
Metal salt assisted electrospray ionization mass spectrometry for the soft ionization of GAP polymers in negative ion mode†
Theoneste
Muyizere‡
a,
Yajun
Zheng‡
a,
Hongni
Liu‡
b,
Jia
Zhao
a,
Jin
Li
a,
Xianming
Lu
b,
Daniel E.
Austin
c and
Zhiping
Zhang
 *a
*a
aSchool of Chemistry and Chemical Engineering, Xi'an Shiyou University, Xi'an 710065, China. E-mail: zhangzp0304@gmail.com; Fax: +8629 8838 2693; Tel: +86 29 8838 2694
bXi'an Modern Chemistry Research Institute, Xi'an 710065, China
cDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA
First published on 30th October 2019
Abstract
Glycidyl azide polymers (GAP) are one of the most important energetic polymers, but it is still a challenge to elucidate their structures using mass spectrometry due to their fragility upon ionization. Herein we developed a soft metal salt assisted electrospray ionization (MSAESI) to characterize directly GAP polymers using mass spectrometry. This technique combines paper spray ionization and the complexing effect of anions from metal salts with GAP in the negative ion mode to softly ionize GAP polymers prior to mass spectrometry analysis. The effects of experimental parameters (e.g., ion mode, applied voltage, and type and concentration of metal salts) have been investigated in detail. In contrast to the positive ion mode, a softer ionization was observed for GAP polymers when the negative ion mode was applied. The radius and average charge of cations and anions in metal salts were found to play crucial roles in determining the performance of the MSAESI analysis of GAP. For a given charge number, a smaller radius of cations favored the soft ionization of GAP polymers (e.g., Na+ > K+ > Rb+), whereas a larger radius of anions led to a preferred performance (e.g., F− < Cl− < Br− < I−) due to variation in dissolution ability. For anions with multiple charges, the ones with fewer charges gave a more favorable ionization to the GAP sample because of their better complexing to GAP molecules than those with more charges in the structure of anions (e.g., NO3− > SO42− > PO43−). According to the experimental observation and evidence from mass spectrometry, we proposed the plausible electrospray mechanisms of MSAESI for GAP analysis with the involvement of metal salts. Moreover, the developed protocol has been applied successfully to the analysis of various GAP samples, and works for other types of sources such as nanoelectrospray ionization.
Introduction
Numerous energetic polymers, including poly(glycidyl nitrate), poly(3-azidomethyl-3-methyl oxetane), poly(3,3-bis-azidomethyl oxetane) and others,1,2 have been demonstrated to be suitable for use as binders in advanced solid propellants, gun propellants, and explosives.3,4 Energetic polymers generally contain nitro, nitrato, and azido groups and release high energy during combustion. Among various energetic polymeric binders, glycidyl azide polymers (GAP, shown in Fig. 1) are considered promising candidates for propellant binders owing to their low glass transition temperature (−45 °C), low detonation tendency, high positive heat of formation (957 J g−1), and good compatibility with high energy oxidizers.5 GAP was reported originally by Vandenburg in 1972 through the reaction of sodium azide in dimethylformamide with polyepichlorohydrin.6 Subsequent studies have been carried out on its synthesis, thermal behavior, physicochemical characteristics, and explosive properties.3,7–11 Its structure, thermal behavior and decomposition kinetics have also been extensively investigated by means of different techniques, including infrared (IR) spectroscopy,12,13 Raman spectroscopy,12,14 planar laser-induced fluorescence,14 ultraviolet/visible (UV-vis) absorption spectroscopy,14 thermal gravimetric analysis,15 fast neutron activation analysis,16 colorimetric method,17 gel permeation chromatography (GPC),18 nuclear magnetic resonance (NMR)19 and others.4 Despite the progress, there is still a high demand for developing novel methods to map out the structures of GAP polymers.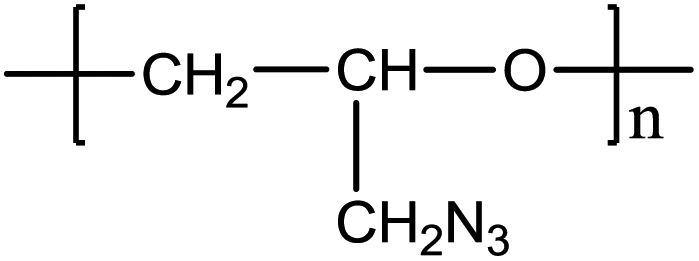 | ||
| Fig. 1 Structure of the glycidyl azide polymer (GAP). | ||
Mass spectrometry, including secondary ion mass spectrometry (SIMS),20 matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS),21 and ambient ionization mass spectrometry,22–24 is a powerful tool for the characterization of polymers due to its high sensitivity, specificity and selectivity. These techniques have been widely applied in polymer analysis and extensively reviewed.25–28 Some researchers have also employed MS for the characterization of thermal decomposition reactions and products of GAP polymers. For example, Farber et al.29 studied the thermal decomposition behaviors of GAP and three other azido polymers with the aid of effusion mass spectrometry to determine their primary and secondary decomposition mechanisms, activation energies, and products of decomposition. The decomposition mechanism of GAP was proposed with the initial elimination of molecular nitrogen from the azide functional group. Secondary decomposition at higher temperatures involved the rupture of the carbon backbone into smaller fragments, consistent with other reports.8,29,30 Tang and co-workers11 analyzed the species products from the thermal decomposition of cured GAP with heat fluxes of 50, 100, and 200 W cm−1 delivered by a CO2 laser through a micro-probe/mass spectrometry system. Kuibida and colleagues31 employed molecular beam mass spectrometry to study the flame structure of composite propellants and sandwiches based on ammonium dinitramide and GAP at pressures ranging from 0.015 to 0.3 MPa. Wang et al.9 explored the thermal decomposition reactions and products of GAP at low pressure by tunable synchrotron vacuum ultraviolet photoionization and molecular beam sampling mass spectrometry. Compared with previous studies on thermal decomposition, several free radicals at the initial degradation of GAP were identified, and the formation mechanisms of important radicals are discussed. These studies are of significance to help understand the energy releasing mechanism of GAP thermal decomposition. However, limited information is available regarding various aspects of GAP based compositions. Detailed studies still need to be carried out to exploit fully the tremendous potential of GAP based solid propellants.
