
- G. Chen and X. Guo, Int. Rev. Neurobiol., 2017, 135, 77–95 CrossRef PubMed.
- A. Bigi, G. Cojazzi, S. Panzavolta, N. Roveri and K. Rubini, Biomaterials, 2002, 23, 4827–4832 CrossRef CAS PubMed.
- G. Yang, Z. Xiao, H. Long, K. Ma, J. Zhang, X. Ren and J. Zhang, Sci. Rep., 2018, 8, 1616 CrossRef PubMed.
- P. Sanchez, J. L. Pedraz and G. Orive, Int. J. Biol. Macromol., 2017, 98, 486–494 CrossRef CAS PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CrossRef CAS PubMed.
- R. Chen, X. Cai, K. Ma, Y. Zhou, Y. Wang and T. Jiang, Biofabrication, 2017, 9, 025028 CrossRef PubMed.
- L. Chronopoulou, Y. Toumia, B. Cerroni, D. Pandolfi, G. Paradossi and C. Palocci, New Biotechnol., 2017, 37, 138–143 CrossRef CAS PubMed.
- C. Y. Zhang, L. E. Parton, C. P. Ye, S. Krauss, R. Shen, C. T. Lin, J. A. Porco Jr. and B. B. Lowell, Cell Metab., 2006, 3, 417–427 CrossRef CAS PubMed.
- K. N. Nam, Y. S. Choi, H. J. Jung, G. H. Park, J. M. Park, S. K. Moon, K. H. Cho, C. Kang, I. Kang, M. S. Oh and E. H. Lee, Int. Immunopharmacol., 2010, 10, 493–499 CrossRef CAS PubMed.
- H. J. Koo, K. H. Lim, H. J. Jung and E. H. Park, J. Ethnopharmacol., 2006, 103, 496–500 CrossRef CAS PubMed.
- F. Li, W. Li, X. Li, F. Li, L. Zhang, B. Wang, G. Huang, X. Guo, L. Wan, Y. Liu, S. Zhang, S. Kang and J. Ma, J. Ethnopharmacol., 2016, 185, 77–86 CrossRef CAS PubMed.
- H. J. Koo, Y. S. Song, H. J. Kim, Y. H. Lee, S. M. Hong, S. J. Kim, B. C. Kim, C. Jin, C. J. Lim and E. H. Park, Eur. J. Pharmacol., 2004, 495, 201–208 CrossRef CAS PubMed.
- Y. Koriyama, K. Chiba, M. Yamazaki, H. Suzuki, K. Muramoto and S. Kato, J. Neurochem., 2010, 115, 79–91 CrossRef CAS PubMed.
- S. Habtemariam, Molecules, 2018, 23(1), 117 CrossRef PubMed.
- S. Habtemariam, Int. J. Mol. Sci., 2018, 19(1), 4 CrossRef PubMed.
- B. R. Kim, Y. A. Jeong, Y. J. Na, S. H. Park, M. J. Jo, J. L. Kim, S. Jeong, S. Y. Lee, H. J. Kim, S. C. Oh and D. H. Lee, Oncotarget, 2017, 8, 101952–101964 Search PubMed.
- R. Wang, K. C. MoYung, Y. J. Zhao and K. Poon, Int. J. Med. Sci., 2016, 13, 507–516 CrossRef CAS PubMed.
- E. S. Kim, C. S. Jeong and A. Moon, Oncol. Rep., 2012, 27, 567–572 CAS.
- H. Y. Hong and B. C. Kim, Biochem. Biophys. Res. Commun., 2007, 362, 307–312 CrossRef CAS PubMed.
- I. Dando, R. Pacchiana, E. D. Pozza, I. Cataldo, S. Bruno, P. Conti, M. Cordani, A. Grimaldi, G. Butera, M. Caraglia, A. Scarpa, M. Palmieri and M. Donadelli, Free Radical Biol. Med., 2017, 113, 176–189 CrossRef CAS PubMed.
- B. C. Kim, H. G. Kim, S. A. Lee, S. Lim, E. H. Park, S. J. Kim and C. J. Lim, Biochem. Pharmacol., 2005, 70, 1398–1407 CrossRef CAS PubMed.
- N. Wang, M. Zhu, S. W. Tsao, K. Man, Z. Zhang and Y. Feng, PLoS One, 2012, 7, e46318 CrossRef CAS PubMed.
