
Manufacturing human pluripotent stem cell derived endothelial cells in scalable and cell-friendly microenvironments†
Haishuang
Lin
 a,
Qian
Du
b,
Qiang
Li
ac,
Ou
Wang
ac,
Zhanqi
Wang
d,
Christian
Elowsky
e,
Kan
Liu
b,
Chi
Zhang
b,
Soonkyu
Chung
f,
Bin
Duan
g and
Yuguo
Lei
*acgh
a,
Qian
Du
b,
Qiang
Li
ac,
Ou
Wang
ac,
Zhanqi
Wang
d,
Christian
Elowsky
e,
Kan
Liu
b,
Chi
Zhang
b,
Soonkyu
Chung
f,
Bin
Duan
g and
Yuguo
Lei
*acgh
aDepartment of Chemical and Biomolecular Engineering, University of Nebraska-Lincoln, Nebraska, USA. E-mail: ylei14@unl.edu; Fax: +402-472-6989; Tel: +402-472-5313
bDepartment of Biological Systems Engineering, University of Nebraska-Lincoln, Nebraska, USA
cBiomedical Engineering Program, University of Nebraska-Lincoln, Nebraska, USA
dDepartment of Vascular Surgery, Beijing Anzhen Hospital of Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Diseases, Beijing, China
eDepartment of Agronomy and Horticulture, University of Nebraska-Lincoln, Nebraska, USA
fDepartment of Nutrition and Health Sciences, University of Nebraska-Lincoln, Lincoln, NE, USA
gMary and Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, Nebraska, USA
hFred & Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, Nebraska, USA
First published on 16th November 2018
Abstract
Human pluripotent stem cell derived endothelial cells (hPSC-ECs) are of great value for studying and treating vascular diseases. However, manufacturing high quantity and quality hPSC-ECs with current cell culture technologies remains very challenging. Here, we report a novel method that can manufacture hPSC-ECs in scalable and cell-friendly microenvironments to address this challenge. Using this method, hPSCs are expanded and differentiated into ECs in microscale alginate hydrogel tubes. The hydrogel tubes protect cells from the highly variable hydrodynamic conditions and critical hydrodynamic stresses in the culture vessel and limit the cell mass less than the diffusion limits (of human tissue) to ensure efficient mass transport. The hydrogel tubes provide uniform and friendly microenvironments for cells to grow. This novel design leads to extremely high production efficiency. We showed that hPSC-ECs could be produced in 10 days with high viability (>90%), high purity (>80%) and high yield (∼5.0 × 108 cells per mL of microspace). The yield is about 250 times that of the current-state-of-the-art. hPSC-ECs made in these hydrogel tubes had similar in vitro and in vivo functions to hPSC-ECs generated by conventional cell culture methods. This simple, scalable, efficient, defined and cost-effective technology will make hPSC-ECs broadly available and affordable for various biomedical applications.
Introduction
Endothelial cells (ECs), which are major components of blood vessels, play an important role in normal vascular functions and are involved in a variety of vascular diseases.1,2 They are of great value for basic research, drug screening, cell therapy and tissue engineering.3–11 However, obtaining large numbers of primary ECs for those applications, in particular for clinical applications, is very challenging due to their limited proliferation capability and quick phenotype change during the in vitro culturing.12–17 Human pluripotent stem cells (hPSCs) provide a potential solution to this challenge.18 hPSCs, including human embryonic stem cells (hESCs)19 and induced pluripotent stem cells (iPSCs),20,21 have unlimited proliferation capability and can be cultured for a long time to generate large numbers of cells. They also have the potential to become all somatic cells of the human body.22,23 In addition, cells derived from patient-specific iPSCs have the patient's genetic information and can model many human diseases. Furthermore, autologous iPSC derived products induce a minimal immune response in vivo.24 These features make them an ideal cell source for various biomedical applications. Over the past decade, scientists have developed efficient protocols to differentiate hPSCs into ECs either through 3D suspension cultures (e.g. as embryonic bodies)18,25–30 or 2D monolayer cultures.4,31–34 These hPSC-ECs can provide unlimited cells for studying and treating diseases.While efficient differentiation protocols have been developed,34–38 methods or bioprocesses utilizing these protocols to produce large numbers of hPSC-ECs are still lacking. Current two dimensional (2D) cell culturing methods (e.g. using cell culturing flasks, in which cells are cultured as adherent cells on 2D surfaces) have low cell yield and are considered only suitable for preparing small-scale cells (e.g. <1010).39,40 3D suspension culturing methods (e.g. using stirred-tank bioreactors, in which cells are suspended in an agitated culture medium) have been widely studied to scale up the cell production and have had encouraging results. However, these studies also revealed the significant challenges.23,39–44 hPSCs have strong cell-to-cell interactions that make them aggregate to form large cell agglomerates in 3D suspension culturing.39 The mass transport to cells located at the center of large agglomerates (e.g. >400 μm diameter) becomes insufficient, leading to slow cell growth, cell death and uncontrolled differentiation.39 While agitating the culture can reduce cell agglomeration, it also generates hydrodynamic stresses that induce significant cell death.39,45,46 As a result, 3D suspension culturing typically results in low cell growth and low volumetric yield.23 For instance, we and others have shown that hPSCs typically expanded about four fold in four days per passage to yield about 2.0 × 106 cells per mL. At this yield, cells only occupy ∼0.4% of the bioreactor volume.41–44 In summary, producing hPSC-ECs in large scales is still a challenge.
To address this challenge, we previously developed a scalable and high-cell-yield method for expanding hPSCs.47 Using this method, cells are processed into and cultured in microscale alginate hydrogel tubes that are suspended in the cell culture medium in a culture vessel. The hydrogel tubes are used to protect cells from hydrodynamic stresses in the culture vessel and limit the cell mass smaller than the diffusion limit of human tissue (e.g. about 400 μm) to ensure efficient mass transport during the entire culturing. Together, the hydrogel tubes provide highly uniform and cell-friendly microenvironments for hPSCs to grow. This novel design led to significant improvements in hPSC culture efficiency and consistency. Under optimized culture conditions, hPSCs could be cultured in these hydrogel tubes for long-term (e.g. >10 passages, >50 days) with high cell viability, high growth rate (1000-fold expansion over 10 days per passage), high purity (>95% OCT4+) and high yield (∼5.0 × 108 cells per mL of microspace). The yield is about 250 times the yield of 3D suspension culturing. As discussed later, this high yield and high expansion per passage make large-scale hPSC production technically and economically feasible. We hypothesize that this technology is also suitable for making ECs from hPSCs (i.e. hPSC-SCs) with high efficiency and yield, and it can address the challenge of producing hPSC-ECs on large scales. However, whether hPSCs can be efficiently differentiated into ECs in the alginate hydrogel tubes and hPSC-ECs made in alginate hydrogel tubes are similar to hPSC-ECs made by conventional culture methods have not been studied. In this paper, we systematically addressed these questions. We found that hPSC-ECs could be made in the hydrogel tubes with extremely high yield and purity and these cells were similar to hPSC-ECs made in 2D cultures. As a result of these studies, a scalable bioprocess for making hPSC-ECs with a high volumetric yield, high viability and high purity was developed. This bioprocess will significantly advance the applications of hPSC-ECs in regenerative medicine.
Experimental
Routine cell culture
H9 hESCs (#WA09, WiCell) were purchased from WiCell Research Institute. Fib-iPSCs and MSC-iPSCs were obtained from Human Embryonic Stem cell core, Harvard Medical School. Fib-iPSCs and MSC-iPSCs were reprogrammed from fibroblasts and mesenchymal stem cells, respectively, by George Q. Daley Lab (Children's Hospital Boston, MA) and are well characterized and described in the literature.48 hPSCs (H9s and iPSCs) were maintained in a 6-well plate coated with Matrigel (#354277, BD Biosciences) in Essential 8™ medium (E8, #A1517001, Invitrogen). Cells were passaged every 4 days with 0.5 mM EDTA (#AM9260G, Invitrogen). The medium was changed daily. Cells were routinely checked for the expression of pluripotency markers, OCT4 and NANOG, their capability to form teratomas in immunodeficient mice, their karyotypes, and bacterial or mycoplasma contamination. Human aortic vascular smooth muscle cells (VSMC, #CC-2571, Lonza) and HUVECs (#00191027, Lonza) were obtained from Lonza.Processing alginate hydrogel tubes
Alginate hydrogel tubes are processed as described in our previous publication.47 Briefly, a custom-made micro-extruder was used to process the alginate tubes. A hyaluronic acid (HA, Lifecore Biomedical, #HA700K-5) solution containing single cells and an alginate (Wako Chemicals, 80-120 cp, #194-13321) solution was pumped into the central and side channel of the home-made micro-extruder, respectively, and extruded into a CaCl2 buffer (100 mM) to make alginate hydrogel tubes. Subsequently, CaCl2 buffer was replaced by the cell culture medium.Culturing hPSCs in alginate hydrogel tubes
The method for hPSC culture in alginate hydrogel tubes has been described in our previous publication.47Making hPSC-ECs in 2D culturing or alginate hydrogel tubes
For EC differentiation in 2D, hPSCs were dissociated with Accutase and plated on a Matrigel-coated plate at a density of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 in E8 medium with 10 μM RI. After 24 hours, the medium was replaced with a differentiation medium, consisting of N2B27 medium (1:1 mixture of DMEM/F12 (#SH30004.04, HyClone) with Glutamax-I (#35050061, Life Technologies) and Neurobasal medium (#21103049, Life Technologies) supplemented with N2 (#17502048, Life Technologies) and B27 minus vitamin A (#12587010, Life Technologies) with 8 μM CHIR99021 (#C6556, LC laboratories) and 25 ng mL−1 BMP4 (#314BP010, R&D Systems). After 3 days, the differentiation medium was replaced by the EC induction medium consisting of the StemPro-34 SFM medium (#10639011, Life Technologies) supplemented with 200 ng mL−1 VEGFA (#100–20, PeproTech) and 2 μM Forskolin (#F3917, Sigma). The induction medium was changed after 1 day. On day 6, ECs were replated on human fibronectin (#F1141, Sigma)-coated dishes at a density of 25000 cells per cm2 in the EC expansion medium, consisting of the EGM-2 medium (#CC3162, Lonza) supplemented with 50 ng mL−1 VEGFA. The EC expansion medium was replaced every other day.
000 cells per cm2 in E8 medium with 10 μM RI. After 24 hours, the medium was replaced with a differentiation medium, consisting of N2B27 medium (1:1 mixture of DMEM/F12 (#SH30004.04, HyClone) with Glutamax-I (#35050061, Life Technologies) and Neurobasal medium (#21103049, Life Technologies) supplemented with N2 (#17502048, Life Technologies) and B27 minus vitamin A (#12587010, Life Technologies) with 8 μM CHIR99021 (#C6556, LC laboratories) and 25 ng mL−1 BMP4 (#314BP010, R&D Systems). After 3 days, the differentiation medium was replaced by the EC induction medium consisting of the StemPro-34 SFM medium (#10639011, Life Technologies) supplemented with 200 ng mL−1 VEGFA (#100–20, PeproTech) and 2 μM Forskolin (#F3917, Sigma). The induction medium was changed after 1 day. On day 6, ECs were replated on human fibronectin (#F1141, Sigma)-coated dishes at a density of 25000 cells per cm2 in the EC expansion medium, consisting of the EGM-2 medium (#CC3162, Lonza) supplemented with 50 ng mL−1 VEGFA. The EC expansion medium was replaced every other day.