As an energetic polymer, GAP is labile upon shock or ionization in mass spectrometry analysis. Due to its instability, there are no reports on directly characterizing the component of GAP using MS to date, which makes it a challenge to analyze rapidly the structure of GAP polymers. To address this issue, herein we report a novel metal salt assisted electrospray ionization-mass spectrometry (MSAESI-MS) to characterize various GAP polymers. This technique combines the ionization of paper spray32–36 and the complexing interaction between GAP molecules and anions in a metal salt to promote the soft ionization of GAP polymers in the negative ionization mode. The effects of experimental parameters (e.g., ion mode, applied voltage, and type and concentration of metal salts) have been investigated in detail. Based on the experimental observation and evidence from mass spectrometry, the ionization mechanism of MSAESI-MS is elucidated. Furthermore, the strategy thus developed has been applied to the analysis of various GAP samples, and has been shown to be suitable to other types of ionization sources such as nanoelectrospray.
Experimental section
Materials
All the necessary chemicals were from commercially available sources. Sodium nitrate (NaNO3) and sodium sulfate (Na2SO4) were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd (Tianjin, China); sodium phosphate (Na3PO4·12H2O), sodium fluoride (NaF), sodium chloride (NaCl), and sodium bromide (NaBr) were from Guangdong Guanghua Technology Co., Ltd (Shantou, China), Tianjin Tianli Chemical Reagents Factory (Tianjin, China), Tianji Guangfu Technology Co., Ltd (Tianjin, China), and Tianjin Baishi Chemical Co., Ltd (Tianjin, China), respectively. Methanol was purchased from Beijing J&K Scientific Ltd (Beijing, China). Filter paper was purchased from Hangzhou Special Paper Co. (Fuyang, China). The GAP samples were prepared following two steps, namely the preparation of polyepichlorohydrin (PECH) and GAP. The detailed procedure was the same as that in the previous report.37Preparation of the GAP sample solution
0.5 μL of GAP samples (e.g., GAP #1, #2, #3, and #4) were first quantitatively transferred into 0.5 mL of dichloromethane and then diluted with 0.5 mL of methanol to make a GAP solution with a concentration of 500 μg mL−1. Afterwards, exactly 200 μL of the above GAP solution and 1 μL of 1 mol L−1 NaNO3 aqueous solution were transferred into 799 μL of methanol, and the final concentration of GAP solution was 100 μg mL−1 containing 1 mmol L−1 NaNO3.MS analysis
All experiments on MSAESI-MS were performed with a TSQ Quantum Access Max mass spectrometer (Thermo Fisher Scientific, San Jose, CA). For paper spray, filter paper was directly cut into a triangle (around 13 mm height and 9 mm base width). For nanoESI analysis, the capillary with a tip orifice of 1.0 μm was pulled from borosilicate glass capillaries with filament (Sutter Instrument, USA, 1.5 mm o.d., 0.86 mm i.d., 10 cm length) using a micropipette puller (Model P-97, Sutter Instrument Co., Novato. CA, USA). The distance between the tip of the paper triangle/nanoESI capillary and the MS inlet capillary was about 15 mm. Mass spectra were recorded in the positive and negative ion modes with a capillary temperature of 270 °C.The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (PDI) were calculated according to eqn (1), (2), and (3),25 respectively.
