
Sensitive determination of psoralen and isopsoralen in Fructus Psoraleae by online solid phase microextraction with a porphyrin-based porous organic polymer modified capillary†
Yuling
Chen
,
Rong
Wang
and
Zilin
Chen
 *
*
Key Laboratory of Combinatorial Biosynthesis and Drug Discovery (Wuhan University), Ministry of Education, Wuhan University School of Pharmaceutical Sciences, Wuhan, 430071, China. E-mail: chenzl@whu.edu.cn; Fax: +86-27-68759850; Tel: +86-27-68759893
First published on 1st November 2018
Abstract
A novel porphyrin based porous organic polymer modified capillary (PPOP-capillary) was prepared for the extraction of psoralen and isopsoralen in Fructus Psoraleae. Fructus Psoraleae is an important traditional Chinese medicine in clinical therapy. Sensitive and effective extraction and determination of psoralen and isopsoralen benefit the quality control in Fructus Psoraleae and related preparations. PPOPs were immobilized on the inner wall of the capillary by a bottom-up approach. The formation of PPOPs has been confirmed by scanning electron microscopy, X-ray diffraction and Fourier transform infrared spectroscopy. An online solid phase microextraction coupled with HPLC method was developed for selective extraction and analysis of psoralen and isopsoralen in aqueous solutions. The proposed method showed a good linear range of 0.05–100 ng mL−1 with a low limit of detection of 0.01 ng mL−1. The method was also applied for the extraction of psoralen and isopsoralen in Fructus Psoraleae, which showed high enrichment efficiency and good clean-up capacity. Recoveries were calculated to be 93.3–96.7% with relative standard deviations less than 1.6%.
1. Introduction
Fructus Psoraleae, known as Buguzhi in traditional Chinese medicine (TCM), is the dried ripe fruit of Psoralea corylifolia L.1,2 It is widely used in the clinical treatment of coronary artery disease,3 asthma, diarrhea,4 osteoporosis,5 alopecia areata and vitiligo.1 A number of active components in this herb have been investigated including coumarins, flavonoids and meroterpenes.6 Psoralen and isopsoralen are two typical coumarin components (ESI Fig. S1†) with extensive pharmacological actions such as antibacterial, antitumor, anti-inflammatory, estrogenic activities, antioxidant activities and so on.7–9 In the National Commission of Chinese Pharmacopoeia (2015), the two components were selected as markers for the quality control of Buguzhi and related preparations.1 Therefore, it is necessary to develop a proper method to determine psoralen and isopsoralen for quality control in Fructus Psoraleae and related preparations.To date, different kinds of analytical methods including capillary electrophoresis (CE),10 high performance liquid chromatography (HPLC)11,12 and high-speed counter current chromatography13 have been successfully applied for determination of psoralen and isopsoralen. Due to their low content and complex sample matrix in natural samples, some extraction techniques were reported for sample treatment before chromatographic analysis, including liquid–liquid extraction,11,14 supercritical fluid extraction13 and solid phase extraction.15 These methods are widely used, but they also have some shortcomings. Some of them are time-consuming or exhaustive, which is not suitable for the purpose of highly sensitive determination. As a result, developing a pretreatment method containing effective extraction and clean-up capacity is definitely desirable.
Solid phase microextraction (SPME) is a versatile and economical sample preparation tool that has been demonstrated to be well-suited for facile and effective analysis according to plenty of applications.16,17 In-tube SPME is introduced as an alternative to fiber-based SPME, where the sorbents are coated, packed or synthesized inside capillaries or tubes.18–20 In-tube SPME, using an open tubular fused-silica capillary column with an inner surface coating as the SPME device, is simple and can be easily coupled online with HPLC.21 The abundant active silanol groups in the inner wall of the tube are good binding sites for immobilization of sorbents. Therefore, new modification materials are still desirable for facile synthesis and efficient sample pretreatment. In this work, porphyrin based porous organic polymers (PPOPs) were immobilized in the capillary as a sorbent.
Porphyrin based porous organic polymers are very attractive due to the presence of basic porphyrin containing ample cavities with 18-π electron systems and aromatic linkers in their skeleton, which endows them with rich porosity, high surface area and solvent stability.22–24 However, the porphyrin monomers are usually pre-synthesized and the condensation process is expensive because of using a Pd-catalyst.25,26 A facile one-pot bottom-up approach to porphyrin chemistry was developed by Bhaumik's group.27 PPOPs were synthesized by an extended aromatic substitution reaction between pyrrole and aromatic dialdehydes in glacial acetic acid medium. Although a plethora of studies have demonstrated their wide applications in catalysis28–32 and gas adsorption,27,33–35 examples of their application in extraction are rare.36 Considering the rich macrocyclic cavities, large surface areas, inherent nanopores, and excellent stability of PPOPs, it makes sense to broaden their application in the extraction field. The abundant porphyrin groups, phenyl rings and pore structure throughout the entire skeleton are expected to form hydrophobic interaction, π–π interaction and size selection with some aromatic compounds.