For EC differentiation in AlgTubes, single hPSCs were encapsulated in AlgTube (1.0 × 106 cells) and cultured in the E8 medium for 5 days. The E8 medium was removed and replaced with the EC differentiation medium. After 3 days, the differentiation medium was replaced by the EC induction medium. The induction medium was changed after 1 day. On day 10, cell masses were collected for the following analysis.
Producing hPSC-ECs in alginate hydrogel tubes in the prototype bioreactor
1 mL of the hPSC suspension in the AlgTube were suspended in a bioreactor. hPSCs were cultured in the E8 medium with 5% CO2 and 21% O2 at 37 °C for 5 days. The E8 medium was removed and replaced with the EC differentiation medium for 3 days, followed by the EC induction medium for 2 days. For the bioreactor, the medium was stored in a bellow bottle that was periodically pressed to flow the medium into, or released to withdraw the medium from the bioreactor. On day 10, hydrogel tubes were dissolved by adding 0.5 mM EDTA buffer. Cell masses were pelleted by centrifugation. Cell masses were dissociated into single cells through incubating in Accutase at 37 °C for 10 minutes. Magnetic beads coated with anti-CD144 antibodies (cat #130-097-857, Miltenyi Biotec) were added to pull down ECs with a magnetic cell separator. The bioprocess was repeated 2 times.Immunocytochemistry and flow cytometry
For 2D immunostaining, the cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 20 minutes, permeabilized with 0.25% Triton X-100 for 30 minutes, and blocked with 5% donkey serum for 1 hour before incubation with primary antibodies (Table S1†) at 4 °C overnight. After extensive washing, secondary antibodies (Table S1†) and 10 μM 4′,6-Diamidino-2-Phenylindole (DAPI) in 2% BSA were added and incubated at room temperature for 4 hours. Cells were washed with PBS 3 times before imaging with an A1 confocal microscope.For cell mass immunostaining, cell masses were fixed for 30 minutes, and then incubated with PBS +0.25% Triton X-100 +5% (vol/vol) goat serum +primary antibodies at 4 °C for 48 hours. After extensive washing, secondary antibodies in 2% BSA were added and incubated at 4 °C for 24 hours. Cells were washed with PBS three times before imaging with a confocal microscope.
For flow cytometry analysis, the harvested cells were dissociated into single cells with Accutase, and then fixed with 4% PFA at room temperature for 20 minutes. Single cells were stained with primary antibodies (Table S1†) at 4 °C overnight. After 3 times washing with 1% BSA in PBS, secondary antibodies were added and incubated at room temperature for 2 hours. Cells were washed with 1% BSA in PBS and analyzed using a Cytek DxP10 flow cytometer. Single-color and isotype controls served as compensation and negative gating.
RNA extraction, cDNA synthesis and quantitative PCR
Total RNA for qPCR and RNA sequencing was extracted from 2D-ECs and AlgTube-ECs on day 5 of the differentiation using Trizol (#15596018, Invitrogen), according to the manufacturer's instructions. Reverse transcription is carried out with the Maxima First Strand cDNA Synthesis Kit (#K1642, Life Technologies). Quantitative real-time PCR was carried out in an Eppendorf MasterCycler RealPlex4 (ThermoFisher Scientific) using the Power SYBR Green PCR Master Mix (#4367659, ThermoFisher), according to the manufacturer's instructions. The data were normalized to the endogenous GAPDH. The primer sequence is listed in Table S2.†Embryoid body (EB) differentiation and teratoma formation in vivo
The Animal Care and Use Committee of the University of Nebraska, Lincoln approved the animal research protocol and all procedures followed the approved protocol. Details of EB differentiation and teratoma assays can be found in our previous publication.47 Three mice (two teratomas per mouse) were used for teratoma assay for each hPSC line.Tube formation
200 μL of Matrigel was aliquoted into each well of a 12-well plate and incubated for 30 minutes at 37 °C to allow the gel to solidify. 80000 hPSC-ECs were then seeded onto the matrix and cultured for 24 hours at 37 °C until image acquisition. Cells were fixed with 2% PFA for 10 minutes and analyzed using a Zeiss fluorescence microscope.
Co-culture assay of hPSC-ECs and VSMCs
For the functional tube formation in vitro, the hPSC-ECs and the VSMCs were prestained with DiI (red) and DiO (green) (#V22889, Vybrant multicolor cell-labeling kit, Fisher Scientific), respectively, according to the manufacturer's instructions. Cells were grown in StemPro-34 SFM supplemented with 50 ng mL−1 VEGF-A. For the experiment, 2.0 × 104 hPSC-ECs per cm2 and 2.0 × 104 VSMCs per cm2 were co-cultured for 24 hours in the incubator. Cells were fixed with 2% PFA for 10 minutes and analyzed using a Zeiss fluorescence microscope.Uptake of Dil-Ac-LDL
Cells were incubated with 2.5 g mL−1 DiI-Ac-LDL-Alexa597 (#L3484, Molecular Probes/Life Technologies) in StemPro-34 medium for 4 hours at 37 °C. Thereafter, cells were washed three times with PBS and fixed with 4% PFA for 10 minutes. DNA was counterstained with DAPI. Cellular uptake of DiI-Ac-LDL-Alexa 597 was visualized with a fluorescence microscope (Zeiss).trans-Endothelial electrical resistance (TEER)
hPSC-ECs were seeded at a density of 1.0 × 106 cells per mL on 24-well cell culture inserts (0.4 μM pore size, cat # 3470, Corning) coated with fibronectin. Cells were grown for 72 hours in the EGM-2 medium. The medium was then changed to the starvation medium (basal EBM medium +0.5% FBS) with or without vascular permeability factors (100 ng mL−1 TNF-α (cat # 300-01A, Peprotech), IL-1β (cat # 200-01B, Peprotech) and 100 ng mL−1 VEGF-A) for 30 hours. trans-Endothelial electrical resistance was recorded using a Digital AC/DC Clamp Meter (cat # MS2101, MASTECH). This experiment was performed in triplicate for each condition and repeated 3 times. Results are depicted as the steady-state TEER values with the blank filter subtracted.Glycolysis
Cell glycolysis was analyzed using the Glycolysis Assay Kit (cat # 600450, Cayman Chemical) according to the manufacturer's instruction. Briefly, on day 5 of the differentiation, 10 μL of the cell culture medium was collected and mixed with 90 μL assay buffer. Then, 100 μL of the reaction solution was added to each well, and the plate was incubated with gentle shaking for 30 minutes at room temperature. A plate reader was used to read the absorbance at 490 nm.Matrigel plug assay