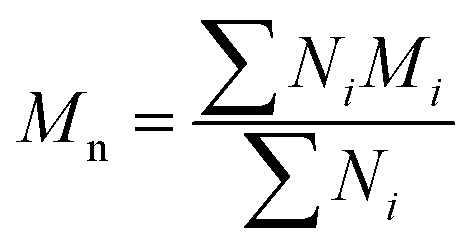 | (1) |
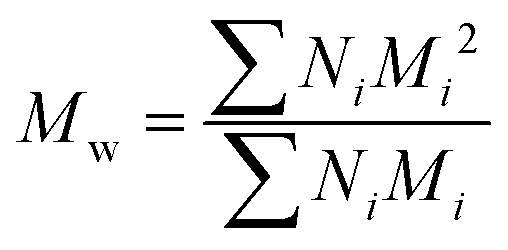 | (2) |
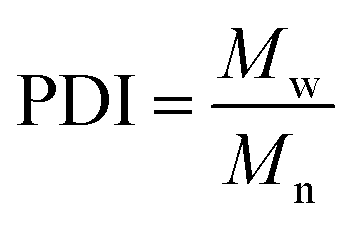 | (3) |
Results and discussion
Development of MSAESI-MS
Fig. 2a shows the procedure to analyze GAP samples using MSAESI-MS. This technique consists of two simple steps, namely the preparation of a GAP solution containing 1.0 mmol L−1 sodium nitrate (NaNO3) and the direct use of the resulting GAP solution as the spray solvent for paper spray ionization. In the process, the applied ion mode and choice of metal salt (such as NaNO3) played crucial roles in determining the performance of the paper spray with GAP samples. As the positive ion mode was applied to analyze GAP solution #1, the addition of NaNO3 had little effect on the analysis. Fig. 2b shows the mass spectrum of GAP sample #1 when no NaNO3 was involved in the system. It is obvious that four series of peaks resulting from the GAP solution are present in the spectrum, namely series i (m/z 504, 603, 702, 801, 900, 999, 1098, 1197, 1296, and 1395), series ii (m/z 432, 531, 630, 729, 828, 927, 1026, 1125, and 1224), series iii (m/z 460, 559, 658, and 757), and series iv (m/z 757, 856, and 955). The mass difference between two adjacent peaks was 99, attributable to the basic unit –CH2–CH(CH2N3)–O– for constructing GAP. Among them, series i and ii demonstrated more abundant peaks compared with others, and the number-average molecular weight (Mn) and weight-average molecular weight (Mw) for both series were 688.7 and 721.6, and 605.4 and 618.5, respectively. As NaNO3 was included in the system, the dominant peaks shifted somewhat to higher mass-to-charge ratios (Fig. 1c). Similar to the system without NaNO3 (Fig. 1b), four series of peaks also were observed in the mass spectra, namely series I (m/z 509, 608, 707, 806, 905, 1004, 1103, 1202, and 1301), series II (m/z 437, 536, 635, 734, 833, 932, and 1031), series III (m/z 465, 564, 663, and 762), and series IV (m/z 779, 878, 977, 1076, 1175, and 1274). After comparing the mass-to-charge values in series I–IV with those in series i–iv, it was interesting to find that the mass difference between series i and I, series ii and II, and series iii and III was 5 Da, and the mass difference between series iv and IV was 22 Da (Table 1). This 5 Da value could be attributed to the typical mass shift between [M + Na]+ and [M + NH4]+ adducts. In the current experiment, NH4+ was not included in the sample, and its occurrence in the current study presumably resulted from residual cations in the electrospray source from previous experiments (i.e., a carryover effect).38,39 The 22 Da value was due to a typical H/Na exchange between [M + H]+ and [M + Na]+ adducts.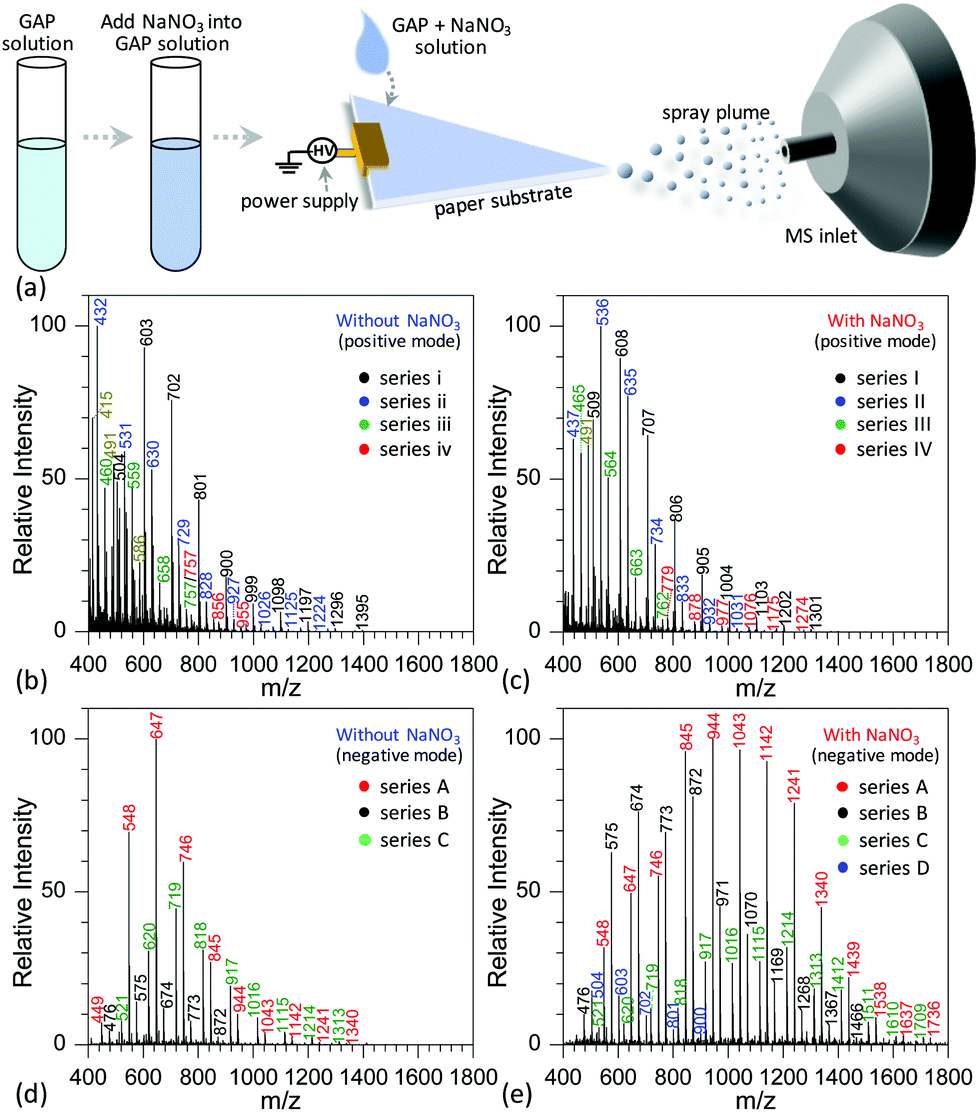 | ||
| Fig. 2 (a) Schematic representation of the procedure for analyzing GAP samples using MSAESI-MS; mass spectra of GAP solution #1 (b) without and (c) with NaNO3 in positive mode (+3.5 kV of applied voltage); mass spectra of GAP solution #1 (d) without and (e) with NaNO3 in negative mode (applied voltage: −3.5 kV). | ||
| No. | NaNO3 or not | Series | Composition | Observed peaks | M n | M w | PDI | |
|---|---|---|---|---|---|---|---|---|
| “P-without” means “positive ion mode and without NaNO3”, “P-with” means “positive ion mode and with NaNO3”, “N-without” means “negative ion mode and without NaNO3”, “N-with” means “negative ion mode and with NaNO3”, and “n/a” means no related signals. | ||||||||