In this study, we aim to develop an online SPME-HPLC method for the extraction and analysis of two coumarins in Fructus Psoraleae. Porphyrin based porous organic polymers were immobilized onto the capillary as sorbents by a facile bottom-up approach. The functional capillary was applied to extract psoralen and isopsoralen. Conditions for synthesis and extraction were systematically investigated to obtain optimum efficiency. The SPME-HPLC method was validated with good analytical performance. As expected, the proposed method was successfully applied for extraction and determination of psoralen and isopsoralen in Fructus Psoraleae.
2. Experimental
Chemicals and materials
The reagents used in the experiments were of analytical or chromatographic grade. Psoralen, isopsoralen, p-phthalaldehyde, and pyrrole were obtained from Aladdin Reagent (Shanghai, China). (3-Aminopropyl)-triethoxysilane (APTES) was purchased from Sigma-Aldrich (MO, USA). Sodium hydroxide (NaOH), hydrochloric acid (HCl), glacial acetic acid (HAc) and sodium phosphate dibasic dodecahydrate (Na2HPO4·12H2O) were commercially available from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Methanol was of HPLC grade and purchased from Tedia (OH, USA). Water was purified using a Milli-Q system (MA, USA). Fused-silica capillaries with a dimension of 200 μm i.d. × 365 μm o.d. were obtained from Ruifeng Chromatographic Devices (Yongnian, Hebei, China). Fructus Psoraleae was purchased from a local pharmacy in Fruit Lake, Wuhan, China.Instrumentation
A Shimadzu HPLC system (Tokyo, Japan) consisting of two 20AD pumps, a 20A3 degasser, a 20A UV detector, and a thermostat controlled column compartment was used for chromatographic analysis. Data were acquired and processed using Shimadzu LC solution software. The analytical column was a 250 mm × 4.6 mm, 5 μm C-18 column from GL Sciences (Tokyo, Japan). The mobile phase consisted of methanol and H2O (55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45, v/v) with a flow rate of 1.0 mL min−1. The detection wavelength was set at 254 nm and column temperature was 30 °C. A precise syringe pump (Longer Pump Company, Baoding, China) was used to push the solutions through capillaries.
:45, v/v) with a flow rate of 1.0 mL min−1. The detection wavelength was set at 254 nm and column temperature was 30 °C. A precise syringe pump (Longer Pump Company, Baoding, China) was used to push the solutions through capillaries.
Preparation of a NH2-terminated capillary
Fig. 1 illustrates the modification of porphyrin based porous organic polymers in the inner surface of the capillary. A 20 cm fused-silica capillary (200 μm i.d. × 365 μm o.d.) was treated sequentially with 1 M NaOH for 2 h, water for 30 min, 1 M HCl for 30 min, water for 30 min, and methanol for 1 h. The pretreated capillary was filled with a solution of APTES in water (10%, v/v) with both ends sealed and kept in an oven at 95 °C for 30 min. The amino coating procedure was repeated twice in order to obtain sufficient amino groups for immobilization of p-phthalaldehyde in the next step. The amino-modified capillary was rinsed with methanol to flush out the residuals and dried with a stream of nitrogen overnight for further use. The flow rate was kept unchanged at 1.0 mL h−1.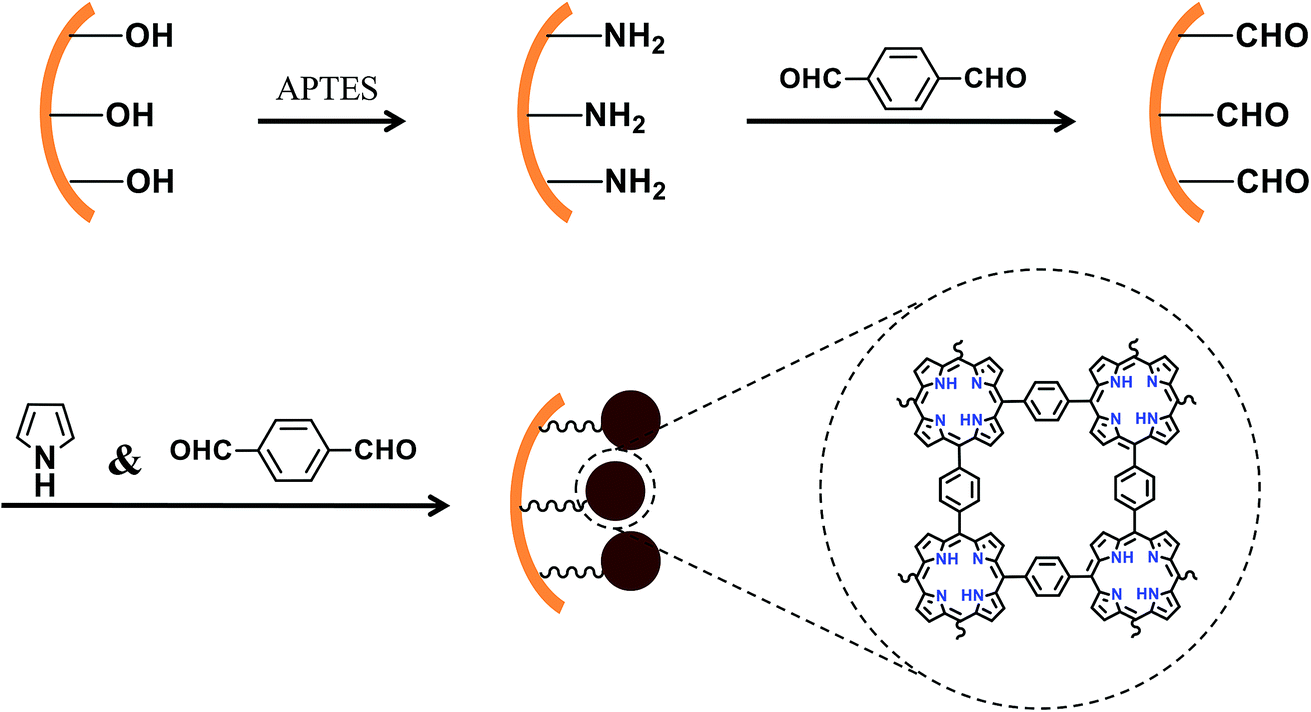 | ||
| Fig. 1 Schematic of modification of PPOPs on the fused-silica capillary. | ||
Preparation of a CHO-terminated capillary
p-Phthalaldehyde was immobilized on the surface of the amino-terminated capillary by Schiff reaction. A methanol solution of p-phthalaldehyde (4 mg mL−1) was continually injected into the amino-terminated capillary with a syringe pump for 20 min at room temperature. Then, the capillary was circled with a Teflon tube and kept in a water bath at 40 °C for 30 min. The immobilization of p-phthalaldehyde was carried out twice. The prepared capillary was rinsed with methanol to remove residuals and then flushed with N2 for 2 h to remove the solvent. The CHO-terminated capillary interface facilitated the growth and formation of PPOPs. The flow rate was kept unchanged at 1.0 mL h−1.Preparation of the PPOP-modified capillary
