
Label-free fluorescence DNA walker for protein analysis based on terminal protection and dual enzyme assisted cleavage induced G-quadruplex/berberine conformation†
Shue
Deng‡
,
Baolong
Zhou‡
,
Wenjing
Li
,
Hui
Li
,
Fenglian
Zhang
and
Jingjing
Ming
 *
*
College of Pharmacy, Weifang Medical University, Weifang 261053, China. E-mail: mingjj@wfmc.edu.cn; Fax: +86-536-8462539; Tel: +86-536-8462539
First published on 29th October 2019
Abstract
Development of a simple, fast, cost-efficient and sensitive approach for accurate protein analysis is of high significance due to its potential application in disease diagnosis and biomedicine research. Thus, we established a label-free fluorescence DNA walker for streptavidin detection based on terminal protection and dual enzyme assisted cleavage induced G-quadruplex/berberine conformation. In this paper, the swing arm probe and report probe were pre-assembled on gold nanoparticles. With the addition of a target, through the high-efficiency affinity between streptavidin and biotin in order to prevent the hydrolysis of exonuclease I, the swing arm probe which contains 8–17 DNAzyme cannot be destroyed and plays a role in the catalytic cleavage of the report probe, and the liberating fragment of the report probe which contains a specific sequence (5′-(TTAGGG)4) of G-quadruplex units can combine with berberine and shows an evident fluorescence signal enhancement. Our method, a sensitive and selective method of protein detection, achieves a 20 pM detection limit toward streptavidin. This developed DNA walker, which combines terminal protection and a dual enzyme assisted strategy, provides a prospective channel for streptavidin detection and should also be used for the design of biosensors in bio-detection and disease diagnosis.
Introduction
Detection of protein biomarkers plays a critical role in food safety regulations, environmental monitoring and disease diagnostics.1,2 Thus, a simple, fast, cost-efficient and sensitive approach to accurate protein analysis is of significance for our life processes.3 Nowadays, with the advances in science and technology, many different protein detection methods have been proposed such as enzyme-linked immunosorbent assays (ELISA),4 chemiluminescence,5 electrochemistry,6 chromatography,7 colorimetry,8 surface plasmon resonance9 and fluorescence.10–12 Among them, fluorescence assays rely on portable spectrometers or the naked eye and are well known for their rapid, simple and sensitive signal feedback, which makes them highly applicable in on-field detection.13 Even though fluorescence spectroscopy is a useful technique for the detection of biomolecules, there is still some insufficiency. First, improvement in sensitivity is needed, such as signal amplification and background reduction. Second, cost-effectiveness is also a problem, which should be worth considering. Therefore, designing an excellent sensor strategy is important.Molecular machines, defined by E. Drexel as “mechanical devices that perform useful functions using components of nanometer-scale and defined molecular structure”, have been known to play a crucial role in physiological functions such as mechanical motion, signal transduction and cellular transport.14,15 Among these molecular machines, the DNA walker, which was first proposed by Seeman in 2004,16 has attracted a lot of interest for the construction of biosensors. Through DNA strand displacement or DNAzyme/endonuclease-mediated DNA hydrolysis, it moves autonomously and progressively along a linear DNA track, in order to convert chemical energy to mechanical motion as a machine. Gold nanoparticles (AuNPs), owing to their distinct physical and chemical attributes such as unique optoelectronic properties, excellent biocompatibility and size controllability, are excellent scaffolds for the design of DNA walkers.17 For example, X. Chris Le developed a DNA walker that operates in living cells for microRNA detection.18 Lei introduced a super DNA walker machine in order to monitor the telomerase activity in humans.19 Li established a dual-enzyme-assisted 3D DNA walking machine for quantitative detection of T4 polynucleotide kinase.20 Fan's group reported a particle stochastic DNA walker for monitoring DNA hybridization, which is powered by exonuclease III.21 Even though these DNA walkers achieved satisfactory results in bioanalysis, there is still large room for improvement in signal output. All these methods are used by molecular beacons, which have a fluorescent modified-DNA probe as a signal output. This may sometimes generate a false positive signal and high background. Thus, more attention should be paid to designing a new DNA walker for sensitive detection, and low background and effective signal amplification are essential to meet the requirements.