| #1 | P-without | i | [M1 + NH4]+ | m/z | 504, 603, 702, 801, 900, 999, 1098, 1197, 1296, 1395 | 688.7 | 721.6 | 1.048 |
| P-with | I | [M1 + Na]+ | m/z | 509, 608, 707, 806, 905, 1004, 1103, 1202, 1301 | 678.0 | 712.9 | 1.051 | |
| basic unit | M1 | 486, 585, 684, 783, 882, 981, 1080, 1179, 1278, 1377 | ||||||
| N-without | A | [M1 + NO3]− | m/z | 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340 | 677.3 | 700.9 | 1.035 | |
| N-with | A | [M1 + NO3]− | m/z | 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538, 1637, 1736 | 1001.2 | 1057.8 | 1.056 | |
| #2 | P-without | ii | [M2 + NH4]+ | m/z | 432, 531, 630, 729, 828, 927, 1026, 1125, 1224 | 605.4 | 618.5 | 1.022 |
| P-with | II | [M2 + Na]+ | m/z | 437, 536, 635, 734, 833, 932, 1031 | 616.6 | 630.8 | 1.023 | |
| basic unit | M2 | 414, 513, 612, 711, 810, 909, 1008, 1107, 1206 | ||||||
| N-without | B | [M2 + NO3]− | m/z | 476, 575, 674, 773, 872 | 647.1 | 664.5 | 1.027 | |
| N-with | B | [M2 + NO3]− | m/z | 476, 575, 674, 773, 872, 971, 1070, 1169, 1268, 1367, 1466 | 827.2 | 877.1 | 1.060 | |
| #3 | P-without | iii | [M3 + NH4]+ | m/z | 460, 559, 658, 757 | 545.5 | 559.4 | 1.025 |
| P-with | III | [M3 + Na]+ | m/z | 465, 564, 663, 762 | 539.9 | 551.7 | 1.022 | |
| basic unit | M3 | 442, 541, 640, 739 | ||||||
| N-without | D | [M3 + NO3]− | m/z | n/a | ||||
| N-with | D | [M3 + NO3]− | m/z | 504, 603, 702, 801, 900 | 624.6 | 644.7 | 1.032 | |
| #4 | P-without | iv | [M4 + H]+ | m/z | 757, 856, 955 | 821.4 | 830.1 | 1.011 |
| P-with | IV | [M4 + Na]+ | m/z | 779, 878, 977, 1076, 1175, 1274 | 928.3 | 953.0 | 1.027 | |
| basic unit | M4 | 756, 855, 954, 1053, 1152, 1251 | ||||||
| N-without | C | [M4 + NO3]− | m/z | 521, 620, 719, 818, 917, 1016, 1115, 1214, 1313 | 773.4 | 803.6 | 1.039 | |
| N-with | C | [M4 + NO3]− | m/z | 521, 620, 719, 818, 917, 1016, 1115, 1214, 1313, 1412, 1511, 1610, 1709 | 1090.0 | 1146.0 | 1.051 | |
When the negative ion mode was used for the analysis of GAP #1, the introduced NaNO3 played a crucial role. As shown in Fig. 2d, when there was no NaNO3 in the system, three series of peaks resulting from GAP were observed in the spectra, namely series A (m/z 449, 548, 647, 746, 845, 944, 1043, 1142, 1241, and 1340), series B (m/z 476, 575, 674, 773, and 872), and series C (m/z 521, 620, 719, 818, 917, 1016, 1115, 1214, and 1313), in which the dominant peak was at m/z 647. After NaNO3 was introduced to GAP solution #1, the above peaks shifted significantly to higher mass-to-charge ratios (Fig. 2e). For example, the peaks from series A turned into m/z 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538, 1637 and 1736, and those from series B turned into m/z 476, 575, 674, 773, 872, 971, 1070, 1169, 1268, 1367, and 1466. By comparing both systems with and without NaNO3, the corresponding Mn values increased from 677.3 to 1001.2 for series A, and 647.1 to 827.2 for series B. These results indicated that the addition of NaNO3 did not change the pattern of obtained peaks from GAP #1, but greatly shifted the peaks to higher mass-to-charge ratios, effectively yielding a soft ionization of this GAP sample. This also suggests that the involvement of NaNO3 in GAP solution #1 is an effective strategy to prevent the structure of GAP #1 from rupture in the negative ion mode.