The growth of PPOPs was carried out on the CHO-terminated capillary according to Modak's method.24 Pyrrole (30 mg, 0.44 mmol) and p-phthalaldehyde (17 mg, 0.13 mmol) were dissolved in glacial acetic acid (5 mL). Ultrasonication was applied for pre-polymerization of PPOPs for 3 min. The CHO-terminated capillary was quickly filled with the pre-polymerization solution with a syringe and incubated in a 150 °C oil bath for 2 h with both ends sealed with rubbers. Under acidic conditions, p-phthalaldehyde is primarily activated through protonation. Then electrophilic aromatic substitution on pyrrole with p-phthalaldehyde is performed to construct a porous porphyrinic network. The PPOP-modified capillary was flushed with methanol for 2 h to remove the residual monomers and dried with N2 overnight for further use.Sample preparation
The Fructus Psoraleae samples were first dried and ground into powder. 0.4 g sample powder was used for extraction with 4 mL methanol under ultrasonication for 30 min. The supernatant was collected after centrifugation of 6932g for 10 min and then filtered through a 0.22 μm nylon membrane to obtain a Fructus Psoraleae extract sample. Then 5 μL of the extract sample was diluted to 40 mL with 10 mM phosphate buffer (pH 7) and acetonitrile for extraction and direct injection, respectively.Online solid phase microextraction
The extraction capillary replaced the loop of a six-port valve of the HPLC (ESI Fig. S2†). The detailed process for sample loading and elution is provided in the ESI.†3. Results and discussion
Characterization of the PPOP-modified capillary
Porphyrin based porous organic polymers were modified on the inner wall of the capillary by polymerization of pyrrole and p-phthalaldehyde through a hydrothermal reaction in glacial acetic acid medium. The prepared PPOP-modified capillary was characterized by scanning electron microscopy (SEM), X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR).As can be seen in the photographs of the capillary in Fig. 2B, after PPOP modification, the inner wall of the capillary becomes dark brown. The morphology of the bare column and PPOP-modified column was investigated by SEM, as shown in Fig. 2. It can be seen that the inner wall of the bare capillary is quite smooth (Fig. 2C and E). After modification, a homogeneous layer is observed, indicating the successful polymerization and immobilization of PPOPs. Fig. 2F reveals that the PPOPs are uniform spherical particles with a dimension of about 1 μm. Such a uniform and interesting morphological feature is beneficial for application in extraction.
 | ||
| Fig. 2 Photographs and scanning electron micrographs (SEM) of the bare capillary (A, C and E) and PPOP-capillary (B, D and F). | ||
X-ray diffraction was used to characterize the formation of porphyrin based porous organic polymers. As shown in ESI Fig. S3A,† there is no apparent peak diffraction in XRD analysis, revealing that the formed polymers contain an amorphous network. Besides, in our synthetic procedure, the polymerization involves simple aromatic electrophilic substitution on pyrrole and p-phthalaldehyde. At acidic pH, the polymerization of pyrrole, which leads to polypyrrole or oligopyrrole, may exist. As we know, the characteristic peak of amorphous polypyrrole polymers is at about 26 degrees.37,38 From the XRD analysis, the absence of any broad peak at about 26 degrees of 2θ shuts out the possibility of polymerization of pyrrole. This result demonstrates the successful formation of porphyrin based porous organic polymers.
