DOI:
10.1039/C8AY02168F
(Paper)
Anal. Methods, 2019,
11, 49-57
Measurement of methylated metabolites using liquid chromatography-mass spectrometry and its biological application†
Received
3rd October 2018
, Accepted 14th November 2018
First published on 15th November 2018
Abstract
Methylation aberrations play an important role in many metabolic disorders including cancer. Methylated metabolites are direct indicators of metabolic aberrations, and currently, there is no liquid chromatography-mass spectrometry (LC-MS) based method available to cover all classes of methylated metabolites at low detection limits. In this study, we have developed a method for the detection of methylated metabolites, and its biological application. In this approach, we used a HILIC based HPLC method combined with MS to measure methylated organic acids, amino acids, and nucleotides. These metabolites were separated from each other by their hydrophobic interactions and analyzed using a targeted metabolomics approach by single reaction monitoring in positive and negative modes of electrospray ionization. These metabolites were quantified, and the interday reproducibility was <10% relative standard deviation. Furthermore, by applying this method, we identified high levels of methylated metabolites in bladder cancer cell lines compared to benign cells. In vitro treatment of cancer cells with a methylation inhibitor, 5-aza-2′-deoxycytidine, showed a decrease in these methylated metabolites. These data indicate that HPLC analysis using this HILIC based method could be a powerful tool for measuring methylated metabolites in biological specimens. This method is rapid, sensitive, selective, and precise to measure methylated metabolites.
Introduction
Methylation represents the addition of a methyl group onto a substrate or the replacement of an atom or a group of atoms by a methyl group. In biological systems, epigenetics describes heritable changes in the phenotype of the cell which are independent of alterations occurring in the DNA sequence.1 To date, there are at least four different DNA modifications2,3 and 16 types of histone modifications4,5 which are altered by chromatin modifying enzymes in a highly regulated manner. One of these modifications is methylation of nucleotides such as cytosine, adenine and amino acids present in histones particularly lysine and arginine.6 Methylation can regulate gene expression, RNA processing, DNA repair, and protein function. Abnormal methylation is one of the hallmarks of cancer development.7
DNA methylation at CpG islands is catalyzed by DNA methyl transferases resulting in the generation of 5-methylcytosine (5mC). It plays an important role in promoter hypermethylation of tumor-suppressor genes, which is a hallmark of human cancer.8 Methylations at CpG are erased by α-ketoglutarate-dependent ten-eleven translocation (TET) dioxygenases, through sequential oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), finally converting to unmethylated cytosine.9–11 The quantitative levels of 5mC and 5hmC reveal the DNA methylation status, and serve as epigenetic markers for cancer and various other diseases.12–14 The methylation of amino acid arginine is known to play a major role in signal transduction, nuclear transport or direct modulation of nucleic acid interactions.15 Methylated organic acids can act as diagnostic markers for vitamin B-12 deficiency in humans, which can lead to excess production of methylmalonic acid.16,17 Despite the biological significance of methylated metabolites, there are very few methods available to identify and quantify methylated metabolites.18–20 However, due to the low sensitivity and inability of the current methods to cover multiple methylated metabolites, there is a need to develop new methods with high sensitivity to understand their biological role.
HPLC coupled to MS provides high sensitivity and selectivity, making these assays superior to conventional detection technologies. Hydrophilic interaction liquid chromatography (HILIC) in conjunction with MS has become a powerful tool for the analysis of a wide variety of analytes. It separates compounds by elution with a strong organic mobile phase against a hydrophilic stationary phase where elution is driven by increasing the water content in the mobile phase. It has been reported that the ionization responses for basic and acidic polar compounds were enhanced by 5–8 fold in the positive ionization mode and up to 20-fold in the negative ionization mode by HILIC LC-MS/MS methods as compared to reverse phase LC-MS/MS methods.21 The application of this technique has increased dramatically over the past decade, especially for the analysis of polar analytes like methylated metabolites where reverse phase chromatography is unsuitable. The major advantage of HILIC is that even with small volumes of samples methylated metabolites are analyzed with high resolution and high sensitivity. LC-ESI-MS based methods are extremely selective and sensitive allowing them to be utilized in applications with limited amounts of materials such as clinical samples.
In this study, we have developed a new HILIC based LC/MS method for the measurement of multiple methylated organic acids, nucleotides and amino acids. This method allows the quantitative determination of these metabolites up to the picogram level. We have validated this method in bladder benign and cancer cells and found that cancer cells have high levels of methylated metabolites. We further observed low levels of these methylated metabolites upon 5-aza-2′-deoxycytidine (AZA) treatment in bladder cancer (BLCA) cells by the same method.
Methods
Reagents and internal standards
HPLC grade ammonium acetate, acetonitrile, methanol, chloroform, ethanol, hexane, and water were procured from Burdick & Jackson (Morristown, NJ). MS grade formic acid, standard compounds and isotope labeled standards, including N-acetyl aspartic acid-d3, tryptophan-15N2, sarcosine-d3, glutamic acid-d5, thymine-d4, gibberellic acid, trans-zeatin, jasmonic acid, 15 N anthranilic acid, and testosterone-d3, were purchased from Sigma-Aldrich (St. Louis, MO).
Preparation of a standard calibration curve
All standard compounds were dissolved as per the vendor's instructions, and 1 mg ml−1 stock solutions were prepared. A standard mixture stock solution was prepared so that the final concentration of each standard was 1000 ng ml−1. The standard curve dilutions were made with (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) methanol:water. The concentration of the lowest dilution is 0.2 pg ml−1, and the injection volume is 5 μL.
:1) methanol:water. The concentration of the lowest dilution is 0.2 pg ml−1, and the injection volume is 5 μL.
Cell culture and AZA treatments
The cells were maintained using standard cell culture procedures in an incubator at 37 °C with 5% CO2 and atmospheric oxygen. The cells were cultured in respective media, SVHUC-1 (F-12K), RT4 and T24 (McCoy's 5A), and 5637, J82, TCCSUP, and UMUC3 (RPMI), supplemented with 10% fetal bovine serum and 1% pen-strep. AZA was resuspended in DMSO, and 5 μM final concentration was added to the culture media for 72 hours. After the time point the cells were harvested and an equal number of cells were collected from control and treatment conditions. The cell pellets were flash frozen and stored at −80 °C until metabolites were extracted.
Sample preparation for mass spectrometry and metabolomics analysis
Metabolites were extracted from benign and BLCA cell lines. A mouse liver pool was used as quality control and the extraction procedure was followed as described previously22–26. We used 5 million cells for the extraction and to each sample 750 μL of ice-cold methanol:water (4:1) containing 20 μL spiked internal standard was added and homogenized. Ice-cold chloroform and water were added in a 3:1 ratio for a final proportion of 4:3:2 methanol:chloroform:water. Both the organic and aqueous layers were transferred into a new tube, dried and resuspended in 50:50 methanol:water. The resuspended samples were deproteinized by using a 3 kDa molecular filter (Amicon ultracel-3K Membrane; Millipore Corporation, Billerica, MA) and the filtrate was dried under vacuum (Genevac EZ-2plus, NY). Prior to MS, the dried extracts were re-suspended in 50:50 methanol:water and were subjected to LC-MS.