To determine the reasons for the higher mass peaks and softer ionization of GAP oligomers in the negative ion mode, the first step is to identify the nature of the observed species. For mass spectrometry coupled to a paper spray or electrospray ionization source, the molecular ion peak with a composition of [M + H]+ generally occurs in the positive ion mode, and that with [M − H]− exists in the negative ion mode. In both ion modes, the basic components of GAP oligomers should be kept constant. Bearing this in mind, the molecular weights of GAP oligomers in different series were calculated according to the related mass-to-charge ratios in the positive ion mode by combining the compositions of [M + Na]+, [M + NH4]+ or [M + H]+ as listed in Table 1. For series i and I (#1) in Fig. 2b and c, the corresponding molecular weights (M1) were 486, 585, 684, 783, 882, 981, 1080, 1179, 1278, and 1377. After comparing this series of data with those in series A (m/z 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, and 1439) as shown in Fig. 2c and d, a 62 Da difference was observed for both series, which was just the molecular weight of the NO3−. For other series of data, a similar phenomenon was observed, which indicates that NO3− played a crucial role in determining the soft ionization of GAP oligomers by the formation of [GAP + NO3]− ions. It should be pointed out here that the observation of [GAP + NO3]− ions in the negative ion mode without the presence of NaNO3 was probably due to the interaction between GAP molecules and residual NO3− ions resulting from the carryover effect in the previous experiments.38,39
According to the literature, numerous cations (i.e., Li+, Na+, K+, Rb+, Cs+, Ag+, Mg2+, and Ca2+) have been widely applied to the analysis of various polymers by the formation of [M + cation]n+ adducts,40–49 whereas no attempts have been made to use anions to induce the soft ionization of polymer oligomers although some [M + anion]n− adducts (anion = F−, Cl−, Br−, and NO3−)50–54 have been observed in measurement of explosives, cucurbit[n]urils, poly(ethylene glycol), polyether, poly(styrene), poly(2-vinylpyridine), and poly(acrylamide). This to a certain degree suggests that the current study is the first report to improve the performance of mass spectrometry for polymer analysis using anions such as NO3−.
Determining factors on the GAP analysis
By examining the influence of experimental conditions on the performance of MSAESI-MS for GAP analysis, the results demonstrated that the ionization efficiency varied with the applied voltage, NaNO3 concentration, and type of involved metal salt. Fig. 3 shows the effect of spray voltage on the analysis of 100 μg mL−1 GAP #1 with the addition of 1.0 mmol L−1 NaNO3. With variation in the applied voltage, four series of peaks resulting from GAP are present in the spectrum, namely series A, B, C and D. Owing to the more intensive peaks in series A and B than those in series C and D, only the former were considered in the current study, namely series A ([M1 + NO3]−, m/z 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538, and 1637), and series B ([M2 + NO3]−, m/z 575, 674, 773, 872, 971, 1070, 1069, 1268, and 1367). It is apparent that with variation in the applied voltage, the absolute ion intensity gradually decreased from 1.2 × 105 to 4.2 × 104 (Fig. 3a–d), and the peak intensity