As shown in ESI Fig. S3B,† the peak at 1694 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) is strongly attenuated. The absence of aldehyde groups suggests the formation of polymers. The other characteristic peaks of pyrrole at 1603 cm−1, 1313 cm−1 and 1049 cm−1 and peaks of phenyl groups at 1503 cm−1, 802 cm−1 and 703 cm−1 confirm the incorporation of pyrrole and p-phthalaldehyde. The FT-IR result also underpins the formation of PPOPs.
O stretching) is strongly attenuated. The absence of aldehyde groups suggests the formation of polymers. The other characteristic peaks of pyrrole at 1603 cm−1, 1313 cm−1 and 1049 cm−1 and peaks of phenyl groups at 1503 cm−1, 802 cm−1 and 703 cm−1 confirm the incorporation of pyrrole and p-phthalaldehyde. The FT-IR result also underpins the formation of PPOPs.
Absorptive behavior of the PPOP-modified capillary
In this work, psoralen and isopsoralen were extracted by means of hydrophobic interaction, π–π interaction and size selection with the PPOP-modified capillary. To investigate the selectivity of the PPOP coating, several polycyclic aromatic hydrocarbon (PAH) compounds, including FLU, PYR, B[a]AN, B[b]FL, B[a]PY and D[a,h]AN, were used for the purpose. The results are shown in ESI Table S1.† The enrichment factor was calculated and served as a measure of the binding affinity of the PPOP coating with PAHs. It was defined as the ratio of analyte peak areas with and without SPME (EF = ASPME/A0). As can be seen in ESI Table S1,† all compounds are efficiently enriched through hydrophobic interaction and π–π interaction between the condensed rings of PAHs and π-conjugated aromatic groups of the PPOP coating. Besides, it seems that the pore sizes of analytes also influence the extraction. The compounds with a similar size to the pore size of PPOPs (ca. 10.8 Å)27 could be more effectively enriched. In this work, psoralen and isopsoralen are selected as the analytes because of their abundant π-conjugated structures and specific molecular sizes (about 10 Å), which give rise to an efficient extraction. The EFs of psoralen and isopsoralen are 413 ± 12 and 314 ± 5. In conclusion, the selective adsorption of the PPOP-modified capillary could be attributed to hydrophobic interaction, π–π interaction and size selection.Optimization of synthetic conditions
The thickness and structure of PPOPs would affect the interaction between the analytes and PPOPs in the capillary. For the sake of effective extraction efficiency, some synthetic conditions that can impact the modification of PPOPs were investigated, including the (a) concentration of p-phthalaldehyde, (b) synthetic time and (c) concentration of pyrrole (as shown in ESI Fig. S4†). Respective details are given in the ESI.† The optimized conditions were as follows: (a) concentration of p-phthalaldehyde: 4 mg mL−1; (b) synthetic time: 2 hours; and (c) concentration of pyrrole: 88 mM.Optimization of extraction conditions
Psoralen and isopsoralen were selected as analytes due to their strong interaction with PPOPs and demands of their low-content quantification. For higher extraction efficiency, the following extraction conditions were investigated.The sample pH value is usually an important parameter in the extraction process, which affects the structure of both the sorbent materials and analytes and further influences the interaction between them. In this work, ester bonds existed in the structure of psoralen and isopsoralen, which may be sensitive to the pH of the sample solution. As shown in Fig. 3A, pH values varying from 3 to 9 were investigated. The medium was composed of 10 mM phosphate buffer and the concentrations of psoralen and isopsoralen were 50 ng mL−1. Sample solutions of 2 mL were loaded into the PPOP-capillary at a constant flow rate (0.3 mL min−1). Changes of the extraction efficiency of psoralen and isopsoralen were not so significant. A slight increase was observed along with the increase of pH in the range of 3–7, and a higher pH value would result in lower extraction efficiency. The possible reasons are that medium with a lower or higher pH value is disadvantageous for the ester structure of psoralen and isopsoralen. However, the short time-consumption of the sampling process did not make a big difference in the extraction. To ensure the best extraction efficiency and simplify the sample preparation procedures, the pH value of sample solution was maintained at about 7 in further studies.