Data acquisition through LC/MS analysis
The MS experiments were performed on a 6495 QQQ mass spectrometer (Agilent Technologies, Santa Clara, CA). The sample was introduced into the ion source using a 1290 HPLC with a degasser, binary pump, thermostatted auto sampler and column oven. The MS source parameters used were as follows: gas temperature 250 °C; gas flow 14 l min−1; nebulizer gas pressure 20 psi; sheath gas temperature 350 °C; sheath gas flow 12 l min−1; capillary voltage 3000 V positive and 3000 V negative; nozzle voltage 1500 V positive and 1500 V negative. Approximately 8–11 data points were acquired per detected metabolite. All the standards were acquired in the scan mode mass range 50–1200 Da in electrospray ionization (ESI) positive mode and negative mode. The collision-induced dissociation mass spectra (CID) of all methylated compounds were recorded by mass selecting the precursor ions using quadrupole 1, colliding them with a collision gas of nitrogen (>99.9% purity) in the collision cell (quadrupole 2), and the resulting ions were analyzed by scanning quadrupole 3. These experiments were performed using an Agilent automated mass hunter optimizer, and the most abundant fragment ions were used as quantifiers. All the metabolites were measured in selected reaction monitoring (SRM) mode. The developed method was validated concerning specificity, sensitivity, linearity, precision, accuracy, and matrix effect.
Separation of metabolites
We used two methods to separate methylated metabolites.
Method A.
In ESI positive mode the HPLC column was a Waters X-bridge amide 3.5 μm, 4.6 × 100 mm (PN: 186004868, Waters Milford, MA). Mobile phases A and B were 0.1% formic acid in water and acetonitrile respectively. Gradient flow: 0–3 min 85% B, 3–12 min 30% B, 12–15 min 2% B, and 16 min 95% B, followed by re-equilibration till the end of the gradient 23 min to the initial starting condition of 85% B. The flow rate of the solvents used for the analysis is 0.3 ml min−1.