- H. Ko, J. M. Kim, S. J. Kim, S. H. Shim, C. H. Ha and H. I. Chang, Bioorg. Med. Chem. Lett., 2015, 25, 4191–4196 CrossRef CAS PubMed.
- J. M. Kim, H. Ko, S. J. Kim, S. H. Shim, C. H. Ha and H. I. Chang, J. Biochem. Mol. Toxicol., 2016, 30, 45–54 CrossRef PubMed.
- J. H. Lee, D. U. Lee and C. S. Jeong, Food Chem. Toxicol., 2009, 47, 1127–1131 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- A. F. Habeeb, Anal. Biochem., 1966, 14, 328–336 CrossRef CAS PubMed.
- W. A. Bubnis and C. M. Ofner 3rd, Anal. Biochem., 1992, 207, 129–133 CrossRef CAS PubMed.
- S.-W. Lee, J.-M. Lim, S.-H. Bhoo, Y.-S. Paik and T.-R. Hahn, Anal. Chim. Acta, 2003, 480, 267–274 CrossRef CAS.
- F. Gelain, D. Silva, A. Caprini, F. Taraballi, A. Natalello, O. Villa, K. T. Nam, R. N. Zuckermann, S. M. Doglia and A. Vescovi, ACS Nano, 2011, 5, 1845–1859 CrossRef CAS PubMed.
- A. P. Hammersley, ESRF Internal Report, 1997 Search PubMed.
- R. Pugliese, F. Fontana, A. Marchini and F. Gelain, Acta Biomater., 2018, 66, 258–271 CrossRef CAS PubMed.
- R. L. Horan, K. Antle, A. L. Collette, Y. Wang, J. Huang, J. E. Moreau, V. Volloch, D. L. Kaplan and G. H. Altman, Biomaterials, 2005, 26, 3385–3393 CrossRef CAS PubMed.
- L. Mazzini, M. Gelati, D. C. Profico, G. Sgaravizzi, M. Projetti Pensi, G. Muzi, C. Ricciolini, L. Rota Nodari, S. Carletti, C. Giorgi, C. Spera, F. Domenico, E. Bersano, F. Petruzzelli, C. Cisari, A. Maglione, M. F. Sarnelli, A. Stecco, G. Querin, S. Masiero, R. Cantello, D. Ferrari, C. Zalfa, E. Binda, A. Visioli, D. Trombetta, A. Novelli, B. Torres, L. Bernardini, A. Carriero, P. Prandi, S. Servo, A. Cerino, V. Cima, A. Gaiani, N. Nasuelli, M. Massara, J. Glass, G. Soraru, N. M. Boulis and A. L. Vescovi, J. Transl. Med., 2015, 13, 17 CrossRef PubMed.
- A. Raspa, G. A. A. Saracino, R. Pugliese, D. Silva, D. Cigognini, A. Vescovi and F. Gelain, Adv. Funct. Mater., 2014, 24, 6317–6328 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- F. Taraballi, M. Campione, A. Sassella, A. Vescovi, A. Paleari, W. Hwang and F. Gelain, Soft Matter, 2009, 5, 660–668 RSC.
- E. Genove, C. Shen, S. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341–3351 CrossRef CAS PubMed.
- F. Taraballi, A. Natalello, M. Campione, O. Villa, S. M. Doglia, A. Paleari and F. Gelain, Front. Neuroeng., 2010, 3, 1 CAS.
- A. Horii, X. Wang, F. Gelain and S. Zhang, PLoS One, 2007, 2, e190 CrossRef PubMed.
- F. Gelain, S. Panseri, S. Antonini, C. Cunha, M. Donega, J. Lowery, F. Taraballi, G. Cerri, M. Montagna, F. Baldissera and A. Vescovi, ACS Nano, 2011, 5, 227–236 CrossRef CAS PubMed.
- M. Alkilzy, R. M. Santamaria, J. Schmoeckel and C. H. Splieth, Adv. Dent. Res., 2018, 29, 42–47 CrossRef CAS PubMed.
- S. Wan, S. Borland, S. M. Richardson, C. L. R. Merry, A. Saiani and J. E. Gough, Acta Biomater., 2016, 46, 29–40 CrossRef CAS PubMed.
- M. E. Davis, P. C. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS PubMed.