Animal procedures were performed in accordance with an IACUC-approved protocol reviewed by the University of Nebraska–Lincoln Animal Care and Use Committee. The female 6–8 week old SCID mice (Charles River Laboratory) were used. Differentiated ECs were added to the Matrigel mixture to a final concentration of 10 million cells per mL. The Matrigel mixture (300 μL) was then immediately engrafted subcutaneously into the dorsal flank of the mouse. Two implants were engrafted per animal. Implants were recovered after 14 days, and then the implants were excised. They were fixed in 4% PFA. Hematoxylin & eosin staining and immunostaining were performed to analyze the tube formation potential in vivo.Western blotting
On day 5 of the differentiation, 2D-ECs or AlgTube-ECs were harvested and cell lysates were prepared using the Pierce IP™ lysis buffer (cat # 87788, ThermoFisher) supplemented with Halt™ Protease & Phosphatase Single-Use Inhibitor Cocktail (100×, cat # 78442, ThermoFisher). 30 μg of total proteins for each sample were subjected to SDS-PAGE analysis on a 4%–12% NuPAGE™ bis–tris protein gel (cat # NP0321BOX, ThermoFisher). GAPDH was used as a loading control.RNA sequencing and data analysis
Total RNA of day 6 ECs cultured in 2D and day 10 ECs cultured in AlgTube was prepared with the RNeasy mini kit (cat # 74104 QIAGEN) according to the manufacturer's instruction. Libraries were prepared with a TruSeq Stranded mRNA Library Prep Kit and sequenced with Illumina NextSeq 500. 20 million 75 bp paired-end reads were generated for each sample. Methods for the data processing, heatmap generating, PCA analysis, and differential gene expression analysis have been described in previous publication.47Statistical analysis
The data are presented as the mean ± standard deviation (SD) from three independent experiments. We used an unpaired t-test to compare two groups and one-way ANOVA to compare more than two groups. A sample size of 3 was selected so that at a significance level of 0.05 there is at least 95% chance of detecting two SD's difference in outcome between the groups. All data were processed using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA).Accession numbers
The accession numbers for the data reported in this paper are GEO: GSE99776 and GSE109683. All other data supporting the findings of this study are available within the paper and its ESI.†Results
Processing cells into alginate hydrogel tubes
The detailed method for designing and fabricating the extruder has been described in our previous publication.47 A micro-extruder was designed and built for processing the cells into alginate hydrogel tubes (or AlgTubes) (Fig. 1A and B). Using two syringe pumps, a cell suspension and an alginate solution are pumped into the central and side channel of the micro-extruder, respectively (Fig. 1A). They form coaxial core–shell flows that pass through a nozzle and into a CaCl2 buffer. The Ca2+ ions instantly crosslink the shell alginate solution into an alginate hydrogel tube (Fig. 1B). Subsequently, cells are grown in the hydrogel tubes that are suspended in a cell culture medium. The hydrogel tubes protect cells from hydrodynamic stresses and confine the cell mass less than 400 μm (in radial diameter) to ensure efficient mass transport (Fig. 1C). In the hydrogel tubes, single hPSCs expand and fill the tubes in about 9 days. To generate massive numbers of hPSCs, the day 9 cell masses can be released by dissolving the hydrogel tubes in 0.5 mM EDTA solution (5 minutes at room temperature), and dissociated into single cells with Accutase and processed into new hydrogel tubes for a second round of expansion. Once the targeted cell number is reached, hPSCs can be differentiated into ECs within 5 days (Fig. 1D). Our following studies showed that using alginate hydrogel tubes led to extremely high efficiency of hPSC-EC production.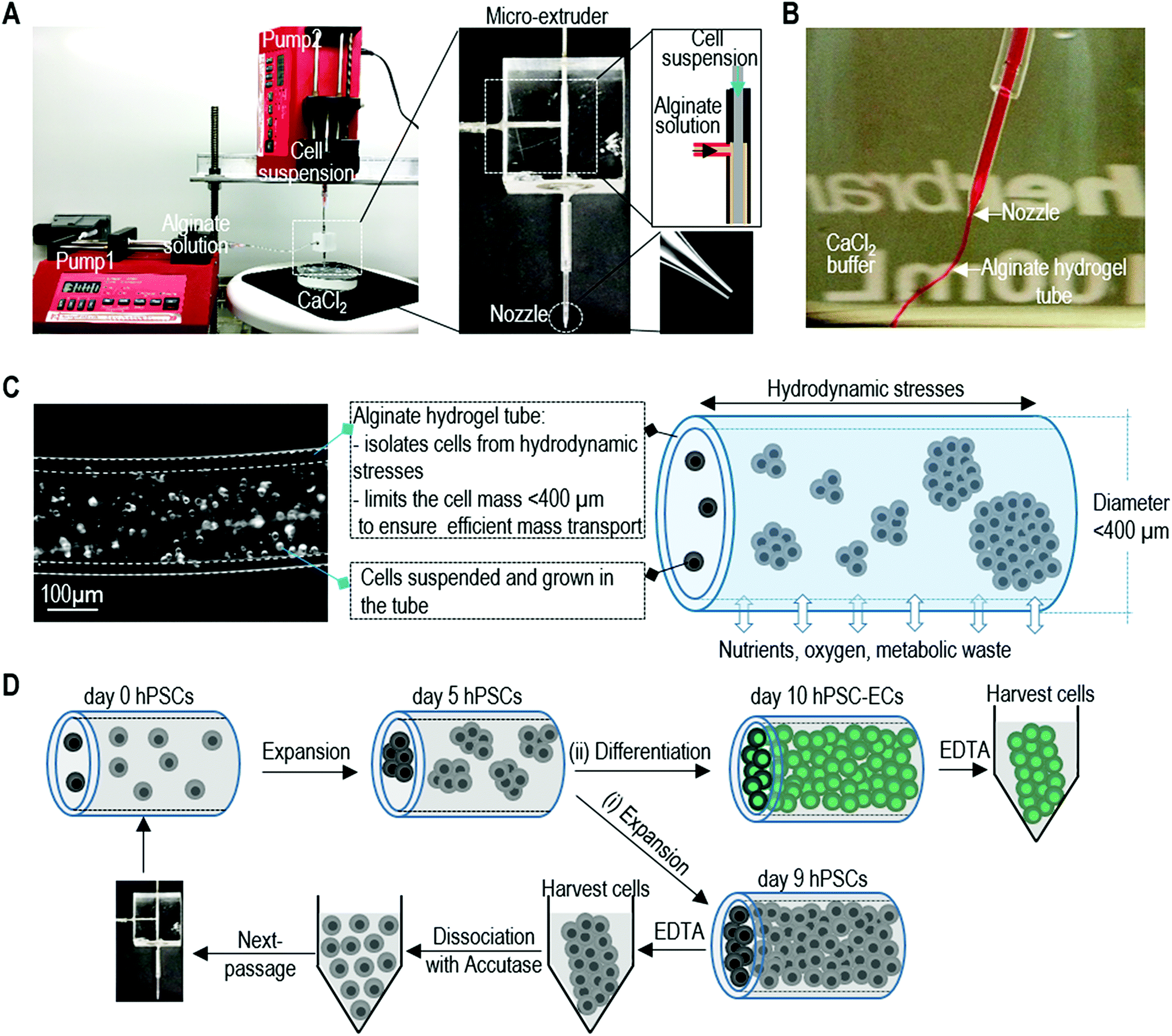 | ||
| Fig. 1 Overview of culturing cells in alginate hydrogel tubes (AlgTubes). (A and B) A micro-extruder is built for processing cells into microscale alginate hydrogel tubes. A cell suspension and an alginate solution are pumped into the central channel and side channel of the micro-extruder, respectively, to form coaxial core–shell flows that are extruded through the nozzle into a CaCl2 buffer. The shell alginate solution is instantly crosslinked by Ca2+ ions to form an alginate hydrogel tube. (C) Subsequently, cells are suspended and cultured in the alginate hydrogel tube that is suspended in a cell culture medium in a culture vessel. The hydrogel tube protects cells from the hydrodynamic stresses in the culture vessel and limits the cell mass <400 μm (in radial diameter) to ensure efficient mass transport. The tube provides uniform and cell-friendly microspace that allows cells to interact with each other and expand. Nutrients, oxygen and metabolic waste can efficiently diffuse through the hydrogel shell. (D) To produce large-scale human pluripotent stem cell derived endothelial cells (hPSC-ECs), hPSC are processed into the hydrogel tubes at low seeding density and expanded for 9 days to fill the tubes. (i) The day 9 cell masses can be released via dissolving the hydrogel tubes in 0.5 mM EDTA solution, dissociated into single cells with Accutase and processed into new hydrogel tubes for a second round of expansion. (ii) Once the targeted cell number of hPSCs is reached, hPSCs (around day 5) can be differentiated into ECs within 5 days. | ||
hPSC expansion in alginate hydrogel tubes
We first checked our starting cells including H9 hESCs, iPSCs reprogrammed from human dermal fibroblasts (Fib-iPSCs), and iPSCs reprogrammed from mesenchymal stem cells (MSC-iPSCs), and confirmed that they were high quality pluripotent stem cells (Fig. S1†). When cultured in hydrogel tubes, single hPSCs associated with each other to form small clusters within 24 hours. These clusters grew in diameter when cells divided, and they filled the tubes to form monodisperse fibrous cell mass in about 9 days (Fig. 2A and B). Very few dead cells were found during the culturing via live/dead cell staining (Fig. 2C). If seeded at 1.0 × 106 cells per mL, hPSCs expanded ∼30, ∼150, ∼480 fold to yield ∼30×, 150×, 480 × 106 cells per mL on days 5, 7 and 9, respectively (Fig. 2D and E). Immunostaining showed that the majority of the cells expressed pluripotent stem cell markers including OCT4, NANOG, SSEA4 and ALP (Fig. 2F). Flow cytometry found that more than 95% of cells were positive for these markers (Fig. 2G and H). Fib-iPSCs and MSC-iPSCs had very similar behaviors to H9s in the hydrogel tubes. The results were also very close to those in our previous publication,47 confirming that expanding hPSCs in alginate hydrogel tubes are robust and reproducible.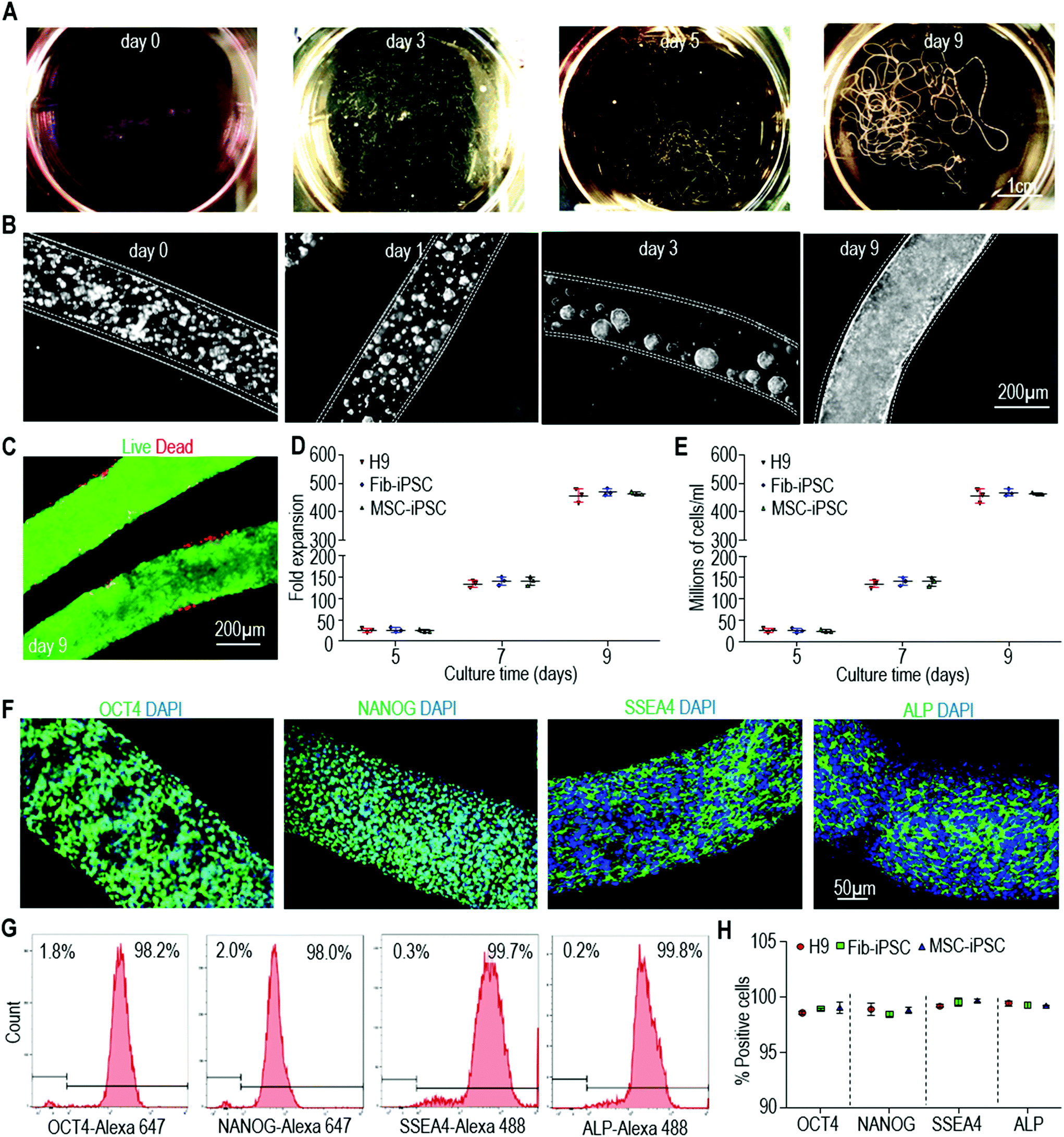 | ||