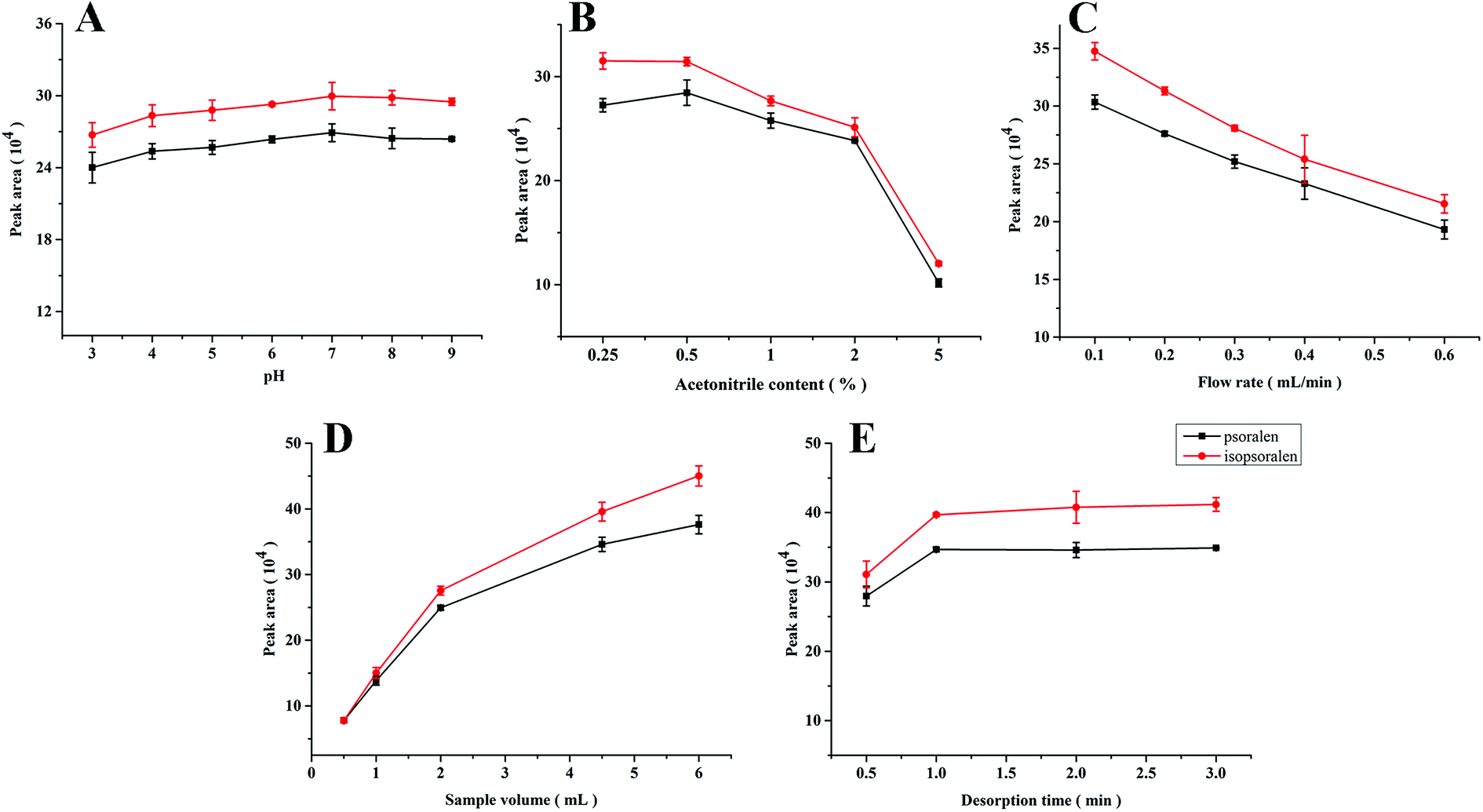 | ||
| Fig. 3 Effects of sample pH value (A), acetonitrile content (B), flow rate (C), sample volume (D) and desorption time (E). | ||
Because of bad solubility of psoralen and isopsoralen in water, acetonitrile was added to the sample solutions to help them dissolve. However, excessive acetonitrile in the aqueous sample solution may not be advantageous for the extraction of psoralen and isopsoralen. Psoralen and isopsoralen sample solutions (2 mL, at a flow rate of 0.3 mL min−1) containing acetonitrile of 0.25–5% (v/v) were loaded onto the PPOP-capillary for SPME. Curves of the peak area of psoralen and isopsoralen extracted by the PPOP-capillary to the acetonitrile content are shown in Fig. 3B. The peak areas of psoralen and isopsoralen were found to increase with acetonitrile content from 0.25% to 0.5% and slightly decreased in the range of 0.5% to 2%. With further increase of acetonitrile content, the extraction efficiency dropped dramatically. This result illustrated that higher acetonitrile content could increase the initial concentration of psoralen and isopsoralen in the sample solution, but their partition coefficient might be severely influenced. In consideration of the extraction efficiency and good solubility of analytes, acetonitrile content of 1% (v/v) was selected.