8–17 DNAzyme, a well-known DNAzyme, owing to its advantages of being more stable and highly catalytic, has been widely employed in signal amplification strategies for sensing a variety of targets such as metal ions,22–25 single nucleotide polymorphisms (SNPs),26 miRNAs,27,28 small molecules,29–31 proteins32 and others.33 Through the introduction of divalent metal ions (such as Pb2+, Mn2+ and Zn2+), it can show strong catalytic ability.34 So far, little research has been reported utilizing 8–17 DNAzyme to construct a DNA walker as a signal amplification strategy. Thus, development of a new DNA walker based on 8–17 DNAzyme is highly desirable for bioanalysis and diagnosis.
Streptavidin is an exocrine protein in the culture of Streptomyces avidinii, which was discovered by Stapley in 1963 when screening antibiotics.35 Its affinity toward biotin is very strong, and the biotin–streptavidin system (BAS) has many advantages, such as high affinity, high sensitivity, strong specificity, good stability, signal amplification and so on.36 It has been widely used in immunology, molecular biology, histochemistry and other fields.37 In this way, it is significant for accurate streptavidin quantification.
Exonuclease I (Exo I), can hydrolyze single stranded DNA from the 3′ terminus to 5′ terminus. In this strategy, we established a DNA walking machine for streptavidin detection to combine dual enzyme assistance and label-free strategies as the signal amplification and output method. The swing arm probe and report probe were pre-assembled on AuNPs. The biotin-modified swing arm probe was easily reacted with streptavidin due to the high affinity between streptavidin and biotin. In view of this, the streptavidin–biotin–DNA swing arm probe was protected from hydrolysis by Exo I; the swing arm probe which contains 8–17 DNAzyme cannot be destroyed and plays a role in the catalytic cleavage of the report probe; the liberating fragment of the report probe which contains a specific sequence (5′-(TTAGGG)4) of G-quadruplex units can combine with berberine and shows intense fluorescence. By multistep cyclic catalysis of each 8–17 DNAzyme, much of the report probe can be cleaved, in order to get an amplified fluorescence signal. To our knowledge, there are very few reports of an unmarked fluorescent probe for the design of DNA walkers in fluorescence sensors. Most of the research groups reported DNA walkers which employed fluorescence modified DNA probes. Even though they may achieve better FL signal values, the background signal may not be satisfactory. By the introduction of an unmarked fluorescent probe in our method, we can achieve a low background signal. In this paper, we have established a label-free fluorescence DNA walker for streptavidin detection based on terminal protection and dual enzyme assisted cleavage induced G-quadruplex/berberine conformation. With this method, we achieved a detection limit of 20 pM. Furthermore, we used our method to detect streptavidin in a complex serum matrix, and the results are satisfactory.
Experimental
Materials and methods
Streptavidin (STA), Exo I (20 units per μL), dithiothreitol (DTT), bovine serum albumin (BSA), fetal calf serum, and thrombin were purchased from Sangon Biotech Co., Ltd (Shanghai, China). Berberine was purchased from Macklin (Shanghai, China). Pepsin and trypsin were obtained from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were obtained from Shanghai Chemical Reagents (Shanghai, China) and used without further purification. Aqua regia was prepared by mixing HCl and HNO3 in the volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. All DNA sequences were synthesized and purified by Sangon Biotechnology Co., Ltd (Shanghai, China), and their sequences are listed in Table S1.† They were dissolved in 10 mM Tris–HCl buffer (10 mM MgCl2, pH 7.4) as stock solutions of 10 μM and stored at 4 °C.
:1. All DNA sequences were synthesized and purified by Sangon Biotechnology Co., Ltd (Shanghai, China), and their sequences are listed in Table S1.† They were dissolved in 10 mM Tris–HCl buffer (10 mM MgCl2, pH 7.4) as stock solutions of 10 μM and stored at 4 °C.
Fluorescence spectra were measured with a Hitachi F-7000 Spectrofluorometer (Tokyo, Japan) with a scan rate of 8000 nm min−1. The excitation wavelength in this experiment was set at 362 nm. Transmission electron microscopy (TEM) images were recorded on a Hitachi 7700 Transmission Electron Microscope. Gel electrophoresis experiments were performed using a DYCZ-24 Electrophoresis Cell (Beijing Liuyi Instrument Factory) equipped with a DYY-12 Electrophoresis Power Supply (Beijing Liuyi Instrument Factory). In the gel electrophoresis assay, 10 μL of the prepared solution was analyzed on a nondenaturing polyacrylamide gel (20%). Electrophoresis was run in 1× TAE (pH 8.3) at 180 V for about 3 h at room temperature. Gel imaging was performed with a Tanon-5500 Gel Imaging System (Shanghai Tanon Science & Technology Co., Ltd, China).