Method B.
In ESI negative mode the HPLC column was a Waters X-bridge amide 3.5 μm, 4.6 × 100 mm (PN: 186004868, Waters, Milford, MA). Mobile phases A and B were 20 mM ammonium acetate in water with pH 9.0 and 100% acetonitrile respectively. Gradient flow: 0–3 min 85% B, 3–12 min 30% B, 12–15 min 2% B, and 15–16 min 85% B followed by re-equilibration till the end of the gradient 23 min to the initial starting condition of 85% B. The flow rate of the solvents used for analysis is 0.3 ml min−1.
Statistical analysis
The limit of detection (LOD) and limit of quantification (LOQ) were calculated by the calibration curve method using the mean slope and standard deviation (SD) of the intercept. Agilent massHunter workstation software quantitative analysis was used for manual review of chromatograms, and the peak area was integrated based on the retention time. The normalization of each metabolite peak area was done by the peak area of the spiked internal standard (zeatin), and then the data were log2 transformed. For every metabolite in the normalized dataset, two sample t-tests were conducted to compare the levels of methylated metabolites between normal and BLCA cell lines. Differential metabolites were identified by adjusting the p-values for multiple testing at a false discovery rate (FDR) threshold of <0.25 for generating the heat maps and box plots.
Results and discussion
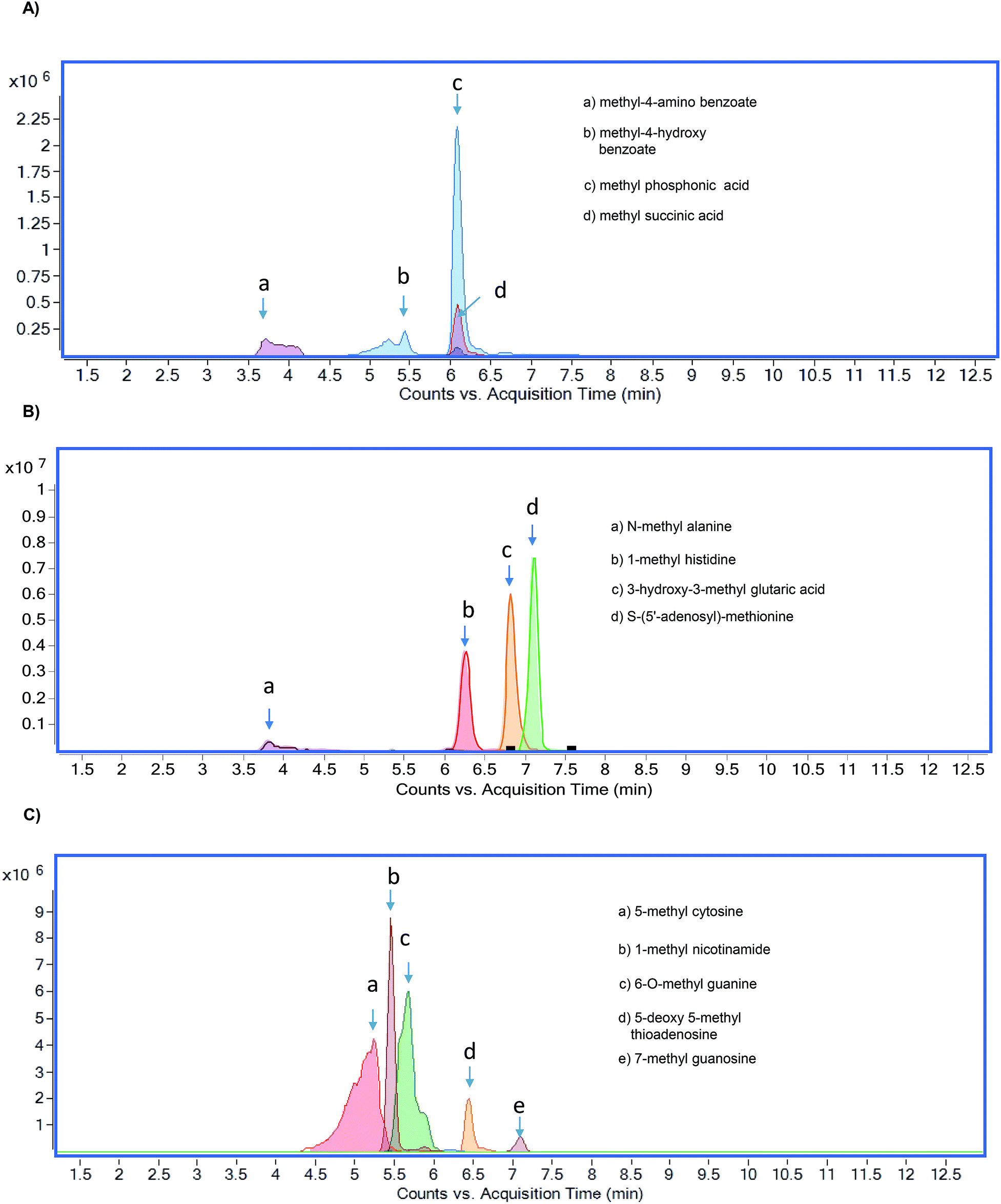
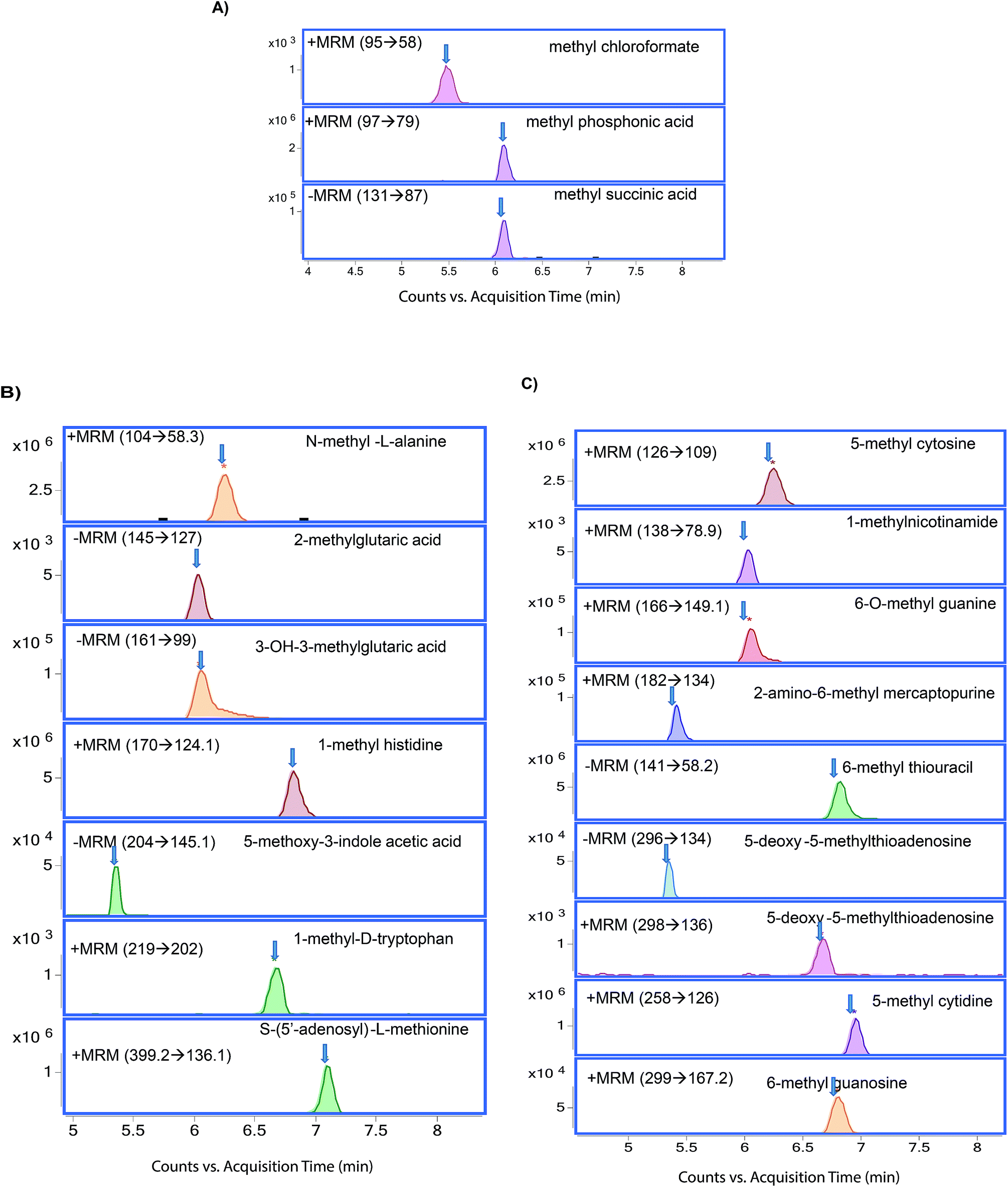
The chromatographic separation of methylated metabolites measured in this study is shown in Fig. 1. The list of metabolites and SRM transitions is shown in Table 1. The individual methylated metabolites and their chromatograms are shown in Fig. 2.