| Fig. 2 hPSC expansion in alginate hydrogel tubes. (A) Photographs of H9 hESCs in hydrogel tubes on days 0, 3, 5 and 9. (B) Phase images of H9 hESCs in hydrogel tubes on day 0, 1, 3 and 9. (C) Live/dead staining of day 9 cells in hydrogel tubes. (D, E) Seeded at 1.0 × 106 cells per mL, H9s, Fib-iPSCs and MSC-iPSCs expanded ∼30-, 150-, 480-fold to yield ∼30×, 150×, 480 × 106 cells per mL on days 5, 7 and 9. Data are represented as mean ± SD (n = 3). (F) Immunostaining of day 9 H9 hESCs for pluripotency markers OCT4, NANOG, SSEA4 and ALP. (G and H) Flow cytometry analysis of day 9 cells for pluripotency markers. Data are represented as mean ± SD (n = 3). The three hPSCs (H9s, Fib-iPSCs, MSC-iPSCs) grew similar in the hydrogel tubes. Fib-iPSCs and MSC-iPSCs: iPSCs made from human fibroblasts and mesenchymal stem cells, respectively. | ||
Differentiating hPSCs into ECs using 2D culturing
Patsch et al. reported an efficient protocol that could differentiate hPSCs into ECs in 6 days in 2D culturing.4 This protocol is simple and quick, and thus is very appealing for making ECs for various biomedical applications, especially for clinical applications (Fig. S2A†). We successfully repeated this protocol with our three hPSCs (Fig. S2†). The produced ECs had the cobblestone morphology typical of ECs (Fig. S2B, F and J†). Immunostaining detected no undifferentiated OCT4+ and NANOG+ hPSCs (Fig. S2C, G and K†). Immunostaining showed that the majority of the generated cells expressed EC markers including PECAM1 (or CD31) and VE-Cadherin (or CD144) (Fig. S2D, H and L†). Flow cytometry analysis found that about 82% ± 1.9% of the cells were PECAM1+/VE-Cadherin+ (Fig. S2E, I and M†). The three hPSCs had similar differentiation outcomes (Fig. S2†). Our results were also very similar to those reported by Patsch et al.,4 indicating the robustness of the differentiation protocol. We termed ECs made in 2D culturing as 2D-ECs.Making hPSC-ECs in alginate hydrogel tubes
We then applied the same differentiation protocol to generate ECs in alginate hydrogel tubes (Fig. 3A). To study if the cell mass size (or diameter) affects the differentiation outcomes, we expanded and differentiated hPSCs into ECs in hydrogel tubes with a diameter of 120, 250 and 330 μm. The diameter of the hydrogel tubes can be precisely controlled through adjusting the nozzle diameter of the micro-extruder (Fig. 3B–D). Single hPSCs were processed into the hydrogel tubes and expanded for 5 days to generate small spheroids. On day 5, the differentiation medium was applied. Cells were harvested for analysis on day 10. hPSCs were successfully differentiated into ECs in all three tubes with similar cell viability, differentiation efficiency and yield (Fig. 3E and F). And, quantitative RT-PCR analysis showed that pluripotency markers (OCT4 and NANOG) had a significant decrease in all three tubes (Fig. 3G). However, quantitative RT-PCR analysis revealed that cells in 250 μm hydrogel tubes had significantly higher expressions for some important EC genes including PECAM1, CD34, VEGFA, FN, COL4A5 and COL4A6 (Fig. 3H and I). We thus decided to use 250 μm hydrogel tubes for the rest of the studies.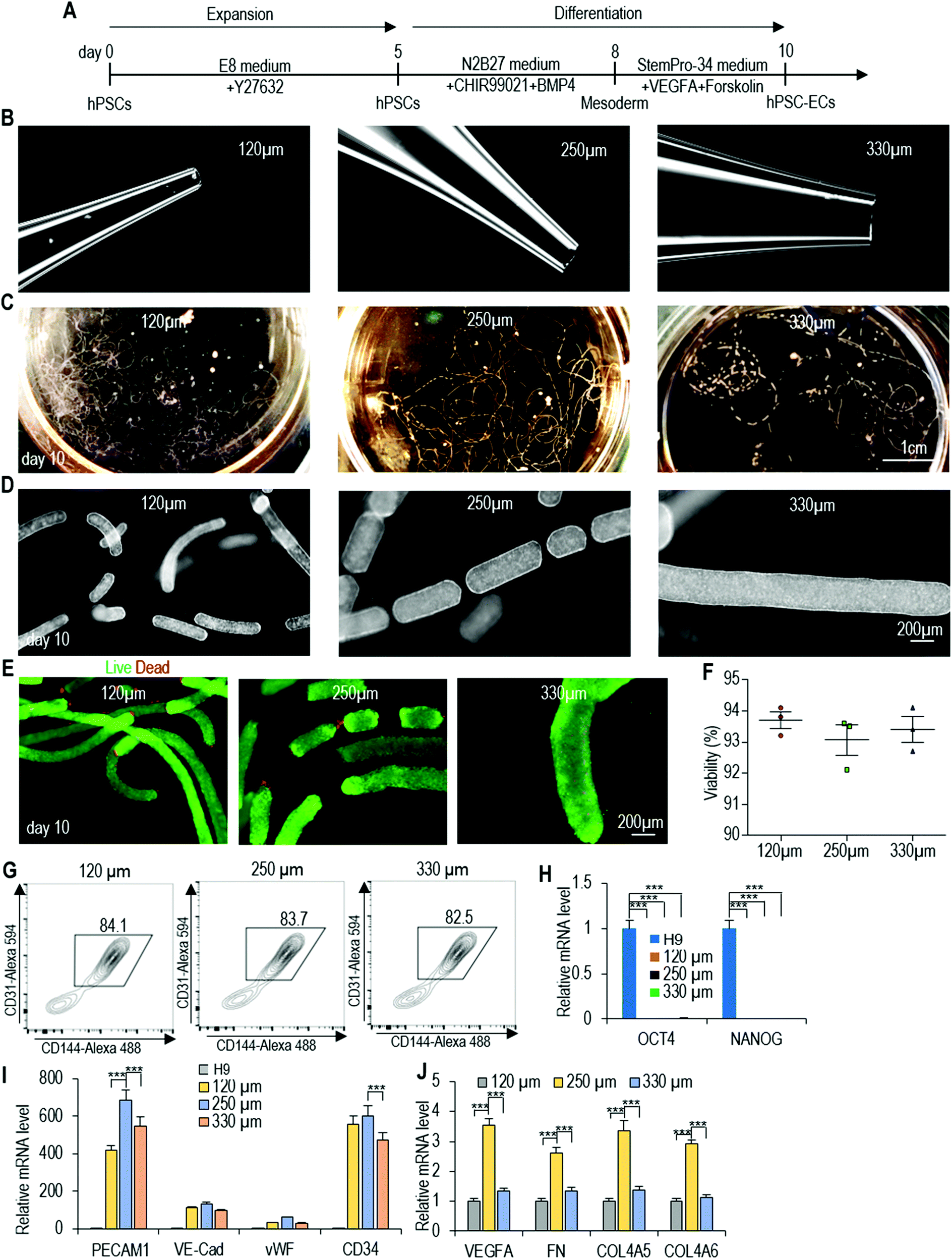 | ||
| Fig. 3 Effect of the cell mass diameter on EC differentiation. (A) The EC differentiation protocol. (B) Nozzles used to process 120 μm, 250 μm and 330 μm (diameter) hydrogel tubes. (C and D) Photographs and phase images of day 10 hPSC-ECs in 120 μm, 250 μm and 330 μm hydrogel tubes. (E–G) Live/dead staining, cell viability and flow cytometry analysis of day 10 hPSC-ECs. Data are represented as mean ± SD (n = 3). (H–J) qRT-PCR analysis of gene expression of H9s and day 10 hPSC-ECs in 120 μm, 250 μm and 330 μm hydrogel tubes, including pluripotency markers OCT4 and NANOG, EC markers PECAM1, VE-Cadherin, vWF and CD34, growth factor VEGFA, and extracellular matrices FN, COL4A5 and COL4A6. Data are represented as mean ± SD (n = 3). H9s were used for making ECs. | ||
Live/dead staining and cell viability analysis showed very high cell viability on day 10 (Fig. 4A–C). About 4.5 × 108 cells per mL of microspace were produced on day 10 (Fig. 4D). Immunostaining and confocal imaging showed that the majority of produced cells were positive for EC markers PECAM1 and VE-Cadherin (Fig. 4E and F). ECs were uniformly distributed, and no cysts were found in the cell mass, indicating no or little cell death during the differentiation. Flow cytometry analysis found that 82.7% ± 1.6% of the cells were PECAM1+ and VE-Cadherin+ (Fig. 4G). When the day 10 cells were released from the tubes and dissociated into single cells and plated on a Matrigel-coated plate at high density, they formed a monolayer with tight cell–cell interactions. Majority of the cells were PECAM1+ and VE-Cadherin+ cells and ∼11% ± 1.8% SM22A+ cells, but no OCT4+ and NANOG+ undifferentiated hPSCs were detected (Fig. 4H–J). H9s, Fib-iPSCs and MSC-iPSCs had similar outcomes (Fig. S3 and S4†). We found that the differentiation efficiency in 2D culturing and in hydrogel tubes was very close. We termed ECs that are made in hydrogel tubes and have not been cultured on any 2D surfaces as AlgTube-ECs.
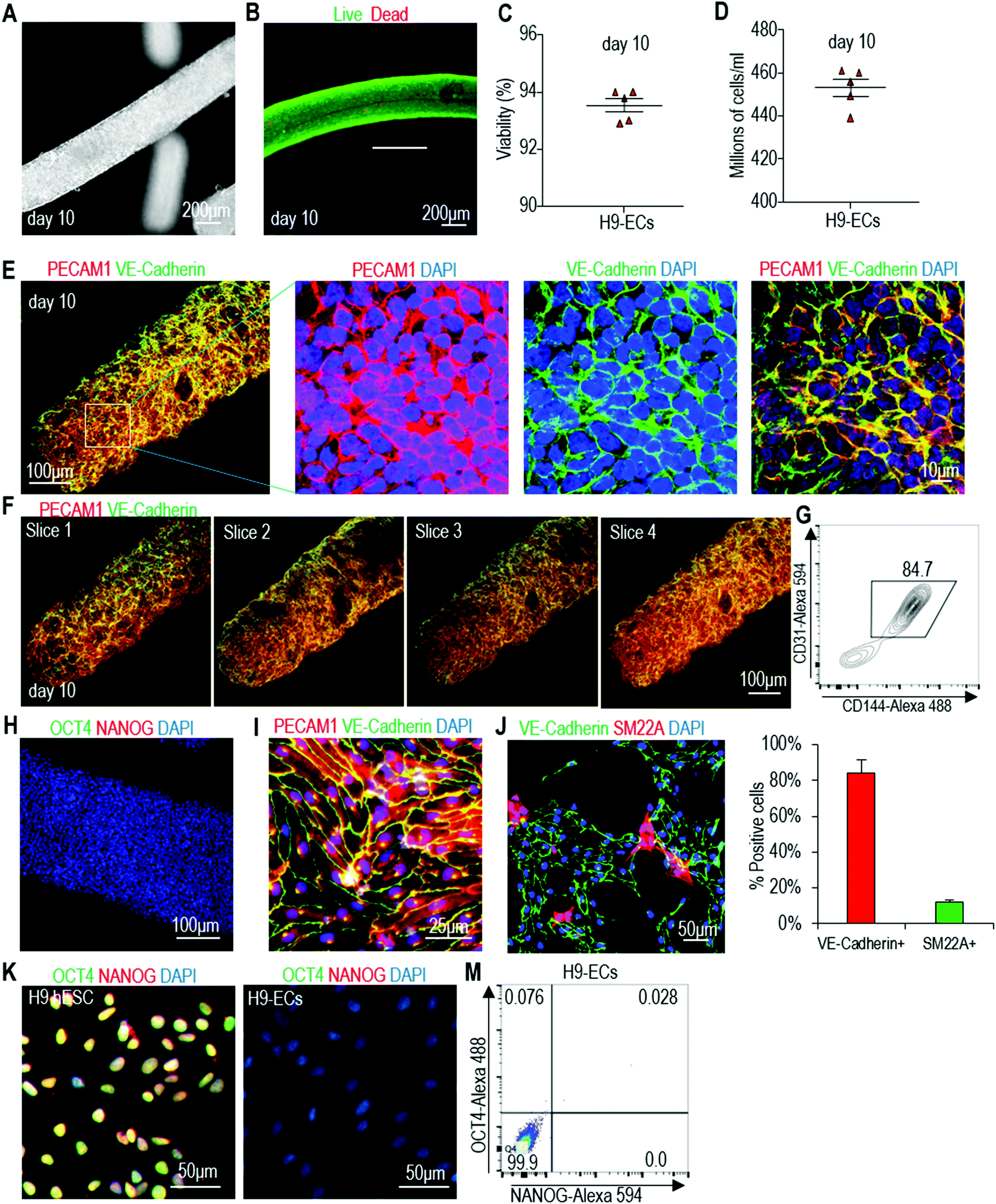 | ||
| Fig. 4 Differentiate H9 hESCs into ECs in alginate hydrogel tubes. (A) Phase images of day 10 cells. (B and C) Live/dead staining and cell viability analysis of day 10 cells. Data are represented as mean ± SD (n = 5). (D) Volumetric yield on day 10. Data are represented as mean ± SD (n = 5). (E–G) Immunostaining and flow cytometry analysis of day 10 cells for EC markers PECAM1 and VE-Cadherin. Confocal microscopy images of 4 slices of one fibrous cell mass are shown in (F). (H) Immunostaining of day 10 cells for pluripotency markers OCT4 and NANOG. (I–M) The day 10 cell masses were dissociated into single cells and plated on 2D surface overnight. Immunostaining showed majority of the cells were PECAM1+/VE-Cadherin+ (I), and detected ∼10% SM22A+ cells (J), but no OCT4+/NANOG+ undifferentiated hPSCs (K, M). Fib-iPSCs and MSC-iPSCs had similar results as shown in ESI figures.† | ||
Properties of hPSC-ECs made in 2D cultures and alginate hydrogel tubes