Preparation of 13 nm AuNPs
AuNPs were synthesized following previously reported methods.38 Briefly, 100 mL of 1 mM HAuCl4 was taken in a round bottomed flask and brought to boiling with rapid stirring; then, 10 mL of 38.8 mM trisodium citrate was added quickly to the above solution. The solution was kept boiling and stirred for additional 20 min and then cooled to room temperature with continuous stirring. The prepared AuNPs were stored at 4 °C for further experiments.Preparation of DNA–AuNP assembly
The swing arm probe and report probe were assembled on AuNPs via Au–S chemistry by adding 5 μL of 1 μM swing arm probe and 10 μL of 10 μM report probe in 160 μL of 2 nM AuNP solution. The solution was incubated at room temperature overnight. Then, NaCl was added to this solution in increments of 0.05 M for the first two times and thereafter in increments of 0.1 M for six times. After each addition of NaCl, the solution was sonicated for 60 s followed by incubation for 60 min at room temperature. After incubation for another 8 h, the solution was centrifuged at 14000g for 20 min to separate the AuNPs from the unconjugated DNA. After washing with 10 mM Tris–HCl three times, the precipitate was resuspended in 160 μL of 20 mM Tris–HCl at a final concentration of 2 nM AuNPs and stored at 4 °C until needed.
Measurement of streptavidin detection
In this assay, 60 μL of the reaction sample containing 50 μL of 2 nM DNA-functional AuNPs and 10 μL of different concentrations of streptavidin were incubated at 37 °C for 20 min. Then, 10 μL 10× reaction buffer of Exo I, 2 μL Exo I (20 units per μL) and 28 μL reaction buffer (10 mM Tris-HCl, pH 7.4) were added to the reaction sample and allowed to react for another 30 min at 37 °C. Subsequently, the solution was heated to 80 °C for 20 min in order to inactivate the character of exonuclease I and then cooled to room temperature. After the cleavage by exonuclease I, 10 μL of 1 mM Pb2+, 20 μL of 1 M K+, 20 μL of 80 μM berberine and 50 μL of the buffer solution (10 mM Tris–HCl, pH 7.4) were introduced into the system. After 50 min of reaction, fluorescence values of the mixture were recorded.Results and discussion
DNA walker for streptavidin analysis strategy
The DNA walker for streptavidin detection strategy is illustrated in Fig. 1. The swing arm probe and report probe were pre-assembled on AuNPs. In this study, 8–17 DNAzyme was employed as a signal amplification avenue for the sensing of streptavidin in view of its excellent catalytic activity and thermal stability. Therefore, the 8–17 DNAzyme sequence should be first embedded into the swing arm probe. The report probe which contains a specific sequence (5′-(TTAGGG)4) of G-quadruplex units can combine with berberine and show a fluorescence signal. In the absence of a target, by the catalysis of Exo I, the swing arm probe can be digested into a single mononucleotide from 3′ to 5′ ends. After the berberine was added to this solution, the AuNPs can quench the fluorescence intensity of FRET (fluorescence resonance energy transfer)-based assays due to their extinction coefficient39 and broad energy banwidth.40 In the presence of streptavidin, due to the high affinity between