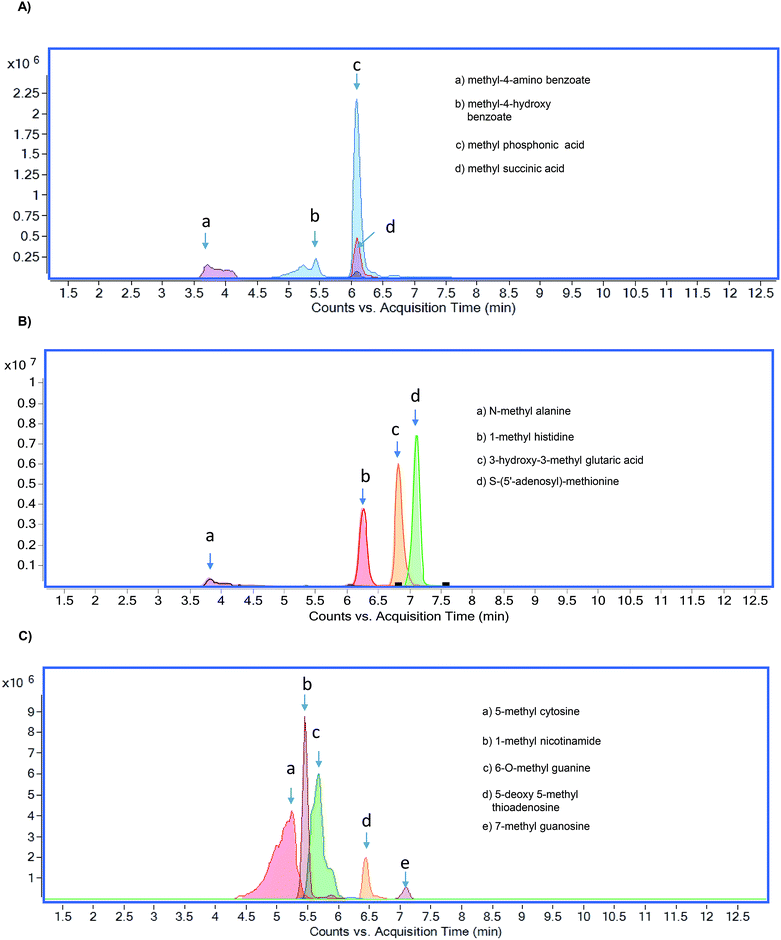 |
| | Fig. 1 Overlaid chromatograms of methylated metabolites (A) methylated organic acids, (B) methylated amino acids, and (C) methylated nucleotides. | |
Table 1 Methylated metabolites and their SRM transitions
| Class |
Metabolite |
Precursor |
SRM (m/z) |
Polarity |
RT |
| ion (m/z) |
(min) |
| Methylated acids |
Methyl chloroformate |
95 |
58 |
Positive |
5.4 |
| Methyl phosphonic acid |
97 |
79 |
6 |
| Methyl-4-amino benzoate |
152 |
120/93 |
3.7 |
| Methyl-4-hydroxy benzoate |
153 |
121/59 |
5.4 |
| Methyl malonic acid |
117 |
73/55 |
Negative |
3.1 |
| Methyl succinic acid |
131 |
87/113 |
6 |
| Methylated amino acids |
N-Methyl-L-alanine |
104 |
58 |
Positive |
6.3 |
| 1-Methy-L-histidine |
170 |
124/83 |
6.8 |
| 1-Methyl-D-tryptophan |
219 |
173/160 |
6.6 |
|
S-(5′-Adenosyl)-L-methionine |
399 |
136/250 |
7 |
| 2-Methyl glutaric acid |
145 |
101 |
Negative |
6 |
| 3-Hydroxy-3-methyl glutaric acid |
161 |
57/99 |
6 |
| 5-Methoxy-3-indole acetic acid |
204 |
160/145 |
5.3 |
| Methylated nucleotides |
5-Methylcytosine |
126 |
109 |
Positive |
5.4 |
| 1-Methylnicotinamide |
138 |
79/93 |
6.3 |
| 6-O-Methyl guanine |
166 |
149/134 |
5.3 |
| 2-Amino-6-methyl mercaptopurine |
182 |
134/107 |
5.1 |
| 5-Methyl cytidine |
258 |
126/109 |
6.4 |
| 7-Methyl guanosine |
299 |
167 |
5.4 |
| 6-Methyl -2-thiouracil |
141 |
58 |
Negative |
5.4 |
| 5-Deoxy-5-methyl thio adenosine |
296 |
134 |
5.3 |
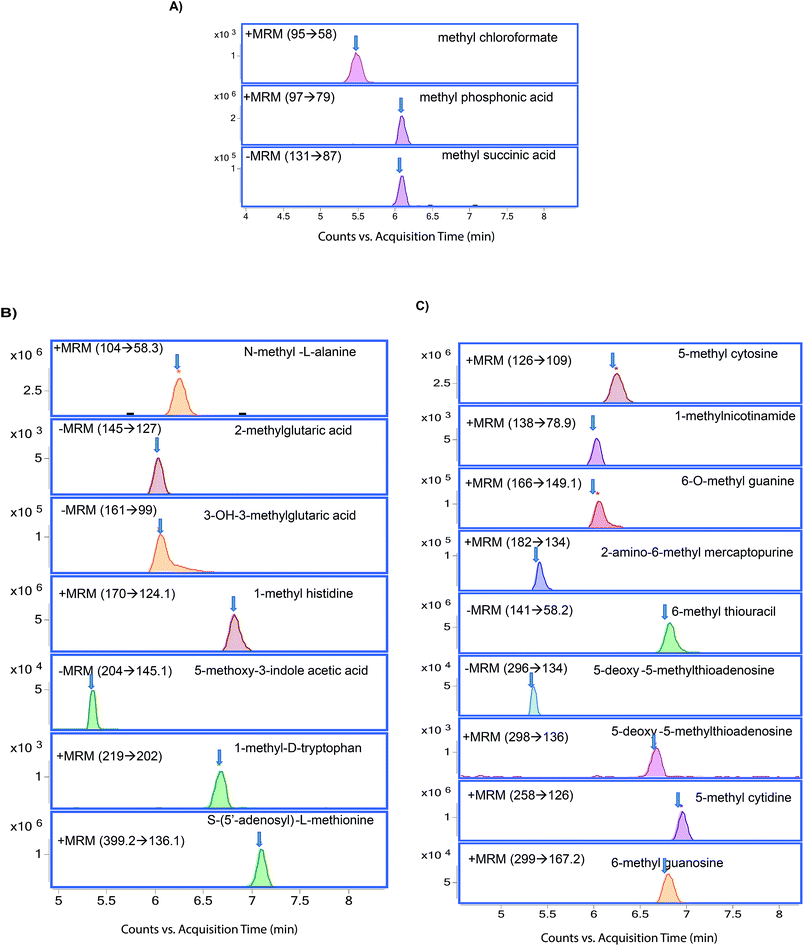 |
| | Fig. 2 Chromatograms of methylated metabolites (A) methylated organic acids, (B) methylated amino acids, and (C) methylated nucleotides. | |
Separation of methylated metabolites
Methylated organic acids, amino acids, and nucleotides were separated by using HILIC (Fig. 1). Selection of HILIC enabled the detection of metabolites at the picogram level (Table 2), and clear separation of methylated organic acids, amino acids, and nucleotides was observed at different retention times (Fig. 2). Using the gradient, we were able to achieve narrow, clear, and reproducible retention times with high sensitivity. Re-equilibration on the column after injection can be useful for effective removal of any contaminants. All methylated metabolites are eluted within four minutes. On the basis of chromatographic results, 0.1% formic acid in acetonitrile, 0.1% formic acid in water and 20 mM ammonium acetate in water by gradient elution at 0.3 ml min−1 were chosen as mobile phases.
Table 2 Methylated metabolite LODs, LOQs, R2, and reproducibility
| Metabolite |
LOD (pg ml−1) |
LOQ (pg ml−1) |
R
2
|
CV% |
Ionization |
| Methyl chloroformate |
421 |
606 |
0.999 |
5.6 |
Positive |
| Methyl phosphonic acid |
25 |
100 |
0.999 |
7.8 |
Positive |
| Methyl-4-hydroxy benzoate |
159 |
390 |
0.999 |
6.3 |
Positive |
| 1-Methyl histidine |
1.0 |
5.0 |
0.999 |
8.9 |
Positive |
| 5-Methyl cytosine |
50 |
152 |
0.997 |
8.7 |
Positive |
| 1-Methyl nicotinamide |
32 |
56 |
0.993 |
9.3 |
Positive |
| 2-Amino-6-methyl mercaptopurine |
2.0 |
6.0 |
0.991 |
5.8 |
Positive |
| 5-Methyl cytidine |
6.0 |
16 |
0.999 |
9.5 |
Positive |
| 5-Deoxy-5-methyl thioadenosine |
2.0 |
6.0 |
0.999 |
7.5 |
Positive |
| 5-Methoxy-3-indole acetic acid |
66 |
119 |
0.999 |
5.3 |
Negative |
Fragmentation pathways of the methylated metabolites and their degradation products
The collision-induced dissociation (CID) spectra of methylated organic acids.