- P. C. Hsieh, M. E. Davis, J. Gannon, C. MacGillivray and R. T. Lee, J. Clin. Invest., 2006, 116, 237–248 CrossRef CAS PubMed.
- J. H. Jeong, T. G. Park and S. H. Kim, Pharm. Res., 2011, 28, 2072–2085 CrossRef CAS PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358(6365), eaam9756 CrossRef PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- G. A. Saracino, D. Cigognini, D. Silva, A. Caprini and F. Gelain, Chem. Soc. Rev., 2013, 42, 225–262 RSC.
- R. Sarroukh, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochim. Biophys. Acta, 2013, 1828, 2328–2338 CrossRef CAS PubMed.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS PubMed.
- H. Yokoi, T. Kinoshita and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS PubMed.
- S. L. Moradi, A. Golchin, Z. Hajishafieeha, M. M. Khani and A. Ardeshirylajimi, J. Cell. Physiol., 2018, 233(10), 6509–6522 CrossRef CAS PubMed.
- A. F. Martins, S. P. Facchi, P. C. F. da Camara, S. E. A. Camargo, C. H. R. Camargo, K. C. Popat and M. J. Kipper, J. Colloid Interface Sci., 2018, 525, 21–30 CrossRef CAS PubMed.
- A. Karimi, S. Karbasi, S. Razavi and E. N. Zargar, Adv. Biomed. Res., 2018, 7, 44 CrossRef PubMed.
- N. Saadatkish, S. Nouri Khorasani, M. Morshed, A. R. Allafchian, M. H. Beigi, M. Masoudi Rad, R. Esmaeely Neisiany and M. H. Nasr-Esfahani, J. Biomed. Mater. Res., Part A, 2018, 106(9), 2394–2401 CrossRef CAS PubMed.
- M. Chen, S. Gao, P. Wang, Y. Li, W. Guo, Y. Zhang, M. Wang, T. Xiao, Z. Zhang, X. Zhang, X. Jing, X. Li, S. Liu, Q. Guo and T. Xi, J. Biomater. Sci., Polym. Ed., 2018, 29, 461–475 CrossRef CAS PubMed.
- N. H. A. Ngadiman, M. Y. Noordin, A. Idris and D. Kurniawan, Proc. Inst. Mech. Eng., Part H, 2017, 231, 597–616 CrossRef PubMed.
- T. G. Kim and T. G. Park, Tissue Eng., 2006, 12, 221–233 CrossRef CAS PubMed.
- J. Yu, A. R. Lee, W. H. Lin, C. W. Lin, Y. K. Wu and W. B. Tsai, Tissue Eng., Part A, 2014, 20, 1896–1907 CrossRef CAS PubMed.
- J. Bockelmann, K. Klinkhammer, A. von Holst, N. Seiler, A. Faissner, G. A. Brook, D. Klee and J. Mey, Tissue Eng., Part A, 2011, 17, 475–486 CrossRef CAS PubMed.
- H. Seren, C. Goksu, C. Asli, U. Tamer, A. B. Tekinay and M. O. Guler, J. Mater. Chem. B, 2017, 5, 517–524 RSC.
- J. R. Paletta, S. Bockelmann, A. Walz, C. Theisen, J. H. Wendorff, A. Greiner, S. Fuchs-Winkelmann and M. D. Schofer, J. Mater. Sci.: Mater. Med., 2010, 21, 1363–1369 CrossRef CAS PubMed.
- A. S. Tayi, E. T. Pashuck, C. J. Newcomb, M. T. McClendon and S. I. Stupp, Biomacromolecules, 2014, 15, 1323–1327 CrossRef CAS PubMed.
- C. Surawut, A. Sirivat and P. Supaphol, Nanotechnology, 2007, 18(14), 145705 CrossRef.
- F. Gelain, A. Horii and S. Zhang, Macromol. Biosci., 2007, 7, 544–551 CrossRef CAS PubMed.
- C. Cunha, S. Panseri, O. Villa, D. Silva and F. Gelain, Int. J. Nanomed., 2011, 6, 943–955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Biomechanics assessment of different cross-linking SAPs; AFM morphological analysis; in vitro degradation of cross-linked hydrogels; QC of electrospun cross-linked nanofibrous scaffolds. See DOI: 10.1039/c8bm00825f |
| This journal is © The Royal Society of Chemistry 2019 |