streptavidin and the biotin-modified swing arm probe, Exo I cannot hydrolyze past the swing arm probe from the 3′ terminus to 5′ terminus because of the obstruction of streptavidin, and the swing arm probe could be preserved and play a role in the catalytic cleavage of the report probe upon the introduction of Pb2+ ions. The liberating fragment which contains a specific sequence (5′-(TTAGGG)4) of G-quadruplex units can combine with berberine and shows intense fluorescence because of their further operating distance from the AuNPs, providing an evident enhancement of fluorescence signal. Finally, by multi-step cyclic catalysis of each 8–17 DNAzyme of the swing arm probe, much of the report probe can be cleaved, in order to get an amplified fluorescence value. Thus, we can achieve the concentration of streptavidin effectively.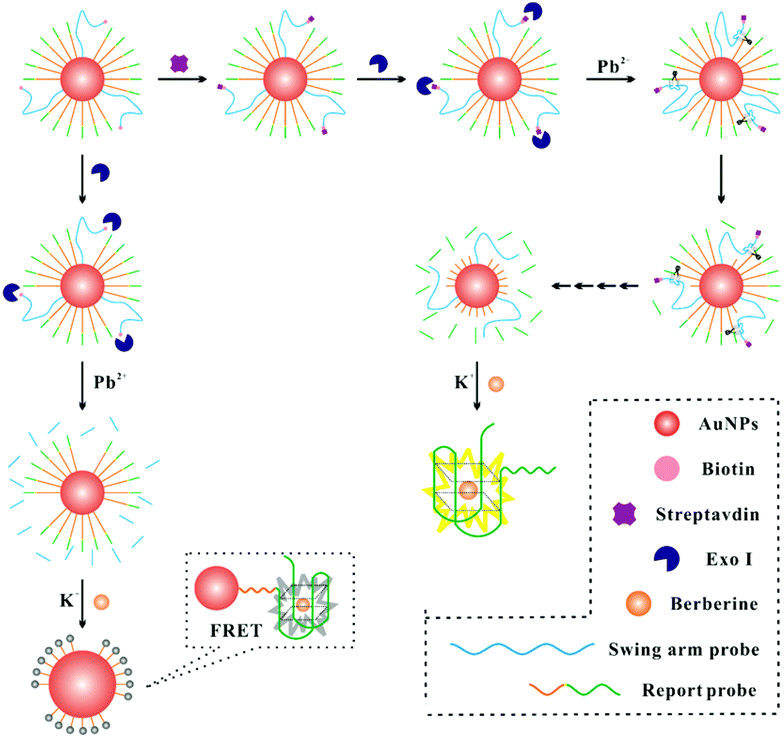 | ||
| Fig. 1 Schematic illustration of the detection of streptavidin. | ||
Determination of the DNA walker properties
To test and verify the feasibility of our strategy in the detection of streptavidin, the movement of the DNA walker should be investigated firstly. TEM images showed good dispersion of the AuNPs with a diameter of 13 nm (Fig. S1A†). On the other hand, the UV-Vis spectrum of AuNP–DNA also demonstrated the successful conjugation of the probe to the gold surface by a characteristic DNA peak at 260 nm (Fig. S1B†). Besides, by monitoring the fluorescence intensity of the DNA walker, we studied the interaction between DNA and AuNPs (Fig. 2A). As shown in Fig. 2B, only in the presence of the report probe and berberine can we observe high fluorescence (curve a). With the addition of the AuNPs, fluorescence signals of both supernatant (curve b) and precipitate (curve c) are greatly diminished. The reduced fluorescence signal of the supernatant can be explained by the fact that most of the report probes were assembled on AuNPs through an Au–S bond interaction. In the case of the precipitate, the excellent quenching of the fluorescence intensity of FRET-based assays by the AuNPs led to a weak fluorescence signal. However, by the addition of 20 mM dithiothreitol (DTT) to the mixture, the report probe assembled on AuNPs can be released. After centrifugation and redispersion, the fluorescence intensity of both supernatant and precipitate was evaluated; the fluorescence signal of the supernatant recovered(curve d), but there was no bright signal for the precipitate (curve e). The fluorescence intensity of the supernatant is similar to that with the addition of Pb2+. Thus, these above experiments confirmed the feasibility of our proposed design of DNA walkers.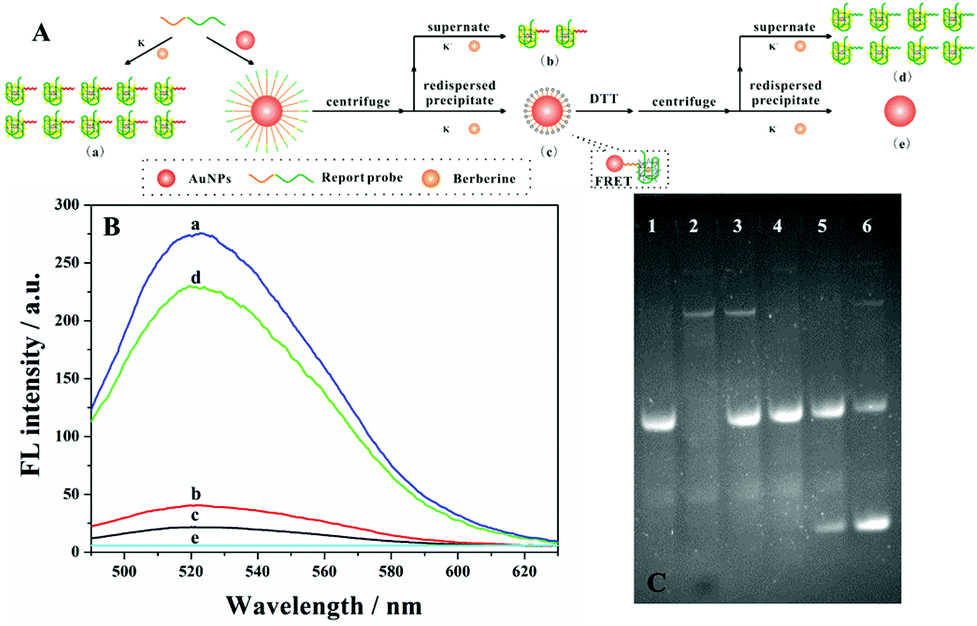 | ||
| Fig. 2 (A) The schematic studies of DNA-functionalized AuNPs. (B) The signal intensity of fluorescence emission spectra corresponding to the modification process under different conditions: a: only report probe; b: fluorescence intensity of the supernatant; c: fluorescence intensity of the re-dispersed precipitate; d: fluorescence intensity of the supernatant after the addition of DTT; e: fluorescence intensity of the precipitate after the addition of DTT. (C) The gel electrophoresis analysis of the terminal protection assay and the cleavage of 8–17 DNAzyme catalysis: lane 1, report probe only; lane 2, swing arm probe only; lane 3, report probe + swing arm probe; lane 4, swing arm probe + Exo I + report probe + Pb2+; lane 5, swing arm probe + 0.05 nM STA + Exo I + report probe + Pb2+; lane 6, swing arm probe + 1 nM STA + Exo I + report probe + Pb2+. | ||