of series B decreased more than others although the relative ion intensity seemed to preserve. As shown in Fig. 3a, when the applied voltage was −2.5 kV, the most dominant peak focused on m/z 773 in series B. As the voltage was more than −3.0 kV, (Fig. 3b–d), the peaks from the GAP solution shifted greatly to higher mass-to-charge ratios compared with those from −2.5 kV, and the leading peaks were centered around m/z 944 from series A. Furthermore, with the increase in the applied voltage from −3.0 to −4.0 kV, the peaks in series A and B presented a steady separation trend. For example, when the voltage was −3.0 kV, the most dominant peaks in series A and B overlapped together (Fig. 3b). When it was −4.0 kV, a separation occurred on both series, and the most intensive peaks in series A and B could be obviously seen (Fig. 3d). In our opinion, this phenomenon could be attributed to the fact that upon the addition of NaNO3 in GAP solution, soft ionization occurred due to the formation of [GAP + NO3]−, and the major peaks from GAP shifted to higher masses. However, with the increase in the applied voltage up to −4.0 kV, the strength of the present electric field should gradually increase, and some labile units were more inclined to be lost from the structures of GAP oligomers, which resulted in the formation of species with a lower molecular weight and the shift of the peaks such as in series B, to lower mass-to-charge ratios. Due to the mass difference of the resulting structures, a separation between series A and B occurred in the mass spectrum. In addition, by increasing the voltage from −3.0 to −4.0 kV, for series A and B, the Mn and Mw all illustrated a very tiny increasing trend as listed in Table S1.† For example, for series A, their corresponding values increased from 949.1 to 990.6, and 989.2 to 1040.6, respectively. These results suggest that when the voltage changed from −2.5 to −3.0 kV, it had a pronounced effect on the ionization performance of [GAP + NO3]− adducts, whereas little influence was observed by further increasing the voltage up to −4.0 kV.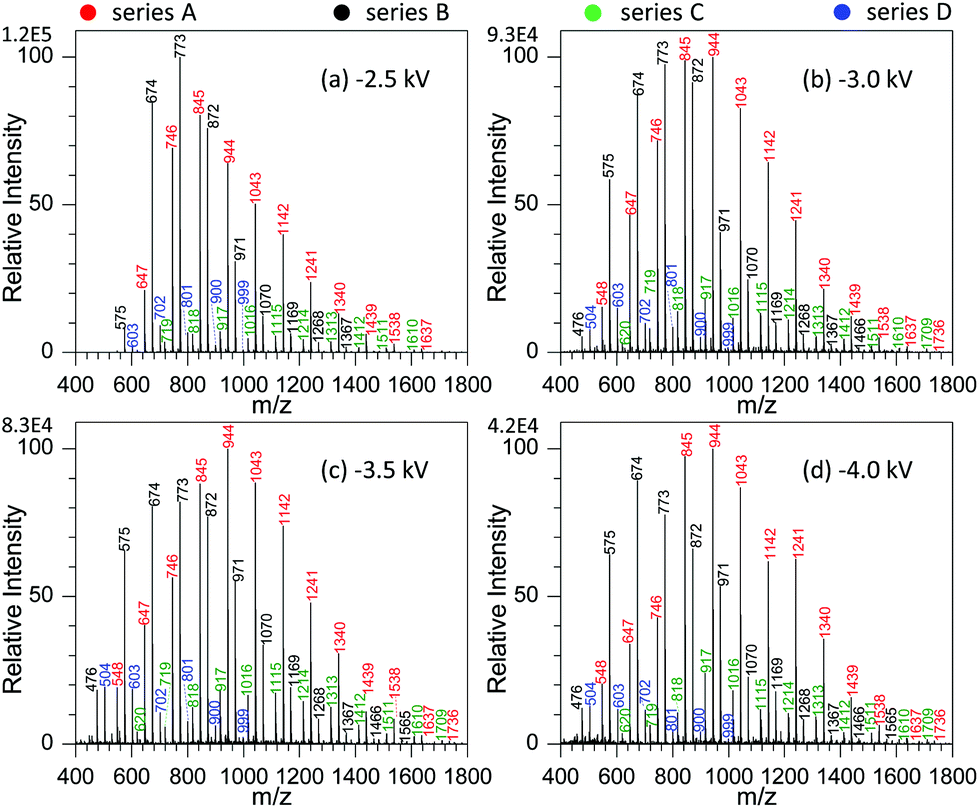 | ||
| Fig. 3 Effect of spray voltage on the analysis of 100 μg mL−1 GAP #1: (a) −2.5 kV, (b) −3.0 kV, (c) −3.5 kV, and (d) 4.0 kV (sample volume: 25 μL for each spray; NaNO3 concentration: 0.5 mmol L−1). | ||
Fig. 4 shows the effect of NaNO3 concentration on the analysis of GAP #1. Apparently, the concentration of NaNO3 in the GAP solution has a pronounced effect on the analysis. When no NaNO3 was included in the spray solution (Fig. 4a), the dominant peaks focused on the one at m/z 647, and three series of peaks appeared in the mass spectrum, namely series A (m/z 548, 647, 746, 845, 944, 1043, 1142, and 1241), series B (m/z 575, 674, 773, and 872), and series C (m/z 620, 719, 818, 917, 1016, 1115, 1214, 1313, and 1412). Among them, the abundance of the peaks in series A overwhelmed those in series B and C, and the peaks in series B demonstrated the least intensity. With the addition of 0.1 mmol L−1 NaNO3 into the sample solution (Fig. 4b), the peaks centered on the one at m/z 674 in series B. In contrast to the mass spectrum from the system without NaNO3 (Fig. 4a), the peak intensity in different series was very different. For example, series B overwhelmed the others, while series C became relatively weaker. In addition, a