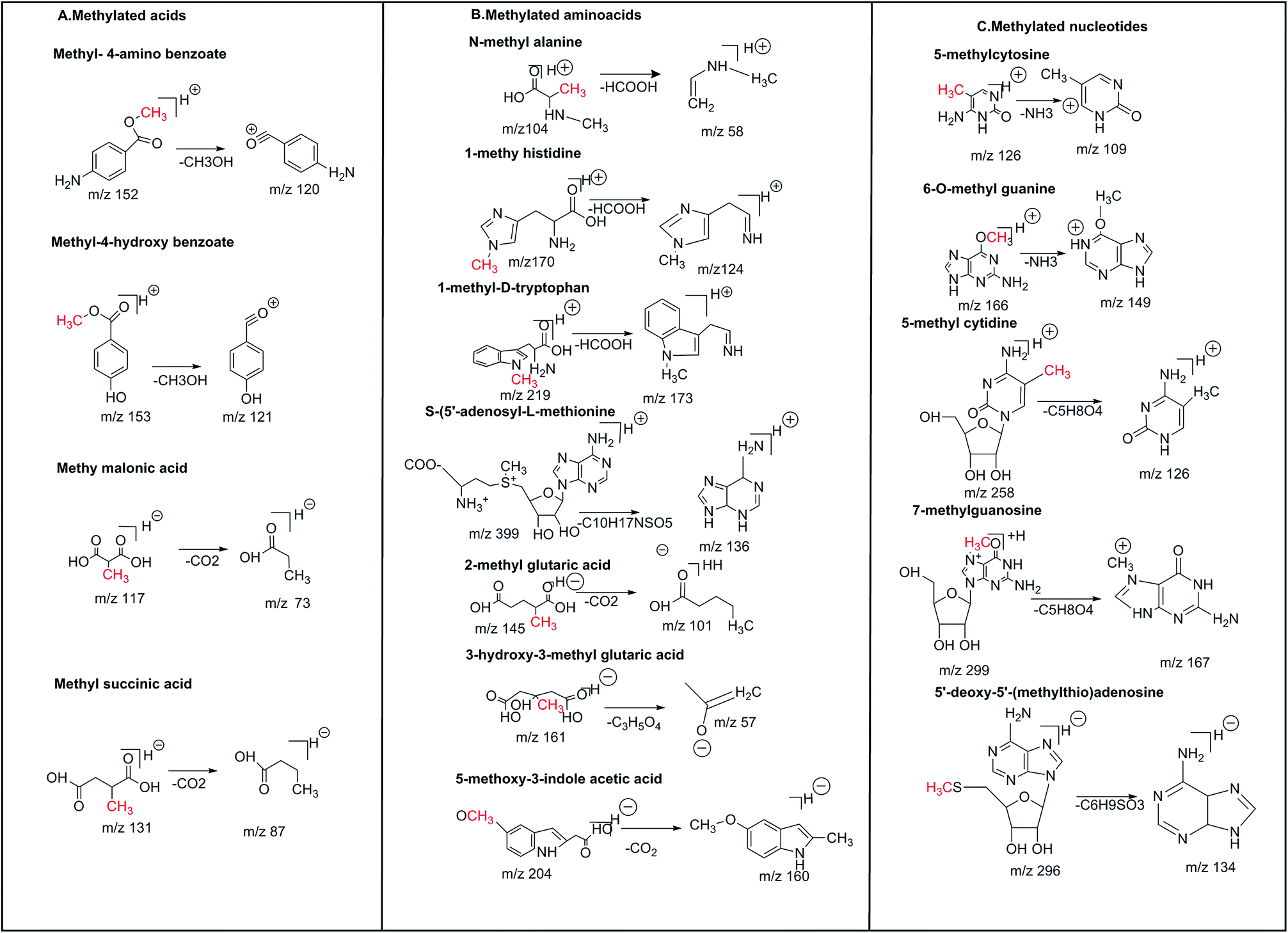
The CID of the methyl chloroformate [M + H]+ ion (m/z 95) resulted in the major fragment ion of m/z 58 corresponding to the loss of HCl. The CID of the methyl phosphonic acid [M + H]+ ion (m/z 97) yielded the major fragment ion of m/z 79 corresponding to the loss of H2O. The CID of the methyl-4-amino benzoate [M + H]+ ion (m/z 152) resulted in the major fragment ion of m/z 93 corresponding to the loss of CH3O. The CID of the methyl-4-hydroxy benzoate [M + H]+ ion (m/z 153) resulted in the major fragment ion of m/z of 120 which resulted in the loss of CH3O. The CID of the methyl malonic acid [M − H]− ion (m/z 117) produced the major fragment ion of m/z 73 corresponding to the loss of CO2. The CID of the methyl succinic acid [M − H]− ion (m/z 131) resulted in the major fragment ion of m/z 87 corresponding to the loss of CO2. The methylated organic acid precursors and fragment ion structures are presented in Fig. 3A and CID spectra are presented in ESI Fig. 1.†
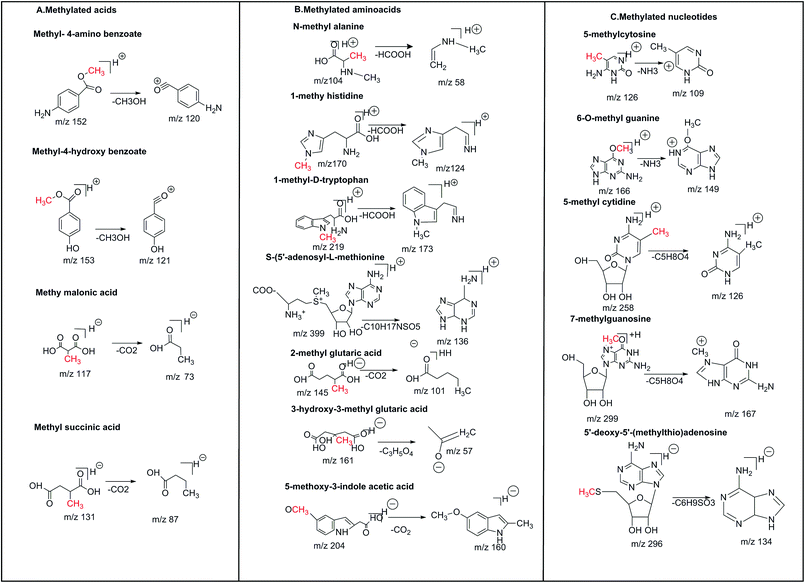 |
| | Fig. 3 Precursor and product ion structures of (A) methylated organic acids, (B) methylated amino acids, and (C) methylated nucleotides. | |
The CID spectra of methylated amino acids.