Determination of the DNA walker with PAGE
In order to investigate the movement of the DNA walker, the binding of streptavidin to the bio-DNA probe, Exo I digestion and 8–17 DNAzyme catalysis were determined by gel electrophoresis. As depicted in Fig. 2C, it could be found that, through the hydrolysis of the swing arm by Exo I, there were no bright bands of the swing arm in the mixture (lane 4), which is comparable with the complete swing arm probe (lane 2). However, by introducing different concentrations of the target, a new band appears in lanes 5 and 6, indicating that the report probe was cleaved by 8–17 DNAzyme, and the more streptavidin added, the brighter the bands of the new cleavage product (lane 6).Optimization of assay conditions
For the best reaction properties, the influence of the amount of the report probe and swing arm probe was critical. The different experimental performances of the report probe and the swing arm probe in solution are shown in Fig. S2A†. When the concentration of the target was 1 nM, the molar ratio of swing arm probe to report probe at 1:20, the best sensing performance was achieved. However, increasing the report probe did not lead to effective interaction with AuNPs and the report probe became saturated; the higher background signal was also unsatisfactory. Therefore, the molar ratio of swing arm probe to report probe was chosen as 1:20 in our experiment. Secondly, the reaction time between biotin and streptavidin should also be optimized. As Fig. S2B† shows, with the increase of the reaction times between biotin and streptavidin, the fluorescence signal increased and then reached saturation in 20 min, indicating that the reaction is complete. It can be found that more interaction between streptavidin and biotin might prevent the hydrolysis of Exo I toward the swing arm. After 20 min, there was no obvious change in the fluorescence value. Thus, we chose 20 min as the optimal reaction time between biotin and streptavidin.
On the other hand, the amount and catalytic time of Exo I were key points in our experiments, which also should be optimized. Firstly, in order to achieve the pure low background signal, the amount of Exo I should be investigated in the absence of the target. As depicted in Fig. S2C,† the fluorescence signal was reduced with the increased amount of Exo I. By the addition of 40 U Exo I, the fluorescence signal reached a minimum, indicating complete hydrolysis. Secondly, from Fig. S2D,† it could be observed that the fluorescence value decreased with the increase of catalytic time of Exo I from 0 to 30 min. After 30 min, the signal reached equilibrium, indicating that the swing arm probes without binding with streptavidin were all completely digested. Thus, the optimal amount and catalytic time of Exo I were 40 units and 30 min.