new series of peaks occurred in the mass spectrum, namely series D (m/z 603, 702, 801, and 900). By increasing the concentration of NaNO3 to 0.5 mmol L−1, series A and B showed an increased abundance and a shift in the mass spectrum to higher mass-to-charge ratios (Fig. 4c). Compared with the system with the concentration of 0.1 mmol L−1, the Mn and Mw values for series A increased from 804.8 to 1010.0 and from 854.0 to 1053.6, respectively, and for series B from 732.4 to 836.9 and 756.0 to 874.4 as listed in Table S2.† Further increasing the concentration of NaNO3 to the range of 1.0–2.0 mmol L−1 (Fig. 4d–f) made the intensity of the peaks from GAP #1 comparable to that from the concentration of 0.5 mmol L−1, but the peaks in series A and B demonstrated a separation trend. The higher the concentration of NaNO3 in GAP solution #1, the less the overlap between the dominant peaks of series A and B. When the concentration of NaNO3 was 3.0 mmol L−1, the peak at m/z 674 in series A became the most dominant one in the mass spectrum (Fig. 4g), and the peaks in series A became weak as a whole. By varying the concentration of NaNO3 from 0.1 to 3.0 mmol L−1, it is noticed that the absolute abundance of ions demonstrated initially an increasing trend followed by a decreasing one (Fig. 4b–g). When the concentration was 0.5 mmol L−1, it was the most intense, and further increasing the concentration led to a gradually decreasing trend, presumably owing to the suppressing effect of excessive metal salts. This was further evidenced by the spectrum with a concentration of NaNO3 as high as 5.0 mmol L−1 (Fig. 4f), which was very similar to that without the addition of NaNO3 (Fig. 4a). The difference between them lies in that the leading peak for the former was at m/z 674 rather than at m/z 647. From these results, it was apparent that the concentration of NaNO3 in GAP solution #1 was a crucial parameter to influence the ionization of GAP #1 during mass spectrometry analysis. When its concentration was less than 0.1 mmol L−1 or higher than 5.0 mmol L−1, the included NaNO3 did not favor the soft ionization of GAP oligomers, and the peaks centered on that at m/z 674 in the mass spectrum. However, when the level of NaNO3 was in the range of 0.1–5.0 mmol L−1, the metal salt promoted the peaks of GAP oligomers shifting to higher mass-to-charge ratios, representing the softer ionization of GAP molecules, and also facilitated the enhancement in the peak intensity at high mass-to-charge ratios.
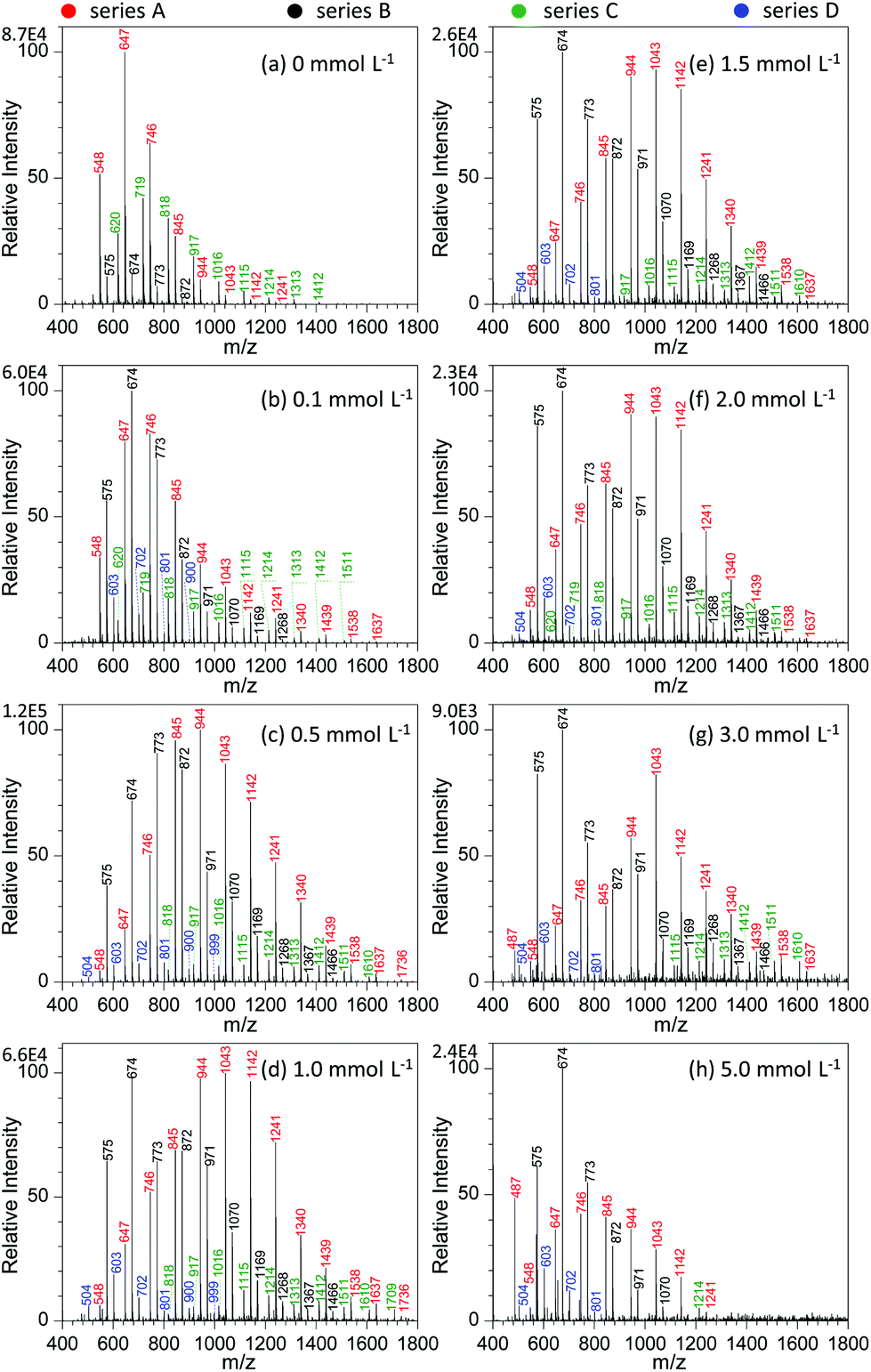 | ||
| Fig. 4 Effect of the concentration of NaNO3 on the analysis of 100 μg mL−1 GAP #1: (a) 0 mmol L−1, (b) 0.1 mmol L−1, (c) 0.5 mmol L−1, (d) 1.0 mmol L−1, (e) 1.5 mmol L−1, (f) 2.0 mmol L−1, (g) 3.0 mmol L−1, and (h) 5.0 mmol L−1 (sample volume: 25 μL for each spray; applied voltage: −3.5 kV). | ||
We also explored the effect of different types of metal salts on the analysis of GAP #1 using MSAESI-MS. To exclude the influence of anions, we selected various metal nitrates, and their concentrations were all 1.0 mmol L−1. The studied metal salts included monovalent salts [e.g., NaNO3 and AgNO3], divalent salts [e.g., Mg(NO3)2, Co(NO3)2, Ni(NO3)2, Cu(NO3)2, Zn(NO3)2, Ba(NO3)2 and Pb(NO3)2], and trivalent salts [e.g., Cr(NO3)3, Al(NO3)3 and Fe(NO3)3]. As shown in Fig. S1,† with variation in the type of involved metal