Our culture system provides a 3D microenvironment for cells. Recent studies found that ECs cultured in 3D hydrogels had different phenotypes and functional properties compared to those cultured in 2D substrates.49,50 We thus asked if AlgTube-ECs and 2D-ECs were similar in phenotypes, functions and gene expression. Using fluorescently labelled acetylated LDL (Ac-LDL) and microscope imaging, we found that they had similar capability to take up lipids (Fig. 5A). They formed similar tubular structures in the tube formation assay, indicating similar angiogenic potential (Fig. 5B and C). Both AlgTube-ECs and 2D-ECs formed tight barriers as shown by the high trans-endothelial electrical resistance (TEER) value.51 The barrier tightness of both AlgTube-ECs and 2D-ECs was disrupted by TNF-α, IL-1β and VEGF-A as shown by a sharp decrease of the TEER values. AlgTube-ECs and 2D-ECs performed very similar to the primary human umbilical vein endothelial cells (HUVECs) (Fig. 5D). To test the angiogenic potential of AlgTube-ECs and 2D-ECs in vivo, we injected them with a Matrigel matrix into the immunodeficient mice. Hematoxylin & eosin-staining and immunostaining revealed a similar blood vessel density and structure in the matrix for AlgTube-ECs and 2D-ECs (Fig. 5E–G). We also used qRT-PCR to quantitatively analyze the expressions of pluripotency markers (OCT4 and NANOG), and a few genes important to ECs including the surface markers (PECAM1, VE-Cadherin, vWF and CD34), growth factors (VEGFA, VEGFB and VEGFC) and extracellular matrices (FN and COL4A). Both ECs had a very low expression level of pluripotency markers (Fig. 5H). AlgTube-ECs had enhanced expressions of the EC related genes (Fig. 5I and J). Similar results were found for iPSC derived ECs (Fig. S5†).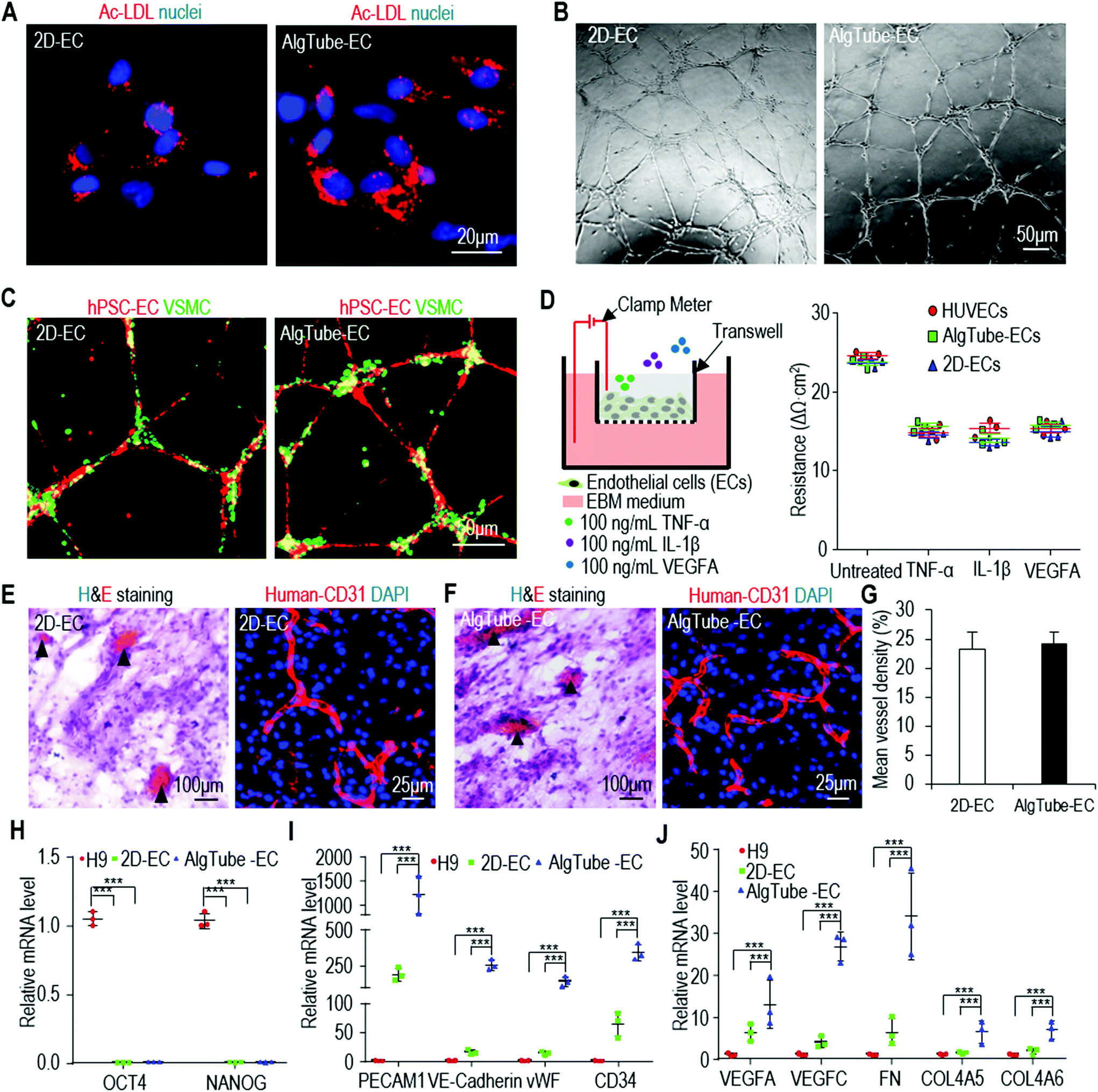 | ||
| Fig. 5 Properties of ECs derived from H9 hESCs in alginate hydrogel tubes (AlgTube-ECs) and 2D culture (2D-ECs). (A) Both ECs efficiently taken up fluorescence-labelled acetylated LDL (Ac-LDL). (B and C) Both ECs formed tubular network when plated on Matrigel for 24 hours. The co-plated vascular smooth muscle cells attached to the EC network. (D) TEER properties, either untreated or treated with 100 ng mL−1 TNF-α, 100 ng mL−1 IL-1β or 100 ng mL−1 VEGF-A, of HUVECs, AlgTube-ECs and 2D-ECs were similar. Data are represented as mean ± SD of three biological replicates (n = 3). (E–G) When transplanted subcutaneously with a Matrigel matrix, both ECs formed nice vascular structures with similar mean vessel density. (H) qRT-PCR of pluripotency markers OCT4 and NANOG for H9s, 2D-ECs and AlgTube-ECs. (I and J) qRT-PCR showed AlgTube-ECs had higher expressions for some key genes related to ECs. Data are represented as mean ± SD of three biological replicates (n = 3). Fib-iPSCs and MSC-iPSCs had similar results as shown in ESI figures.† ***P < 0.001. | ||
Transcriptome analysis of AlgTube-ECs and 2D-ECs
The above qRT-PCR findings drove us to study the genome-wide gene expression difference between AlgTube-ECs and 2D-ECs. We sequenced the mRNAs of the undifferentiated H9s, AlgTube-ECs and 2D-ECs derived from H9s (3 biological replicates for each). Hierarchical clustering analysis showed that AlgTube-ECs and 2D-ECs clustered closely and were very different from H9s (Fig. 6A). The genome-wide gene expression profile correlation coefficients between the AlgTube-ECs and 2D-ECs were >0.78, indicating that they had similar global gene expressions (Fig. 6B). Principal components analysis (PCA) showed slightly different gene expression between AlgTube-ECs and 2D-ECs (Fig. 6C). Differential gene expression analysis identified 800 genes upregulated in AlgTube-ECs, and 802 genes upregulated in 2D-ECs. GO term analysis showed that genes enriched in AlgTube-ECs are related to vasculature development and vascular network formation, ECM assembly and cell adhesion, integrin-mediated signaling, and Notch signaling (Fig. 6D and Fig. S6A†), and genes enriched in 2D-ECs are related to the cell cycle and proliferation and oxidative stress response (Fig. 6D and Fig. S6B†). These results indicated that a 3D environment (AlgTubes) promoted vascular morphogenesis, while 2D culturing induced ECs to have a proliferative phenotype. Our results agree well with the literature.49,50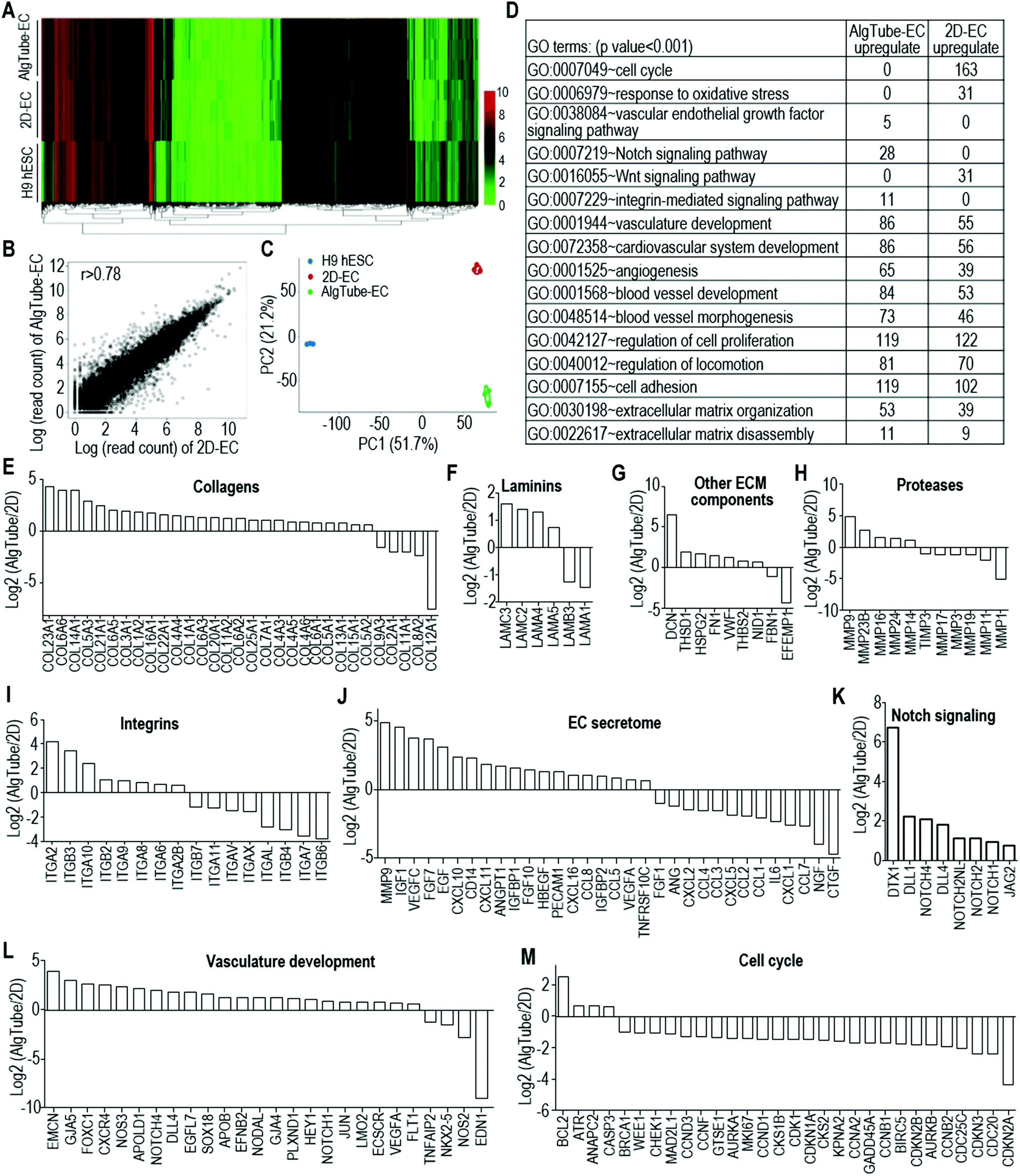 | ||
| Fig. 6 The whole transcriptome analysis of AlgTube-ECs and 2D-ECs derived from H9s. (A and B) Global heat map and global gene expression correlation coefficients (scatterplot in log scale) of AlgTube-ECs and 2D-ECs. (C) The principal component analysis (PCA) showing slightly different gene expression between AlgTube-ECs and 2D-ECs. Three biological replicates are used for each sample. (D) GO terms that have significant differentially expressed genes between AlgTube-ECs and 2D-ECs. (E–M) Log2 (expression level in AlgTube-ECs/expression level in 2D-ECs) of genes related to extracellular matrices (E–G), proteases (H), integrins (I), EC secretome (J), Notch signaling (K), vasculature development (L) and cell cycle (M). | ||
Detailed gene expression analysis with the RNA-Seq data showed the following differences between AlgTube-ECs and 2D-ECs: (a) AlgTube-ECs had higher expressions of ECM genes including collagens: COL23A1, COL6A6, COL14A1, COL5A3, COL21A1, COL6A5, COL3A1, COL1A2, COL16A1, COL22A1, COL4A4, COL1A1, COL6A3, COL20A1, COL11A2, COL6A2, COL25A1, COL7A1, COL4A3, COL4A5, COL4A6, COL6A1, COL5A1, COL13A1, COL15A1 and COL5A2; laminins: LAMC3, LAMC2, LAMA4 and LAMA5; other ECM components: DCN, THSD1, HSPG2, FN1, VWF, THBS2 and NID1; proteases: MMP9, MMP23B, MMP16, MMP24 and MMP14, MMP17 and integrins: ITGA2, ITGB3, ITGA10, ITGB2, ITGA9, ITGA8, ITGA6 and ITGA2B; (Fig. 6E–I and Fig. S7A†); 2D-ECs had higher expressions of ECM genes including collagens: COL12A1, COL8A2, COL11A1, COL2A1, COL9A3; laminins: LAMA1 and LAMB3; other ECM components: EFEMP1 and FBN1; proteases: MMP1, MMP11, MMP19, MMP3, MMP17 and TIMP3 and integrins: ITGB6, ITGA7, ITGB4, ITGAL, ITGAX, ITGAV, ITGA11 and ITGB7 (Fig. 6E–I and Fig. S7A†). (b) AlgTube-ECs had higher expressions of genes for EC secreted factors including MMP9, IGF1, VEGFC, FGF7, EGF, CXCL10, CD14, CXCL11, ANGPT1, IGFBP1, FGF10, HBEGF, PECAM1, CXCL16, CCL8, IGFBP2, CCL5, VEGFA and TNFRSF10C (Fig. 6J and Fig. S7B†); 2D-ECs had higher expressions of genes for EC secreted factors including CTGF, NGF, CCL7, CXCL1, IL6, CCL1, CCL2, CXCL5, CCL3, CCL4, CXCL2, ANG and FGF1 (Fig. 6J and Fig. S7B†). (c) AlgTube-ECs had higher expressions of genes for Notch signaling (Fig. 6K and Fig. S7C†), vasculature development (Fig. 6l and Fig. S7D†), glycolysis including PGAM2, ALDOB, ENO2 and PGM2 (Fig. S8A†), angiogenesis (Fig. S8B and C†), hypoxia signaling (Fig. S8D†), and ephrin receptor signaling (Fig. S8E†); (d) 2D-ECs had higher expressions of genes for the cell cycle and proliferation (Fig. 6M and Fig. S7E†), and Wnt signaling including FOSL1, MYC, WISP1, CCND1, KREMEN1 and DKK3 (Fig. S8F†).