Generally, the sampling rate is a vital factor in in-tube SPME because it has potential to affect the contact between analytes and the polymer sorbent. A faster sampling rate would lead to high pressure and damage of the sorbent, but a slow sampling rate would prolong the pretreatment process. Therefore, low time consuming and good extraction efficiency are desired in the process of sampling. The sampling rate was investigated from 0.1 to 0.6 mL min−1 controlled using an auxiliary pump. As shown in Fig. 3C, the peak areas of psoralen and isopsoralen decreased along with the increase of the sampling rate, which means that the sampling rate has a big influence on the interaction of the analytes and sorbent. In order to balance the extraction efficiency and time consumption, a sampling rate of 0.3 mL min−1 was maintained for further studies.
The extraction efficiency can be enhanced by increasing the volume of the sample solution to some extent. In this work, standard solutions (50 ng mL−1, pH 7) from 0.5 to 6 mL were loaded for extraction. As shown in Fig. 3D, the peak areas of the two analytes increased rapidly along with the increase of sample volumes from 0.5 to 4.5 mL, and increased slowly above 4.5 mL. However, even at a volume of 6 mL, the adsorption procedure still did not reach equilibrium. In consideration of enrichment factors and analysis time, a sample volume of 4.5 mL was selected for further studies.
In in-tube SPME, the analyte was eluted with the mobile phase and simultaneously injected into the LC column for analysis. Rapid and sufficient desorption is taken into consideration. In this work, desorption times from 0.5 to 3 min were investigated and the results are shown in Fig. 3E. The peak areas of psoralen and isopsoralen increased within 0.5 to 1 min and remained almost unchanged until 3 minutes. It seems that desorption could be finished in a short time. In consideration of sufficient desorption and protection of the modification layer in the capillary, 2 min was chosen as the desorption time.
Stability of the PPOP-capillary
The stability of the PPOP layer on the surface of the capillary is essential for SPME. In the process of extraction, the PPOP-capillary was filled with sample solutions or washing solvents. Long-time immersion and pressure of flow could inevitably cause damage to the structure of modification layers, resulting in poor stability and low extraction efficiency. The stability of the PPOP-capillary was investigated from the aspect of consecutive use. In order to evaluate the stability of the PPOP layer, the conservation rate (C%), which evaluates the peak area ratio of after use (for more than 50 times) to before use, is introduced and calculated as:| C% = A1/A0 × 100% |
Method validation
Under the optimum conditions mentioned above, a PPOP-capillary-based online SPME-HPLC method was developed for quantification of psoralen and isopsoralen. Analytical performances of the proposed method were investigated, including the calibration equation, linearity range, correlation coefficients, limit of detection (LOD), limit of qualification (LOQ) and repeatability, under the optimized conditions. The results are listed in Table 1. A good linearity in the range of 0.05–100 ng mL−1 with R ≥ 0.9961 was obtained. The limits of detection (LODs) of this method can reach 0.01 ng mL−1 both for psoralen and isopsoralen. The limits of qualification (LOQs) were 0.03 ng mL−1, which is sufficient for determination of psoralen and isopsoralen in traditional Chinese medicine Fructus Psoraleae. By loading five replicates of standard sample solutions (50 ng mL−1), the RSD values of precision for psoralen and isopsoralen were lower than 5.96%, indicating good reproducibility of the method.| Analyte | Range (ng mL−1) | Regression equation | R | LOD (ng mL−1) | LOQ (ng mL−1) | Precision (RSD, %) | ||