Based on the above reaction conditions, the amount of Pb2+, K+ and berberine and the incubation time between G-quadruplex and berberine are shown in Fig. S3.† As shown in the figure, 50 μM of Pb2+, 100 mM of K+, 8 μM of berberine and 50 min of incubation time were chosen for the best performance of our method.
Detection sensitivity of the proposed biosensor
Fig. 3A depicts the relationship between fluorescence values and different target concentrations under the optimized conditions. It could be found that with the increasing streptavidin concentration from 0.02 nM to 5 nM, the fluorescence signal increases gradually. This is because at the higher amount of streptavidin, more 8–17 DNAzyme of the swing arm probe could be conserved and its catalyzed cleavage toward the report probe triggers a gradual increase in the fluorescence signal. Fig. 3B shows the calibration curve of the proposed method at the fluorescence values of 524 nm. It could be found that, with the target concentration ranging from 0.02 nM to 1 nM, the fluorescence intensity increased linearly. The detection limit of 20 pM was achieved. Even though our detection limit is slightly higher than those in some of the previous reports (Table S2 in the ESI†), with the introduction of an unmarked fluorescence probe, we can achieve the low background signal and our method becomes cost-effective. On the other hand, this advanced strategy provides a good repeatability for the detection of streptavidin. These findings confirm that our sensor is reliable and has prospective applications.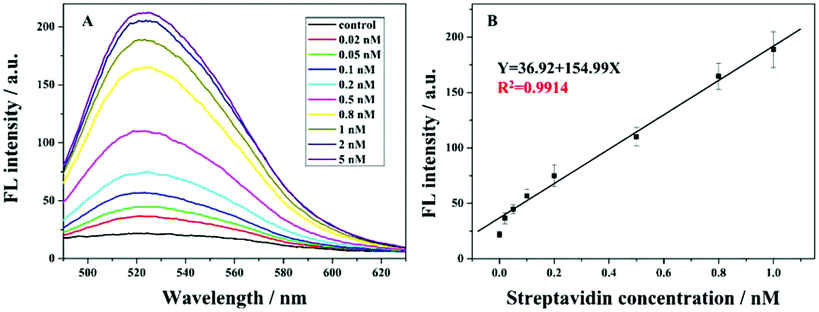 | ||
| Fig. 3 (A) Fluorescence emission spectra of the sensing system when exposed to different concentrations of STA (from bottom to top: 0, 0.02, 0.05, 0.1, 0.2, 0.5, 0.8, 1, 2, 5 nM). (B) The linear relationship between the fluorescence intensity and the concentration of STA from 0.02 to 1 nM. Each data point represents an average of 3 measurements (each error bar indicates the standard deviation). | ||
Selectivity, stability and feasibility in a complex sample
In order to demonstrate the selectivity of our method, irrelevant proteins such as bovine serum albumin (BSA), thrombin, pepsin, and trypsin were employed as the target. As shown in Fig. 4, no bright fluorescence values appeared with the addition of BSA, thrombin, pepsin, and trypsin. This is because these reagents cannot interact with biotin, the swing arm probe might be destroyed by Exo I, and the report probe cannot be cleaved to release. When streptavidin was added, through the high affinity between streptavidin and biotin, in order to prevent from the hydrolysis by Exo I, the 8–17 DNAzyme of swing arm probe can be conserved and show its catalyzed cleavage toward the report probe to induce an evident increase of fluorescence intensity than these of other kinds of protein, indicating the excellent selectivity of our proposed strategy of streptavidin detection.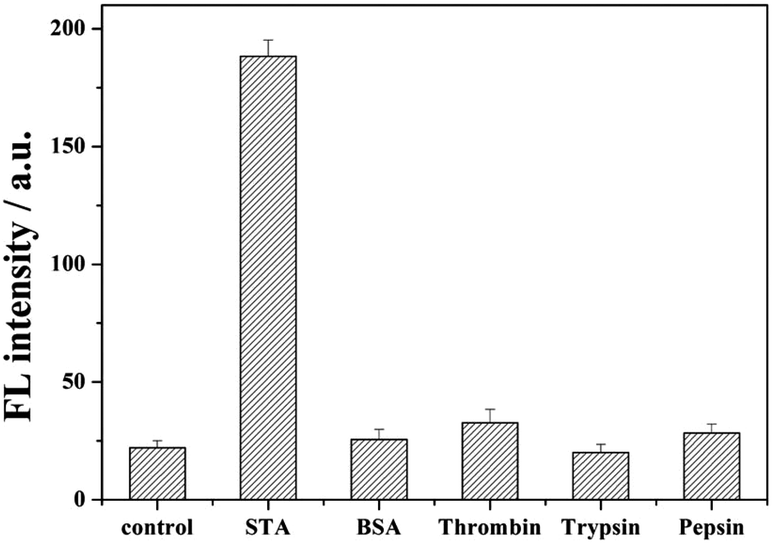 | ||
| Fig. 4 Detection selectivity measured by the fluorescence intensity. The concentration of STA was 1 nM, and the amount of BSA, thrombin, pepsin, and trypsin was also 1 nM. | ||