We carried out further tests to study if the differences found in the RNA-Seq data analysis could also be found in the cell phenotype or function assays. Using Ki67 immunostaining and flow cytometry analysis, we found that more cells were proliferating in 2D-ECs than in AlgTube-ECs (Fig. 7A and B). Glycolysis analysis showed that AlgTube-ECs had a higher glycolytic rate than 2D-ECs (Fig. 7C). Western blotting analysis showed that AlgTube-ECs had higher expression for CD31, CD144, FN, NOTCH4 and ITGA2 than 2D-ECs (Fig. 7D). The results agree well with the RNA-Seq data.
 | ||
| Fig. 7 Functional comparison of 2D-ECs and AlgTube-ECs. (A and B) Immunostaining and flow cytometry of Ki67 showed more proliferation cells in 2D-ECs than in AlgTube-ECs. (C) Glycolysis analysis showed AlgTube-ECs produced more L-lactates than 2D-ECs. ***P < 0.001. (D) Western blotting showed that AlgTube-ECs had higher expression of CD31, CD144, FN, NOTCH4 and ITGA2 than 2D-ECs. | ||
A prototype bioreactor for scalable hPSC-EC production
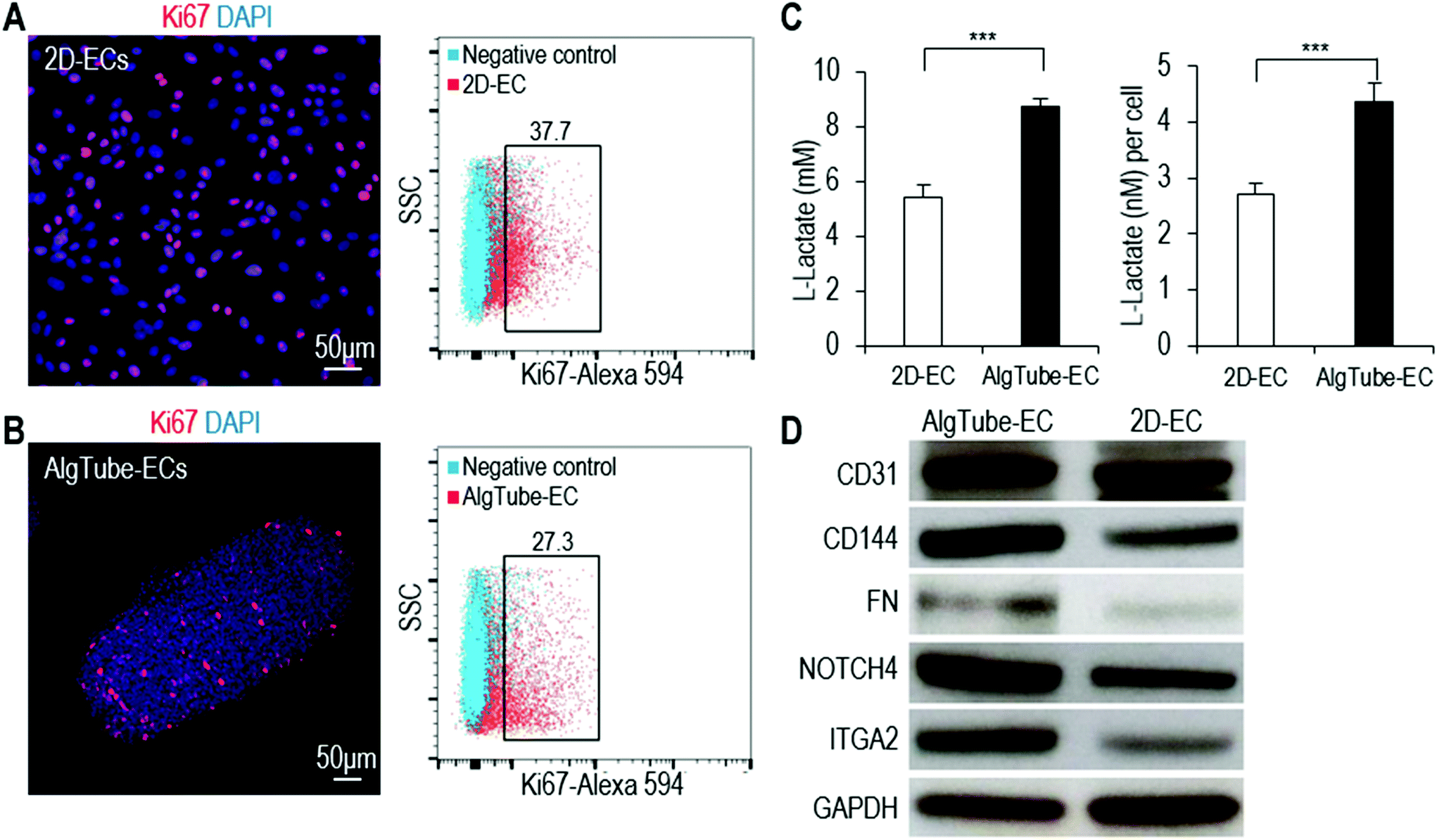
Using the alginate hydrogel tubes, we built a prototype bioreactor for scalable hPSC-EC production (Fig. 8A and B). We found that suspending hydrogel tubes in the conventional stirred tank bioreactor was not possible since the stirring ruptured the hydrogel tubes, leading to extremely low culture efficiency. Thus, we designed a new method for supplying and mixing the cell culture medium (Fig. 8B). The culture medium was stored in a bellow bottle and periodically pumped into and out of the bioreactor. Since hydrogel tubes have a density similar to the cell culture medium, they are uniformly dispersed when the medium is in the bioreactor. They become collected when the medium is withdrawn from the bioreactor. This periodic dispersion and collection mix the medium, but are gentle and do not rupture hydrogel tubes. In the bioreactor, cells were expanded for 5 days and differentiated for 5 days (Fig. 8C). On day 10, hydrogel tubes were dissolved in EDTA and cells were harvested (Fig. 8D). Phase images and live/dead cell staining showed very few dead cells (Fig. 8E and F). Flow cytometer analysis and immunostaining showed that 82.6% ± 1.1% of the produced cells were positive for EC markers (Fig. 8G and H). When transplanted subcutaneously with a Matrigel matrix, these cells formed nice vascular structures (Fig. 8I). We could produce ∼5 × 108 cells per mL of hydrogel microspace. Taking the spaces occupied by hydrogel materials and medium into account, we could produce ∼2 × 108 cells per mL bioreactor volume.47 Future optimization of the bioreactor design may further increase the cell yield per bioreactor volume. This prototype can be scaled up for large-scale production in the future. | ||
| Fig. 8 A prototype bioreactor for scalable hPSC-EC production. (A and B) The prototype consists of a mechanical stage, a controller, a bellow bottle and a bioreactor. Medium was stored in the plastic bellow bottle that could be pressed to flow the medium into, or released to withdraw the medium from the bioreactor. The controller could be programmed for the pressing and releasing speed, as well as the duration of the interval between the pressing and releasing. hPSCs were expanded for 5 days and differentiated into ECs for 5 days. (C and D) The day 10 cell masses. (E–H) Phase, live/dead cell staining, flow cytometry and immunostaining analysis of day 10 cells. (I) When transplanted subcutaneously with a Matrigel matrix, the produced hPSC-ECs formed nice vascular structures. | ||
Discussion
Currently, hPSC-ECs are mainly made in 2D cultures, which provide culturing conditions that are very different from the in vivo 3D microenvironment where cells reside.19,52–55 Additionally, 2D culturing is labor-, space- and reagent-consuming, and considered only suitable for preparing low-scale cells.39 3D suspension culturing has been widely studied to scale up the production of hPSCs and their derivatives.39,45,46 For instance, in very recent research, scientists showed that hPSCs could be efficiently differentiated into ECs as aggregates in a suspension culturing bioreactor.56 However, hPSCs in suspension culturing suffer from severe cellular agglomeration.39,45,46 The strong cell–cell interactions cause hPSCs to form large agglomerates that lead to culture inhomogeneity, impaired mass transport and low culture efficiency.39,45,46 Agitating (stirring or shaking) the culture can reduce agglomeration and enhance mass transport.39,45,46 However, agitation generates complicate hydrodynamic conditions (e.g. the medium flow direction, velocity, pressure, shear force and chemical environment) that vary spatially and temporally, resulting in critical stresses in some locations (e.g. near vessel wall and impeller tip) that induce significant cell death and phenotype changes.23,39–43,45,46,57Additionally, the hydrodynamic conditions are sensitive to many factors, such as the bioreactor design (e.g. impeller geometry, size & position, vessel geometry & size, positions of probes for pH, temperature, oxygen), the medium viscosity and agitation rate.39,57 They are currently not well understood and hard to control.39,45,46,57 These knowledge and technology gaps lead to large culture variations between batches as well as difficulty in scaling up. For instance, in recent studies to make cardiomyocytes from hPSCs in 3D suspension culturing, the yield varied from 40 million to 125 million cells and the product purity varied from 28% to 88% in six different batches (100 mL culture volume).58,59 When the culture was scaled from ∼100 mL to ∼1000 mL, the yield and differentiation efficiency were significantly altered, and re-optimization was required.58,59 These indicate the challenge of further scaling up (e.g., hundreds of liters), since optimizing multiple factors in a large volume is costly. To the best of our knowledge, the largest demonstrated culture volume for hPSCs is <10 liters to date.39,60 In summary, the uncontrolled cell aggregation and hydrodynamic conditions, and the critical hydrodynamic stresses make it difficult to culture hPSCs and their derivatives in large scales with 3D suspension culturing.