|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | Column-to column | ||||||
| a x: concentration of psoralen or isopsoralen (ng mL−1). y: peak areas of psoralen or isopsoralen. | ||||||||
| Psoralen | 0.05–100 | y = 7064x + 1795 | 0.9961 | 0.01 | 0.03 | 2.81 | 3.82 | 3.93 |
| Isopsoralen | 0.05–100 | y = 8408x + 1216 | 0.9970 | 0.01 | 0.03 | 1.70 | 2.71 | 5.96 |
Application in real samples
In this work, the proposed PPOP-capillary-based online SPME-HPLC method was applied to extract and determine the contents of psoralen and isopsoralen in Fructus Psoraleae. Chromatograms of the samples with and without extraction are shown in Fig. 4. Both psoralen and isopsoralen were well detected after extraction. The contents of psoralen and isopsoralen in Fructus Psoraleae are calculated to be 2.72 ± 0.03 mg g−1 and 2.16 ± 0.01 mg g−1, respectively. Recoveries were also tested by addition of psoralen and isopsoralen standards at 2.4 mg g−1. On calculating the mean value of three duplicates, recoveries were in the range of 93.3–96.7% with RSD values less than 1.6%. The results also showed the good accuracy of the proposed method (Table 2).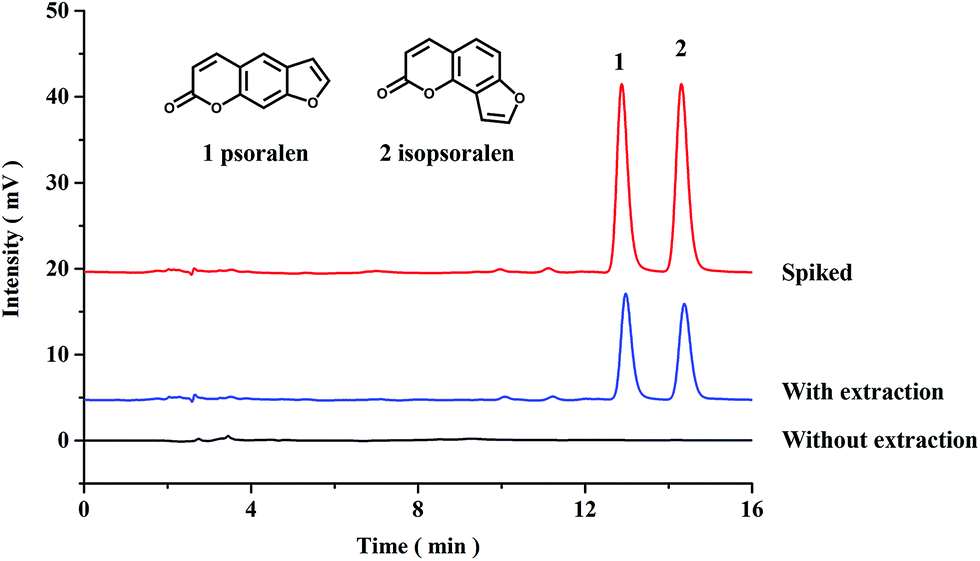 | ||
| Fig. 4 Chromatograms of Fructus Psoraleae samples. | ||
| Sample | Content (mg g−1) | Added (mg g−1) | Found (mg g−1) | Recovery (%) | RSD (%, n = 3) |
|---|---|---|---|---|---|
| Psoralen | 2.72 ± 0.03 | 2.40 | 4.96 ± 0.08 | 93.33 | 1.51 |
| Isopsoralen | 2.16 ± 0.01 | 2.40 | 4.48 ± 0.08 | 96.67 | 1.65 |
Comparison
Compared to reported methods, this work has a lower LOD and a wider linear range, indicating the efficient extraction and sensitive determination by this method (as shown in ESI Table S2†).4. Conclusions
In this work, we fabricated a novel PPOP-capillary-based online SPME-HPLC system for analysis of two coumarins in aqueous and Fructus Psoraleae samples. Porphyrin based porous organic polymers were successfully immobilized on the surface of the capillary. The modified capillary exhibits good stability and excellent extraction efficiency toward psoralen and isopsoralen. The method showed high sensitivity, a wide linear range, and good reproducibility. Application for extraction and determination of the contents of two coumarins in crude extracts of Fructus Psoraleae samples also shows high enrichment efficiency, good repeatability, and good recoveries. The results demonstrated that PPOPs can be potential sorbents for SPME and the proposed method is promising for sample pretreatment and quantitative analysis of psoralen and isopsoralen in traditional Chinese medicine administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 81872828 and 81573384).Notes and references
- National Commission of Chinese Pharmacopoeia, Pharmacopoeia of the People's Republic of China, Chinese Medical Science and Technology Press, Beijing, 2015, vol. 1, pp. 187–188 Search PubMed.
- H. Wagner, R. Bauer, D. Melchart and A. Staudinger, Chromatographic Fingerprint Analysis of Herbal Medicines, Springer, Cham, 2016, vol. 4, pp. 47–57 Search PubMed.
- X. Li, Y. J. Lee, Y. C. Kim, G. S. Jeong, H. Z. Cui, H. Y. Kim, D. G. Kang and H. S. Lee, Phytother. Res., 2011, 25, 1574–1578 CrossRef CAS.
- Y. G. Li, J. Hou, S. Y. Li, X. Lv, J. Ning, P. Wang, Z. M. Liu, G. B. Ge, Y. J. Ren and L. Yang, Fitoterapia, 2015, 101, 99–106 CrossRef CAS.
- W. D. Li, C. P. Yan, Y. Wu, Z. B. Weng, F. Z. Yin, G. M. Yang, B. C. Cai and Z. P. Chen, Phytomedicine, 2014, 21, 400–405 CrossRef CAS PubMed.
- L. H. Zhao, C. Y. Huang, Z. Shan, B. R. Xiang and L. H. Mei, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 821, 67–74 CrossRef CAS PubMed.
- A. H. Yang, J. X. Chen, Y. T. Ma, L. L. Wang, Y. W. Fan and X. He, J. Pharm. Biomed. Anal., 2017, 141, 200–209 CrossRef CAS PubMed.
- Y. F. Wang, Y. N. Liu, W. Xiong, D. M. Yan, Y. Zhu, X. M. Gao, Y. T. Xu and A. D. Qi, J. Ethnopharmacol., 2014, 151, 609–617 CrossRef CAS PubMed.