In order to explore the stability of our method, in this assay, STA with concentrations of 0.5 nM and 1 nM was added and the fluorescence signal was recorded at different times. As shown in Table S3,† the fluorescence intensity was recorded at 1 h, 2 h, 5 h, 8 h after the reaction of the standard sample. The average coefficients of variation were 2.35% and 2.14%, and fluorescence values achieved equilibrium in 8 h. Besides, the reproducibility of our method toward streptavidin is shown in Table S4.† With the addition of 0.2 and 0.8 nM streptavidin by three different experimenters (A, B and C), RSDs of 7.93% and 4.10% were achieved, suggesting that our strategy is stable and satisfactory.
To further demonstrate the feasibility of our method in a complex biological matrix, we also evaluated streptavidin in diluted fetal calf serum. The results of fluorescence responses at different streptavidin concentrations spiked into the real sample, are depicted in Table S5.† The recovery tests of the spiked target were obtained in the range from 87.4% to 95.0%. This shows that this excellent sensor could be applicable for quantitative assays in relatively complex biological samples with a satisfactory accuracy.
Conclusions
In summary, we have established a label-free fluorescence DNA walker for streptavidin detection based on terminal protection and dual enzyme assisted cleavage induced G-quadruplex/berberine conformation. The 8–17 DNAzyme sequence of the swing arm probe and the 8–17 DNAzyme substrate sequence of the report probe were pre-assembled on AuNPs. Under the excellent conditions of quenching the fluorescence intensity by AuNPs to reduce the background signal and 8–17 DNAzyme as signal amplification, sensitive and selective detection of proteins was achieved, with a 20 pM detection limit toward streptavidin. Furthermore, by changing the terminal modified molecule or the sequence of the DNA probe, our method could also be used for the design of biosensors of extensive application in bio-detection and disease diagnosis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ph.D. Foundation of Weifang Medical University.Notes and references
- C. A. Borrebaeck, Nat. Rev. Cancer, 2017, 17, 199–204 CrossRef CAS PubMed.
- S. A. Jayanthi, A. B. Das and U. Saxena, Biosens. Bioelectron., 2017, 91, 15–23 CrossRef PubMed.
- S. B. Nimse, M. D. Sonawane, K. S. Song and T. Kim, Analyst, 2016, 141, 740–755 RSC.
- S. V. Gupta, R. M. McGowen, D. M. Callewaert, T. R. Brown, Y. Li and F. H. Sarkar, J. Immunoassay Immunochem., 2005, 26, 125–143 CrossRef CAS.
- A. T. Hashem, D. G. Binger, J. J. Blackwood, C. Ying, R. S. Creager, D. S. Renuka, R. A. Eickholt, J. E. Gaibor, R. S. Handley and K. P. Kapsner, J. Am. Chem. Soc., 2013, 135, 4191–4194 CrossRef PubMed.
- Y. Fang, Y. Li, M. Zhang, B. Cui, Q. Hu and L. Wang, Analyst, 2019, 144, 2186–2194 RSC.
- G. C. Terstappen, C. Schlupen, R. Raggiaschi and G. Gaviraghi, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS.
- J. Hahn, E. Kim, Y. You and Y. J. Choi, Analyst, 2019, 144, 4439–4446 RSC.