The CID of the N-methyl alanine [M + H]+ion (m/z 104) produced the major fragment ion of m/z 58 corresponding to the loss of HCOOH. The CID of the 1-methyl histidine [M + H]+ ion (m/z 170) resulted in the major fragment ion of m/z 124 corresponding to the loss of HCOOH. The CID of the 1-methyl-D-tryptophan [M + H]+ ion (m/z 219) produced the major fragment ion of m/z 173 corresponding to the loss of HCOOH. The CID of the S-(5′-adenosyl)-L-methionine [M + H]+ ion (m/z 399) produced the major fragment ion of m/z 136 corresponding to the loss of C10H17NSO5. The CID of the 2-methyl glutaric acid [M − H]− ion (m/z 145) yielded a major fragment ion of m/z 101 corresponding to the loss of CO2. The CID of the 3-hydroxy-3-methyl-glutaric acid [M − H]− ion (m/z 160) produced the major fragment ion of m/z 57 corresponding to the loss of C3H5O4. The CID of the 5-methoxy-3-indole acetic acid [M − H]− ion (m/z 204) produced the major fragment ion of m/z 160 corresponding to the loss of CO2. The methylated amino acid precursors and fragment ion structures are shown in Fig. 3B and CID spectra are presented in ESI Fig. 2.†
The CID spectra of methylated nucleotides.
The CID of the 5-methyl cytosine [M + H]+ ion (m/z 125) produced the major fragment ion of (m/z 109) corresponding to the loss of NH3. The CID of the 1-methyl nicotinamide [M + H]+ion (m/z 138) yielded the major fragment ion of m/z 78 corresponding to the loss of CH3COOH. The CID of 6-O-methyl guanine [M + H]+ (m/z 166) produced the major fragment ion of m/z 149 with the loss of NH3. The CID of the 2-amino-6-methyl mercaptopurine [M + H]+ ion (m/z 182) produced the major fragment ion of m/z 134 corresponding to the loss of CH3SH. The CID spectra of the 5-methyl cytidine [M + H]+ ion (m/z 258) resulted in the major fragment ion of m/z 126 corresponding to the loss of C5H8O4. The CID of the 7-methylguanosine [M + H]+ ion (m/z 299) yielded the major fragment ion of m/z 167 corresponding to the loss of C5H8O4. The CID of the 6-methyl-2-thiouracil [M − H]− ion (m/z 141) produced the major fragment ion of m/z 58 corresponding to the loss of NCS. The CID of the 5′-deoxy-5′-methyl thioadenosine [M − H]− ion (m/z 296) produced the major fragment ion of m/z 134 corresponding to the loss of C6H9SO3. The methylated nucleotide fragment ion structures are shown in Fig. 3C. CID spectra are presented in ESI Fig. 3.†
Method validation, limit of detection, limit of quantitation and an accurate calibration curve
The most vital criterion of a quantitative analytical method is the accuracy of detection and sensitivity to quantitation of the results produced. The parameters obtained by LC-MS/MS analysis of methylated metabolites are the coefficient of linearity (R2), LOD, LOQ, and repeatability (CV%), and are presented in Table 2. The LOD of methyl chloroformate is 421 ± 2 pg ml−1, methyl phosphonic acid is 25 ± 3 pg ml−1, methyl-4-hydroxy benzoate is 159 ± 1 pg ml−1, 1-methyl histidine is 1 ± 0.5 pg ml−1, 5-methyl cytosine is 50 ± 2 pg ml−1, 1-methyl nicotinamide is 32 ± 0.4 pg ml−1, 2-amino-6-methyl mercaptopurine is 2 ± 0.7 pg ml−1, 5-methyl cytidine is 6 ± 0.7 pg ml−1, 5-deoxy-5-methyl thioadenosine is 2 ± 0.6 pg ml−1, and 5-methoxy-3-indole acetic acid is 66 ± 0.8 pg ml−1. The linear range of the LOQ of methylated organic acids, amino acids and nucleotides was found to be 25–1 × 106 pg ml−1, 5–1 × 106 pg ml−1, and 5–1 × 106 pg ml−1, respectively.
Measurement of methylated metabolites in BLCA cell lines
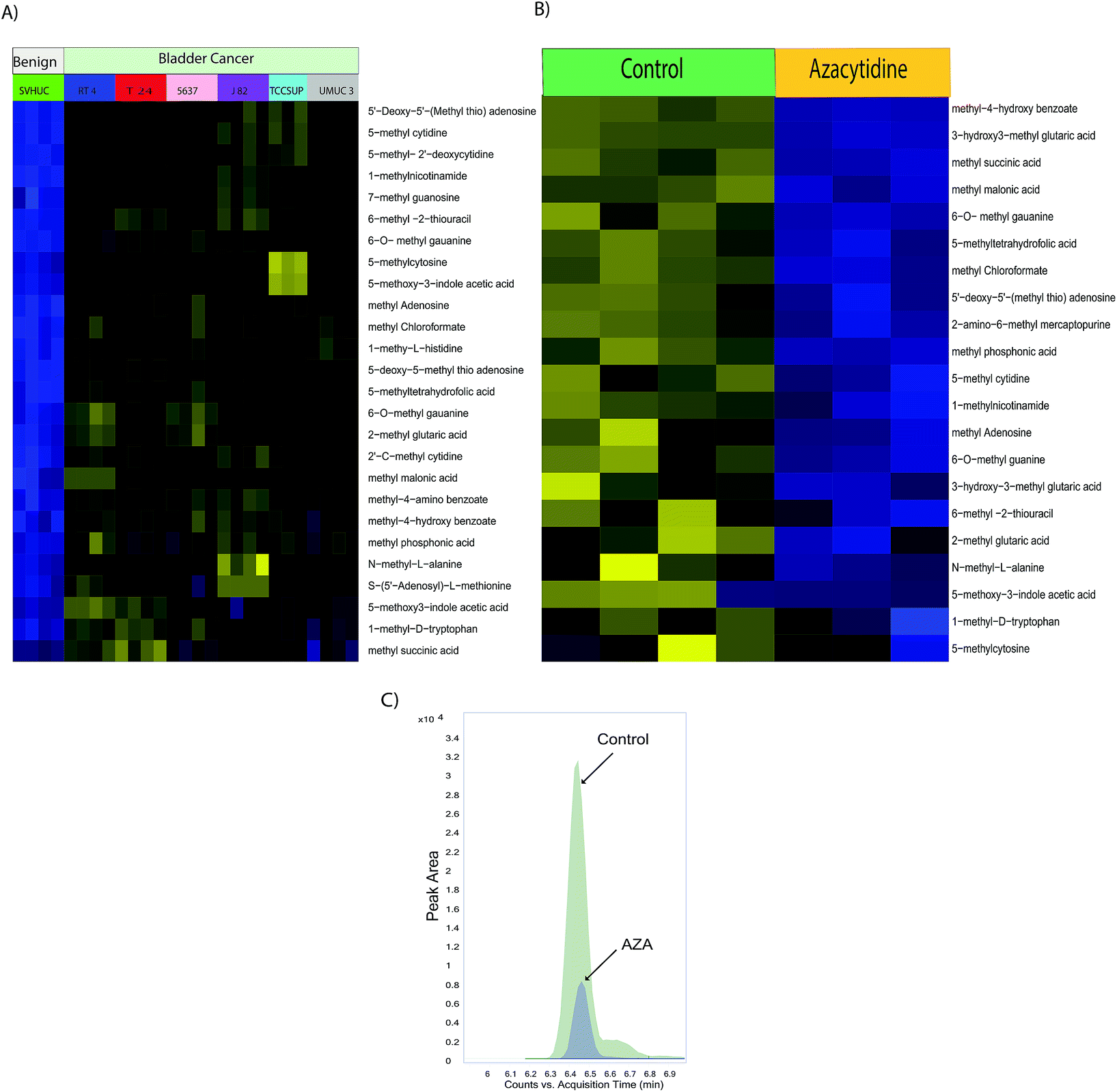