- Q. Q. Gao, C. P. Yan, Z. S. Xu, Y. Wu, Z. B. Weng, G. H. Zhao, L. J. Zhang, J. Y. He, B. C. Cai, Z. P. Chen and W. D. Li, Biomed. Chromatogr., 2016, 30, 528–535 CrossRef CAS PubMed.
- F. Wu, A. F. Wang, Y. Zhou, T. S. Zhou, H. L. Jiang and Y. Z. Fang, Chromatographia, 2005, 61, 157–160 CrossRef CAS.
- Y. Chen, Q. Y. Xiang and Z. L. Chen, Anal. Methods, 2014, 6, 269–275 RSC.
- L. J. Luan, X. Y. Shen, X. S. Liu, Y. J. Wu and M. L. Tan, Biomed. Chromatogr., 2018, 32, e4059 CrossRef.
- X. Wang, Y. Q. Wang, J. P. Yuan, Q. L. Sun, J. H. Liu and C. C. Zheng, J. Chromatogr. A, 2004, 1055, 135–140 CrossRef CAS.
- W. Yang, C. Feng, D. Z. Kong, X. W. Shi, Y. Cui, M. Liu, Q. Wang, Y. L. Wang and L. T. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 575–582 CrossRef CAS.
- C. Feng, J. L. Ruan and Y. L. Cai, J. Braz. Chem. Soc., 2010, 21, 2272–2277 CrossRef CAS.
- N. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyaci, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef.
- W. P. Zhang, W. Zhou and Z. L. Chen, J. Sep. Sci., 2014, 37, 3110–3116 CrossRef CAS.
- X. Q. Wang, J. J. Feng, Y. Tian, C. N. Luo and M. Sun, J. Chromatogr. A, 2018, 1550, 1–7 CrossRef CAS.
- X. Q. Wang, L. Pan, J. J. Feng, Y. Tian, C. N. Luo and M. Sun, J. Chromatogr. A, 2017, 1522, 16–22 CrossRef CAS PubMed.
- J. J. Feng, H. Mao, X. Q. Wang, Y. Tian, C. N. Luo and M. Sun, J. Sep. Sci., 2018, 41, 1839–1846 CrossRef CAS PubMed.
- A. Ishizaki, K. Saito, N. Hanioka, S. Narimatsu and H. Kataoka, J. Chromatogr. A, 2010, 1217, 5555–5563 CrossRef CAS PubMed.
- X. S. Wang, J. Liu, J. M. Bonefont, D. Q. Yuan, P. K. Thallapally and S. Q. Ma, Chem. Commun., 2013, 49, 1533–1535 RSC.
- L. Chen, Y. Yang and D. J. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS.
- Z. H. Xiang, Y. H. Xue, D. P. Cao, L. Huang, J. F. Chen and L. M. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433–2437 CrossRef CAS.
- Z. D. Ding, W. Zhu, T. Li, R. Shen, Y. X. Li, Z. J. Li, X. H. Ren and Z. G. Gu, Dalton Trans., 2017, 46, 11372–11379 RSC.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2011, 2, 686–689 RSC.
- A. Modak, M. Nandi, J. Mondal and A. Bhaumik, Chem. Commun., 2012, 48, 248–250 RSC.
- K. N. Zhang, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 4859–4866 CrossRef CAS.
- Y. G. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- G. L. Lu, Y. L. Zhu, K. L. Xu, Y. H. Jin, Z. Y. Ren, Z. N. Liu and W. Zhang, Nanoscale, 2015, 7, 18271–18277 RSC.
- A. R. Antonangelo, C. G. Bezzu, S. S. Mughal, T. M. Malewschik, N. B. McKeown and S. Nakagaki, Catal. Commun., 2017, 99, 100–104 CrossRef CAS.
- A. R. Oveisi, K. N. Zhang, A. Khorramabadi-zad, O. K. Farha and J. T. Hupp, Sci. Rep., 2015, 5, 10621 CrossRef CAS.
- V. S. P. K. Neti, X. F. Wu, S. G. Deng and L. Echegoyen, Polym. Chem., 2013, 4, 4566–4569 RSC.
- A. Modak, M. Pramanik, S. J. Inagaki and A. Bhaumik, J. Mater. Chem. A, 2014, 2, 11642–11650 RSC.
- X. M. Liu, A. Sigen, Y. W. Zhang, X. L. Luo, H. Xia, H. Li and Y. Mu, RSC Adv., 2014, 4, 6447–6453 RSC.
- J. T. Wang, C. N. Jiao, M. H. Li, X. L. Wang, C. Wang, Q. H. Wu and Z. Wang, Microchim. Acta, 2018, 185, 36, DOI:10.1007/s00604-017-2542-3.
- A. Liu, C. Li, H. Bai and G. Q. Shi, J. Phys. Chem. C, 2010, 114, 22783–22789 CrossRef CAS.
- X. Fan, Z. W. Yang and N. He, RSC Adv., 2015, 5, 15096–15102 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay01885e |
| This journal is © The Royal Society of Chemistry 2019 |