- J. Masson, ACS Sens., 2016, 2, 16–30 CrossRef.
- L. Guo, Y. Shi, X. Liu, Z. Han, Z. Zhao, Y. Chen, W. Xie and X. Li, Biosens. Bioelectron., 2018, 99, 368–374 CrossRef CAS PubMed.
- Y. Tan, Q. Guo, X. Zhao, X. Yang, K. Wang, J. Huang and Y. Zhou, Biosens. Bioelectron., 2014, 51, 255–260 CrossRef CAS PubMed.
- X. Chen, C. Lin, Y. Chen, F. Luo, Y. Wang and X. Chen, Biosens. Bioelectron., 2016, 83, 97–101 CrossRef CAS PubMed.
- J. Sun, X. Zhang, T. Li, J. Xie, B. Shao, D. Xue, X. Tang, H. Li and Y. Liu, Anal. Chem., 2019, 91, 6454–6461 CAS.
- E. K. Drexler, Comput. Stand. Inter., 1993, 15, 319–320 Search PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- W. B. Sherman and N. C. Seeman, Nano Lett., 2004, 4, 1203–1207 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS.
- H. Peng, X. F. Li, H. Zhang and X. C. Le, Nat. Commun., 2017, 8, 14378 CrossRef.
- J. Huang, L. Zhu, H. Ju and J. Lei, Anal. Chem., 2019, 91, 6981–6985 CrossRef CAS.
- C. Feng, Z. Wang, T. Chen, X. Chen, D. Mao, J. Zhao and G. Li, Anal. Chem., 2018, 90, 2810–2815 CrossRef CAS.
- X. Qu, D. Zhu, G. Yao, S. Su, J. Chao, H. Liu, X. Zuo, L. Wang, J. Shi, L. Wang, W. Huang, H. Pei and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS.
- J. Ming, W. Fan, T. Jiang, Y. Wang and Z. Lv, Sens. Actuators, B, 2017, 240, 1091–1098 CrossRef CAS.
- F. Wang, R. Orbach and I. Willner, Chem. – Eur. J., 2012, 18, 16030–16036 CrossRef CAS.
- X. B. Zhang, Z. Wang, H. Xing, Y. Xiang and Y. Lu, Anal. Chem., 2010, 82, 5005–5011 CrossRef CAS.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062–5066 CrossRef CAS.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed.
- T. Tian, H. Xiao, Z. Zhang, Y. Long, S. Peng, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem. – Eur. J., 2013, 19, 92–95 CrossRef CAS.
- Y. Wen, Y. Xu, X. Mao, Y. Wei, H. Song, N. Chen, Q. Huang, C. Fan and D. Li, Anal. Chem., 2012, 84, 7664–7669 CrossRef CAS.
- F. Liu, J. Zhang, R. Chen, L. Chen and L. Deng, Chem. Biodivers., 2011, 8, 311–316 CrossRef CAS.
- P. Song, Y. Xiang, H. Xing, Z. Zhou, A. Tong and Y. Lu, Anal. Chem., 2012, 84, 2916–2922 CrossRef CAS.
- F. Wang, L. Freage, R. Orbach and I. Willner, Anal. Chem., 2013, 85, 8196–8203 CrossRef CAS.
- J. Ming, T. Jiang, Y. Wang and Z. Lv, Sens. Actuators, B, 2017, 252, 450–457 CrossRef CAS.
- T. Tian, H. Xiao, Y. Long, X. Zhang, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem. Commun., 2012, 48, 10031–10033 RSC.
- H. Kim, J. Liu, J. Li, N. Nagraj, M. Li, C. M. B. Pavot and Y. Lu, J. Am. Chem. Soc., 2007, 129, 6896–6902 CrossRef CAS.
- E. O. Stapley, J. M. Mata, I. M. Miller, T. C. Demny and H. B. Woodruff, Antimicrob. Agents Chemother., 1963, 161, 20–27 CAS.
- J. H. Kim, C. S. Lee and B. G. Kim, Biochem. Biophys. Res. Commun., 2005, 331, 210–214 CrossRef CAS PubMed.
- V. Peters and B. H. Rehm, J. Biotechnol., 2008, 134, 266–274 CrossRef CAS PubMed.
- K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735–743 CrossRef CAS.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4589 CrossRef CAS.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an01853k |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2020 |