Epigenetic alterations such as DNA and histone methylations influence the chromatin states and impact gene expression during cancer progression.27 Methylated nucleotides and amino acids are intermediates of these methylation reactions. We further investigated the developed method in bladder benign and cancer cells and found that methylated organic acids, methylated amino acids, and methylated nucleotides in BLCA cells (RT4, 5637, T24, UMUC3, and TCCSUP) were upregulated compared to benign cells (SVHUC-1) (Fig. 4A, ESI Fig. 4†). Interestingly, the central methyl donor s-adenosyl methionine (SAM) is upregulated in BLCA cells, suggesting its role in providing methyl groups for epigenetic modifications. DNA methyl transferases execute the genomic methylation process that plays a crucial role in epigenetic modifications in the promoter regions of specific genes which in turn contribute to cancer development. EZH2 and nicotinamide N-methyltransferase (NNMT) are overexpressed in many cancers, alter protein (histone) methylation profiles and thereby promote carcinogenesis.28–30 Drug development focusing on the inhibition of methylation has been shown to be promising in the treatment of advanced cancers.
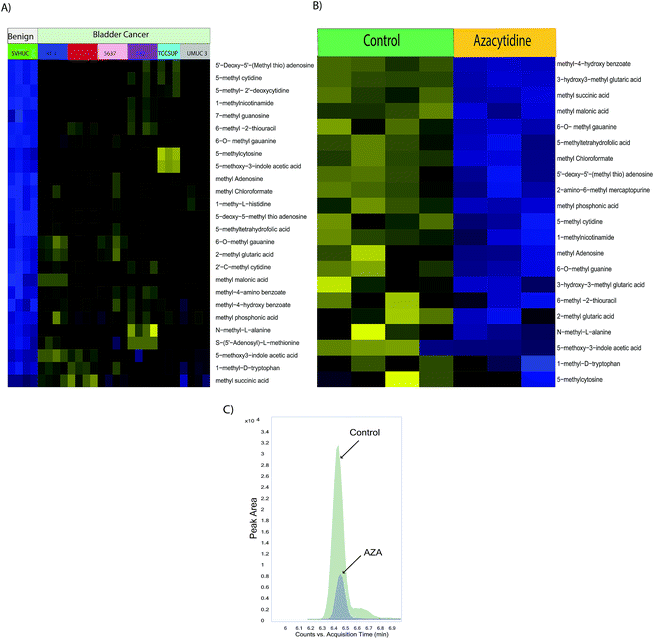 |
| | Fig. 4 (A) Heat map of hierarchical clustering of differential metabolites in benign and BLCA cells. (B) Heat map of hierarchical clustering of differential metabolites in BLCA cells upon AZA treatment. (C) Overlaid chromatogram of 1-methyl nicotinamide in control and AZA treated bladder cancer cells. | |
AZA treatments and expression of methylated metabolites in BLCA cell lines
Drugs that inhibit methylation, most notably the DNA methyltransferase inhibitor AZA, are used to treat cancer.31 We treated UMUC3 BLCA cells with AZA which is a known inhibitor of methylation. We found a significant reduction of methylated metabolites such as methyl succinic acid, methyl malonic acid, methyl chloroformate, methyl phosphonic acid, methylated amino acids such as 2-methyl glutaric acid and 1-methyl-D-tryptophan, and methylated nucleotides such as 6-O-methyl guanine, 5′-deoxy-5′-(methyl thio) adenosine, 5-methyl cytidine, methyl adenosine, 6-O-methyl guanine, and N-methyl-L-alanine upon AZA treatment supporting its therapeutic potential for BLCA (Fig. 4B). 1-Methyl nicotinamide is downregulated upon AZA treatment which is represented in Fig. 4C.
Conclusion
In this study, for the first time, we developed a very robust, accurate, and precise LC-MS-based method to measure 24 methylated metabolites including organic acids, amino acids, and nucleotides. This method can be applied to solid tissues, blood and urine samples with a sensitivity at picogram levels. We have validated this method in bladder benign and cancer cells and found a significant elevation of methylated metabolites in cancer compared to benign cells. Also, treatment of BLCA cells with methylation inhibitor AZA resulted in down-regulation of these methylated metabolites. This LC-MS method proves to be useful for qualitative and quantitative analysis of methylated organic acids, amino acids, and nucleotides which are deregulated in any disease conditions.
Author contributions
C. S. R., V. V., S. R. D., V. P., C. S. A., S. S. R. and N. P. conceived the project and wrote the manuscript with editorial input from all of the authors. C. S. R., A. S. K., V. V., and N. P. designed the experiments. A. M. and M. P. assisted with the dataset analysis.
Conflicts of interest
The authors do not have any conflict of interest.
Acknowledgements
This research was supported by the CPRIT Proteomics and Metabolomics Core Facility (N. P.), (RP170005), P30CA125123 Metabolomics Shared Resources, and the Dan L. Duncan Cancer Center and partially supported by the following grants American Cancer Society (ACS) Award 127430-RSG-15-105-01-CNE (N. P.), NIH R01CA220297 (N. P) NIH R01CA216426 (N. P.), U01CA179674-01A1 (A. S. K.) and NIH/NCI U01 CA214263 (N. P.). This project was also supported by the Agilent Technologies Center of Excellence in Mass Spectrometry, the Prostate Cancer Foundation (A. S. K.), the Brockman Foundation at Baylor College of Medicine, Metabolomics Core.