Others61 and we23 have tried to expand and differentiate hPSCs in hydrogel scaffolds with the intention of using hydrogels as physical barriers to eliminate cell agglomeration and shear stress. However, hPSCs usually have slow growth and moderate yield in hydrogels.47 We speculate that it is due to several factors: (1) the hydrogels hinder the initial cellular interactions required for their survival after cell passaging, and (2) cells must degrade or deform the scaffold to create spaces for the new cells. We recently found that hPSCs could be differentiated into ECs in a very soft thermoreversible hydrogel, yielding ∼2.0 × 107 cells per mL.62 The produced ECs had very similar phenotypes to those made in alginate hydrogel tubes. However, the hydrogel material is expensive and highly variable between batches. Additionally, the gel is very soft and can be easily broken during the culture, leaking cells to the medium. Furthermore, the yield is moderate. Together, the thermoreversible hydrogel system is appropriate for culturing moderate numbers of cells, but not suitable for large-scale cell production.
The alginate hydrogel tubes eliminate both the cell agglomeration and hydrodynamic stresses and their related adverse effects. Consequently, the culture efficiency and consistency are significantly improved. hPSC-ECs could be produced in alginate hydrogel tubes with high viability (>90%), high purity (>80%) and high yield (∼5.0 × 108 cells per mL). This yield is about 250 times the yield of 3D suspension culturing and about 25 times the yield of the thermoreversible hydrogel culture system.23,47 As discussed later, this high yield has large impact on large scale cell manufacturing. We have tested many materials and find that alginates are probably the only suitable materials for making hydrogel tubes for large-scale cell production.47,63–65 Alginates are non-toxic to cells and have been used in clinics, thus unlikely causing any regulatory concerns on the produced cells;66 they can be instantly crosslinked with Ca2+ ions to quickly extrude large numbers of tubes without harming cells; they are affordable and available in large quantities at pharmaceutical-grade from multiple vendors.66 Additionally, the resultant hydrogel tubes can be easily dissolved in EDTA to release the product without hurting cells; allow quick nutrient diffusion through the hydrogel shell; are mechanically and chemically stable that allow long-term and large-scale cell cultures; and are transparent, allowing for optical monitoring.66 Extrusion is one of the very few technologies suitable for quickly processing large numbers of hydrogel tubes. Both the crosslinking reagent CaCl2 and hydrogel dissolving reagent EDTA are biocompatible and affordable. We have not observed any toxicity of these materials to cells. cGMP-grade alginate, CaCl2 and EDTA are all available. Using an affordable, widely-available, safe and cGMP compliant raw material, as well as the simple extrusion technology, ensures the robustness, supply chain and commercial success of our technology.
hPSC-ECs made in hydrogel tubes had similar in vitro and in vivo functions to hPSC-ECs generated by conventional cell culture methods. The high cell viability, expansion, and yield make this technology very attractive for large-scale cell production. For instance, to produce ∼1.1 × 1014 hPSC-ECs (from ∼108 hPSC seeds), our calculation shows that it requires ∼32753 liters of total culture volume, 10 passaging operations, and 49 days using 3D suspension culturing such as stirred-tank bioreactors (Fig. S9A and C†), which is technically and economically challenging. As aforementioned, culturing hPSCs for >10 liters is still a challenge.39,60 Passaging is highly unwanted for large-scale cell production since it requires large amounts of time, labor, and reagent; increases the risk of contamination and culture failure; loses large numbers of cells; and causes difficulties during automation. The same production can be performed with 320 liters of alginate hydrogel tubes in 20 days and 1 passaging (Fig. S9B and C†). The reductions in the culture volume, time, and passaging not only make the production technically feasible, but also lead to enormous cuts in labor, reagents, equipment, cGMP facility size, and overall production cost.
Conclusion
In summary, through using alginate hydrogel tube to create a cell-friendly microenvironment, we were able to produce high quality hPSC-ECs at extremely high density. We believe that the method developed in this paper will make hPSC-ECs broadly available for various applications.Author contributions
Y.L. and H.L conceived the idea and designed the study. H.L., Q.L. and O.W. performed experiments and analyzed data. Q.D., K.L. and C.Z. analyzed RNA sequencing data. C.E. contributed to confocal images. Y.L. and H.L. wrote the manuscript. S.C., B.D. and Z.W. revised the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The Morrison Microscopy Core Research Facility at the University of Nebraska, Lincoln for confocal microscope imaging, the Flow Cytometry core of University of Nebraska, Lincoln for flow cytometry and the UNMC deep sequencing core for RNA sequencing are acknowledged.References
- O. C. Richards, S. M. Raines and A. D. Attie, Endocr. Rev., 2010, 31, 343–363 CrossRef CAS PubMed.
- P. Carmeliet, Nature, 2001, 412, 868–869 CrossRef CAS PubMed.
- O. M. Leung, B. Zhou and K. O. Lui, J. Cardiovasc. Dev. Dis., 2016, 3, 23 CrossRef PubMed.
- C. Patsch, L. Challet-Meylan, E. C. Thoma, E. Urich, T. Heckel, J. F. O'Sullivan, S. J. Grainger, F. G. Kapp, L. Sun, K. Christensen, Y. Xia, M. H. C. Florido, W. He, W. Pan, M. Prummer, C. R. Warren, R. Jakob-Roetne, U. Certa, R. Jagasia, P.-O. Freskgård, I. Adatto, D. Kling, P. Huang, L. I. Zon, E. L. Chaikof, R. E. Gerszten, M. Graf, R. Iacone and C. A. Cowan, Nat. Cell Biol., 2015, 17, 994–1003 CrossRef CAS PubMed.
- R. J. Medina, C. L. O. Neill, M. W. Humphreys, T. A. Gardiner and A. W. Stitt, Retina, 2010, 51, 5906–5913 Search PubMed.
- C. Moubarik, B. Guillet, B. Youssef, J. Codaccioni, M. Piercecchi, F. Sabatier, P. Lionel, L. Dou, A. Foucault-bertaud, L. Velly, F. Dignat-george and P. Pisano, Stem Cell Rev. Rep., 2011, 7, 208–220 CrossRef PubMed.
- T. M. Schwarz, S. F. Leicht, T. Radic, I. Rodriguez-arab, P. C. Hermann, F. Berger, J. Saif, W. Bo, J. W. Ellwart, A. Aicher and C. Heeschen, Arterioscler. Thromb. Vasc. Biol., 2012, 32, e13–e21 CrossRef CAS PubMed.
- K. Kang, M. Coggins, C. Xiao, A. Rosenzweig and J. Bischoff, Angiogenesis, 2013, 16, 773–784 CrossRef CAS PubMed.
- X.-T. Huang, Y.-Q. Zhang, S.-J. Li, S.-H. Li, Q. Tang, Z.-T. Wang, J.-F. Dong and J.-N. Zhang, J. Neurotrauma, 2013, 30, 2080–2088 CrossRef PubMed.
- S. C. Heo, Y. W. Kwon, I. H. Jang, G. O. Jeong, J. W. Yoon, C. D. Kim, S. M. Kwon, Y.-S. Bae and J. H. Kim, Stem Cell, 2014, 32, 779–790 CrossRef CAS PubMed.
- J. Stroncek, L. Ren, B. Klitzman and W. Reichert, Acta Biomater., 2012, 8, 201–208 CrossRef CAS PubMed.
- L. Hayflick, Exp. Cell Res., 1965, 37, 614–636 CrossRef CAS PubMed.
- M. Gumbleton and K. L. Audus, J. Pharm. Sci., 2001, 90, 1681–1698 CrossRef CAS PubMed.
- H. Augustin-Voss, A. Voss and B. Pauli, J. Cell. Physiol., 1993, 157, 279–288 CrossRef CAS PubMed.
- J. R. Van Beijnum, M. Rousch, K. Castermans, E. Van Der Linden and A. W. Griffioen, Nat. Protoc., 2008, 3, 1085–1091 CrossRef CAS PubMed.
- L. Gui, A. Muto, S. A. Chan, C. K. Breuer and L. E. Niklason, Tissue Eng., Part A, 2009, 15, 2665–2676 CrossRef CAS PubMed.
- J. L. De Carvalho, A. Zonari, A. Cláudia, C. De Paula, T. Maria, D. A. Gomes and A. M. Goes, BioMed Res. Int., 2015, 2015, 652474 Search PubMed.
- S. Levenberg, J. Zoldan, Y. Basevitch and R. Langer, Blood, 2007, 110, 806–814 CrossRef CAS PubMed.
- J. A. Thomson, J. Itskovitz-eldor, S. S. Shapiro, M. A. Waknitz, J. J. Swiergiel, V. S. Marshall and J. M. Jones, Science, 1998, 282, 1145–1147 CrossRef CAS PubMed.
- J. Yu, M. A. Vodyanik, K. Smuga-otto, J. Antosiewicz-bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS PubMed.
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, Cell, 2007, 131, 861–872 CrossRef CAS PubMed.
- G. Chen, D. R. Gulbranson, Z. Hou, J. M. Bolin, V. Ruotti, M. D. Probasco, K. Smuga-Otto, S. E. Howden, N. R. Diol, N. E. Propson, R. Wagner, G. O. Lee, J. Antosiewicz-Bourget, J. M. C. Teng and J. A. Thomson, Nat. Methods, 2011, 8, 424–429 CrossRef CAS PubMed.
- Y. Lei and D. V. Schaffer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E5039–E5048 CrossRef CAS PubMed.
- P. A. Lalit, D. J. Hei, A. N. Raval and T. J. Kamp, Circ. Res., 2014, 114, 1328–1345 CrossRef CAS PubMed.
- M. B. Nourse, D. E. Halpin, M. Scatena, D. J. Mortisen, N. L. Tulloch, K. D. Hauch, B. Torok-storb, B. D. Ratner, L. Pabon and C. E. Murry, Arterioscler. Thromb. Vasc. Biol., 2010, 30, 80 CrossRef CAS PubMed.
- Z. J. Li, Z. C. Han and J. C. Wu, J. Cell. Biochem., 2009, 106, 194–199 CrossRef CAS PubMed.
- Z. Li, K. D. Wilson, B. Smith, D. L. Kraft, F. Jia, M. Huang, R. C. Robbins, S. S. Gambhir, I. L. Weissman and J. C. Wu, PLoS One, 2009, 4, e8443 CrossRef PubMed.
- D. James, H. Nam, M. Seandel, D. Nolan, T. Janovitz, M. Tomishima, L. Studer, G. Lee, D. Lyden, R. Benezra, N. Zaninovic, Z. Rosenwaks, S. Y. Rabbany and S. Rafii, Nat. Biotechnol., 2010, 28, 161–166 CrossRef CAS PubMed.
- S. Levenberg, J. S. Golub, M. Amit, J. Itskovitz-Eldor and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4391–4396 CrossRef CAS PubMed.
- G. Condorelli, U. Borello, L. De Angelis, M. Latronico, D. Sirabella, M. Coletta, R. Galli, G. Balconi, A. Follenzi, E. Dejana and G. Cossu, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10733–10738 CrossRef CAS PubMed.
- N. M. Kane, M. Meloni, H. L. Spencer, M. A. Craig, R. Strehl, G. Milligan, M. D. Houslay, J. C. Mountford, C. Emanueli and A. H. Baker, Arterioscler. Thromb. Vasc. Biol., 2010, 30, 1389–1397 CrossRef CAS PubMed.
- M. A. Vodyanik, J. A. Bork, J. A. Thomson and I. I. Slukvin, Blood, 2005, 105, 617–627 CrossRef CAS PubMed.
- N. Cao, H. Liang, J. Huang, J. Wang, Y. Chen, Z. Chen and H. Yang, Cell Res., 2013, 23, 1119–1132 CrossRef CAS PubMed.
- N. J. Palpant, L. Pabon, C. E. Friedman, M. Roberts, B. Hadland, R. J. Zaunbrecher, I. Bernstein, Y. Zheng and C. E. Murry, Nat. Protoc., 2016, 12, 15–31 CrossRef PubMed.
- E. Giacomelli, M. Bellin, L. Sala, B. J. Van Meer, L. G. J. Tertoolen, V. V. Orlova and C. L. Mummery, Development, 2017, 144, 1008–1017 CrossRef CAS PubMed.
- J. Zhang, L. Chu, Z. Hou, M. P. Schwartz, T. Hacker and V. Vickerman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6072–E6078 CrossRef CAS PubMed.
- X. Lian, X. Bao, A. Al-Ahmad, J. Liu, Y. Wu, W. Dong, K. K. Dunn, E. V. Shusta and S. P. Palecek, Stem Cell Rep., 2014, 3, 804–816 CrossRef CAS PubMed.
- V. V. Orlova, F. E. van den Hil, S. Petrus-Reurer, Y. Drabsch, P. Ten Dijke and C. L. Mummery, Nat. Protoc., 2014, 9, 1514–1531 CrossRef CAS PubMed.
- C. Kropp, D. Massai and R. Zweigerdt, Process Biochem., 2017, 59, 244–254 CrossRef CAS.
- M. J. Jenkins and S. S. Farid, Biotechnol. J., 2015, 10, 83–95 CrossRef CAS PubMed.
- Y. Lei, D. Jeong, J. Xiao and D. V. Schaffer, Cell. Mol. Bioeng., 2014, 7, 172–183 CrossRef CAS PubMed.
- D. Steiner, H. Khaner, M. Cohen, S. Even-Ram, Y. Gil, P. Itsykson, T. Turetsky, M. Idelson, E. Aizenman, R. Ram, Y. Berman-Zaken and B. Reubinoff, Nat. Biotechnol., 2010, 28, 361–364 CrossRef CAS PubMed.
- M. Serra, C. Brito, C. Correia and P. M. Alves, Trends Biotechnol., 2012, 30, 350–358 CrossRef CAS PubMed.
- F. M. Wurm, Nat. Biotechnol., 2004, 22, 1393–1398 CrossRef CAS PubMed.
- M. A. Kinney, C. Y. Sargent and T. C. Mcdevitt, Tissue Eng., Part B, 2011, 17, 249–262 CrossRef PubMed.
- K. M. Fridley, M. A. Kinney and T. C. Mcdevitt, Stem Cell Res. Ther., 2012, 3, 45 CrossRef CAS PubMed.
- Q. Li, H. Lin, Q. Du, K. Liu, O. Wang, C. Evans, H. Christian, C. Zhang and Y. Lei, Biofabrication, 2018, 10, 025006 CrossRef PubMed.
- I. H. Park, R. Zhao, J. A. West, A. Yabuuchi, H. Huo, T. A. Ince, P. H. Lerou, M. W. Lensch and G. Q. Daley, Nature, 2008, 451, 141–146 CrossRef CAS PubMed.
- J. Zhang, M. P. Schwartz, Z. Hou, Y. Bai, H. Ardalani, S. Swanson, J. Steill, V. Ruotti, A. Elwell, B. K. Nguyen, J. Bolin, R. Stewart, J. A. Thomson and W. L. Murphy, Stem Cell Rep., 2017, 8, 907–918 CrossRef CAS PubMed.
- D. Zujur, K. Kanke, A. C. Lichtler and H. Hojo, Sci. Adv., 2017, 3, e1602875 CrossRef PubMed.
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, J. Lab. Autom., 2015, 20, 107–126 CrossRef CAS PubMed.
- C. C. Wong, K. E. Loewke, N. L. Bossert, B. Behr, C. J. De Jonge, T. M. Baer and R. A. R. Pera, Nat. Biotechnol., 2010, 28, 1115–1121 CrossRef CAS PubMed.
- T. P. Kraehenbuehl, R. Langer and L. S. Ferreira, Nat. Methods, 2011, 8, 731–736 CrossRef CAS PubMed.
- K. G. Chen, B. S. Mallon, K. R. Johnson, R. S. Hamilton, R. D. G. Mckay and P. G. Robey, Stem Cell Res., 2014, 12, 610–621 CrossRef CAS PubMed.
- K. G. Chen, B. S. Mallon, R. D. G. McKay and P. G. Robey, Cell Stem Cell, 2014, 14, 13–26 CrossRef CAS PubMed.
- R. Olmer, L. Engels, A. Usman, S. Menke, M. N. H. Malik, F. Pessler, G. Göhring, D. Bornhorst, S. Bolten, S. Abdelilah-Seyfried, T. Scheper, H. Kempf, R. Zweigerdt and U. Martin, Stem Cell Rep., 2018, 10, 1657–1672 CrossRef CAS PubMed.
- M. Ismadi, P. Gupta, A. Fouras, P. Verma and S. Jadhav, PLoS One, 2014, 9, e106493 CrossRef PubMed.
- M. Jara-avaca, H. Kempf, R. Olmer, C. Kropp, M. Ru, D. Robles-diaz, A. Franke, D. A. Elliott, D. Wojciechowski, M. Fischer, A. R. Lara, G. Kensah, I. Gruh, A. Haverich, U. Martin and R. Zweigerdt, Stem Cell Rep., 2014, 3, 1132–1146 CrossRef PubMed.
- V. C. Chen, J. Ye, P. Shukla, G. Hua, D. Chen, Z. Lin, J. Liu, J. Chai, J. Gold, J. Wu, D. Hsu and L. A. Couture, Stem Cell Res., 2015, 15, 365–375 CrossRef CAS PubMed.
- H. Kempf, B. Andree and R. Zweigerdt, Adv. Drug Delivery Rev., 2016, 96, 18–30 CrossRef CAS PubMed.
- P. Kerscher, I. C. Turnbull, A. J. Hodge, J. Kim, D. Seliktar, C. J. Easley, K. D. Costa and E. A. Lipke, Biomaterials, 2016, 83, 383–395 CrossRef CAS PubMed.
- H. Lin, Q. Du, Q. Li, O. Wang, Z. Wang, N. Sahu, C. Elowsky, K. Liu, C. Zhang, S. Chung, B. Duan and Y. Lei, Stem Cell Rep., 2018, 11, 454–469 CrossRef CAS PubMed.
- H. Lin, Q. Li, O. Wang, J. Rauch, B. Harm and H. J. Viljoen, Adv. Healthcare Mater., 2018, 7, e1701297 CrossRef PubMed.
- H. Lin, Q. Du, Q. Li, O. Wang, Z. Wang, K. Liu, C. Elowsky, C. Zhang and Y. Lei, ACS Appl. Mater. Interfaces, 2018, 10, 29238–29250 CrossRef CAS PubMed.
- Q. Li, H. Lin, J. Rauch, L. P. Deleyrolle, B. A. Reynolds, H. J. Viljoen, C. Zhang, C. Zhang, L. Gu, E. Van Wyk and Y. Lei, Sci. Rep., 2018, 8, 3531 CrossRef PubMed.
- K. Yong and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01095a |
| This journal is © The Royal Society of Chemistry 2019 |