References
- S. L. Berger,
et al., An operational definition of epigenetics, Genes Dev., 2009, 23(7), 781–783 CrossRef CAS.
- S. B. Baylin and P. A. Jones, A decade of exploring the cancer epigenome - biological and translational implications, Nat. Rev. Cancer, 2011, 11(10), 726–734 CrossRef CAS.
- H. Wu and Y. Zhang, Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation, Genes Dev., 2011, 25(23), 2436–2452 CrossRef CAS.
- T. Kouzarides, Chromatin modifications and their function, Cell, 2007, 128(4), 693–705 CrossRef CAS PubMed.
- M. Tan,
et al., Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification, Cell, 2011, 146(6), 1016–1028 CrossRef CAS.
- M. A. Dawson and T. Kouzarides, Cancer epigenetics: from mechanism to therapy, Cell, 2012, 150(1), 12–27 CrossRef CAS.
- M. Esteller,
et al., DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis, Hum. Mol. Genet., 2001, 10(26), 3001–3007 CrossRef CAS PubMed.
- K. D. Robertson, DNA methylation and human disease, Nat. Rev. Genet., 2005, 6(8), 597–610 CrossRef CAS PubMed.
- S. Ito,
et al., Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine, Science, 2011, 333(6047), 1300–1303 CrossRef CAS.
- M. Tahiliani,
et al., Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1, Science, 2009, 324(5929), 930–935 CrossRef CAS.
- H. Yang,
et al., Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation, Oncogene, 2013, 32(5), 663–669 CrossRef CAS PubMed.
- B. Chowdhury,
et al., Quantification of 5-methylcytosine, 5-hydroxymethylcytosine and 5-carboxylcytosine from the blood of cancer patients by an enzyme-based immunoassay, Anal. Chim. Acta, 2014, 852, 212–217 CrossRef CAS PubMed.
- M. L. Chen,
et al., Quantification of 5-methylcytosine and 5-hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic-interaction liquid chromatography/quadrupole TOF mass spectrometry, Clin. Chem., 2013, 59(5), 824–832 CrossRef CAS PubMed.
- Y. Cheng,
et al., 5-Hydroxymethylcytosine: A new player in brain disorders?, Exp. Neurol., 2015, 268, 3–9 CrossRef CAS PubMed.
- J. D. Gary and S. Clarke, RNA and protein interactions modulated by protein arginine methylation, Prog. Nucleic Acid Res. Mol. Biol., 1998, 61, 65–131 CAS.
- A. Sobczynska-Malefora,
et al., An audit of holotranscobalamin (“Active” B12) and methylmalonic acid assays for the assessment of vitamin B12 status: application in a mixed patient population, Clin. Biochem., 2014, 47(1–2), 82–86 CrossRef CAS PubMed.
- D. J. Harrington, Laboratory assessment of vitamin B12 status, J. Clin. Pathol., 2017, 70(2), 168–173 CrossRef PubMed.
- J. Martens-Lobenhoffer and S. M. Bode-Boger, Chromatographic-mass spectrometric methods for the quantification of L-arginine and its methylated metabolites in biological fluids, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 851(1–2), 30–41 CrossRef CAS PubMed.
- C. W. Hu,
et al., Optimization of global DNA methylation measurement by LC-MS/MS and its application in lung cancer patients, Anal. Bioanal. Chem., 2013, 405(27), 8859–8869 CrossRef CAS PubMed.
- H. Wang and Y. Wang, LC-MS/MS coupled with stable isotope dilution method for the quantification of 6-thioguanine and S(6)-methylthioguanine in genomic DNA of human cancer cells treated with 6-thioguanine, Anal. Chem., 2010, 82(13), 5797–5803 CrossRef CAS.
- L. Zhang,
et al., Simultaneous determination of global DNA methylation and hydroxymethylation levels by hydrophilic interaction liquid chromatography-tandem mass spectrometry, J. Biomol. Screening, 2012, 17(7), 877–884 CrossRef PubMed.
- V. Vantaku,
et al., Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer, Oncotarget, 2017, 8(56), 95620–95631 CrossRef PubMed.
- F. Jin,
et al., Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, and DNA Damage in Bladder Cancer, Cancer Prev. Res., 2017, 10(10), 588–597 CrossRef CAS PubMed.
- D. W. B. Piyarathna,
et al., Distinct Lipidomic Landscapes Associated with Clinical Stages of Urothelial Cancer of the Bladder, Eur. Urol. Focus, 2017, EUF 3211-9 Search PubMed.
- F. C. von Rundstedt,
et al., Integrative Pathway Analysis of Metabolic Signature in Bladder Cancer: A Linkage to The Cancer Genome Atlas Project and Prediction of Survival, J. Urol., 2016, 195(6), 1911–1919 CrossRef PubMed.
- N. Putluri,
et al., Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression, Cancer Res., 2011, 71(24), 7376–7386 CrossRef CAS PubMed.
- Y. J. Park,
et al., Genome-wide epigenetic modifications in cancer, Prog. Drug Res., 2011, 67, 25–49 CAS.
- K. H. Kim and C. W. Roberts, Targeting EZH2 in cancer, Nat. Med., 2016, 22(2), 128–134 CrossRef CAS PubMed.
- M. Yamagishi and K. Uchimaru, Targeting EZH2 in cancer therapy, Curr. Opin. Oncol., 2017, 29(5), 375–381 CrossRef CAS PubMed.
- T. Shlomi and J. D. Rabinowitz, Metabolism: Cancer mistunes methylation, Nat. Chem. Biol., 2013, 9(5), 293–294 CrossRef CAS PubMed.
- S. Sharma, T. K. Kelly and P. A. Jones, Epigenetics in cancer, Carcinogenesis, 2010, 31(1), 27–36 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02168f |
| ‡ Authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Venkatrao
Vantaku‡
b,
Sri Ramya
Donepudi
a,
Chandra Sekhar
Amara
b,
Shiva Shankar
Ravi
b,
Akhil
Mandalapu
b,
Maharajni
Perla
b,
Vasanta
Putluri
a,
Arun
Sreekumar
ab and
Nagireddy
Putluri
a,
Venkatrao
Vantaku‡
b,
Sri Ramya
Donepudi
a,
Chandra Sekhar
Amara
b,
Shiva Shankar
Ravi
b,
Akhil
Mandalapu
b,
Maharajni
Perla
b,
Vasanta
Putluri
a,
Arun
Sreekumar
ab and
Nagireddy
Putluri