salts, the patterns of the obtained mass spectra were comparable (Table S3†), indicating that the involved metal ions had little effect on the analysis of GAP #1 using the MSAESI-MS method. However, careful observation found that for monovalent and divalent salts (Fig. S1a–h†), the dominant peaks mainly focused on the one at m/z 944 in series A except for Ni(NO3)2 (Fig. S1d†). When trivalent salts were employed (Fig. S1i–k†), the leading peaks focused on the one at m/z 773 in series B with an exception for Cr(NO3)3 (Fig. S1i†). In contrast to the mass spectra with involved monovalent and divalent metal salts, the main peaks of GAP #1 shifted a little bit to lower mass-to-charge ratios. The variation could be due to the different radii or charge number of added metal ions or the higher concentration of NO3− in GAP solution #1 with the addition of trivalent salts than those with monovalent and divalent metal salts.
To get a better understanding on the shift of those peaks with variation in metal salts, we investigated systematically the effects of different types of metal salts on the analysis. Fig. 5a–c show the effect of alkali metal chlorides (e.g., NaCl, KCl and RbCl) on the mass spectra of GAP #1. It was obvious that with the increase in the radii of alkali metal ions from Na+ to Rb+, four series of peaks appeared in the mass spectra, namely series A (m/z 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, and 1538), series B (m/z 477, 576, 675, 774, 873, 972, and 1071), series C (m/z 521, 620, 719, 818, 917, 1016, 1115, 1214, 1313, and 1412, 1511, 1510, and 1709), and series D (m/z 791, 890, 989, 1088, 1187, 1286, 1385, 1484, 1583, and 1682). From these series of data, it can be seen that the peaks of series A and C were greater in abundance than those of series B and D. More importantly, by comparing those series of peaks with the series shown in Fig. 2e, 3 and 4, it could be seen that the mass-to-charge ratios in series A and C were the same as those in the systems with NaNO3, but the patterns of those peaks had much difference. For example, the abundance of the peaks in series C was relatively weak with the involvement of NaNO3, whereas a contrary case was observed as alkali metal chlorides were involved. This case could be ascribed to either the same mass-to-charge ratios for those peaks from the systems with alkali metal chlorides and with NaNO3, or the cross-contamination in both experiments resulting from the carryover effect.38,39 To get direct evidence on the above assumptions, we calculated the mass-to-charge ratios of different series of peaks by coupling the basic units (i.e., M1, M2, M3 and M4) of GAP oligomers (Table 1) to Cl− by the formation of [M + Cl]−. As listed in Table 2, it is noticeable that the calculated values matched very well with the observed mass-to-charge ratios in different series from the addition of alkali metal chlorides, and the isotopic ratio between the intensity of 35Cl and 37Cl involved peaks was in the range of 2.2–2.7 (Fig. S2a†), which was close to their naturally isotopic abundance ratio (3.1). These results suggested that the same mass-to-charge ratios as to those in the system with NaNO3 was just a coincidence rather than cross-contamination. Also, it indicated that besides NO3−, Cl− could also couple to the structure of GAP oligomers by the formation of [M + Cl]− adducts.
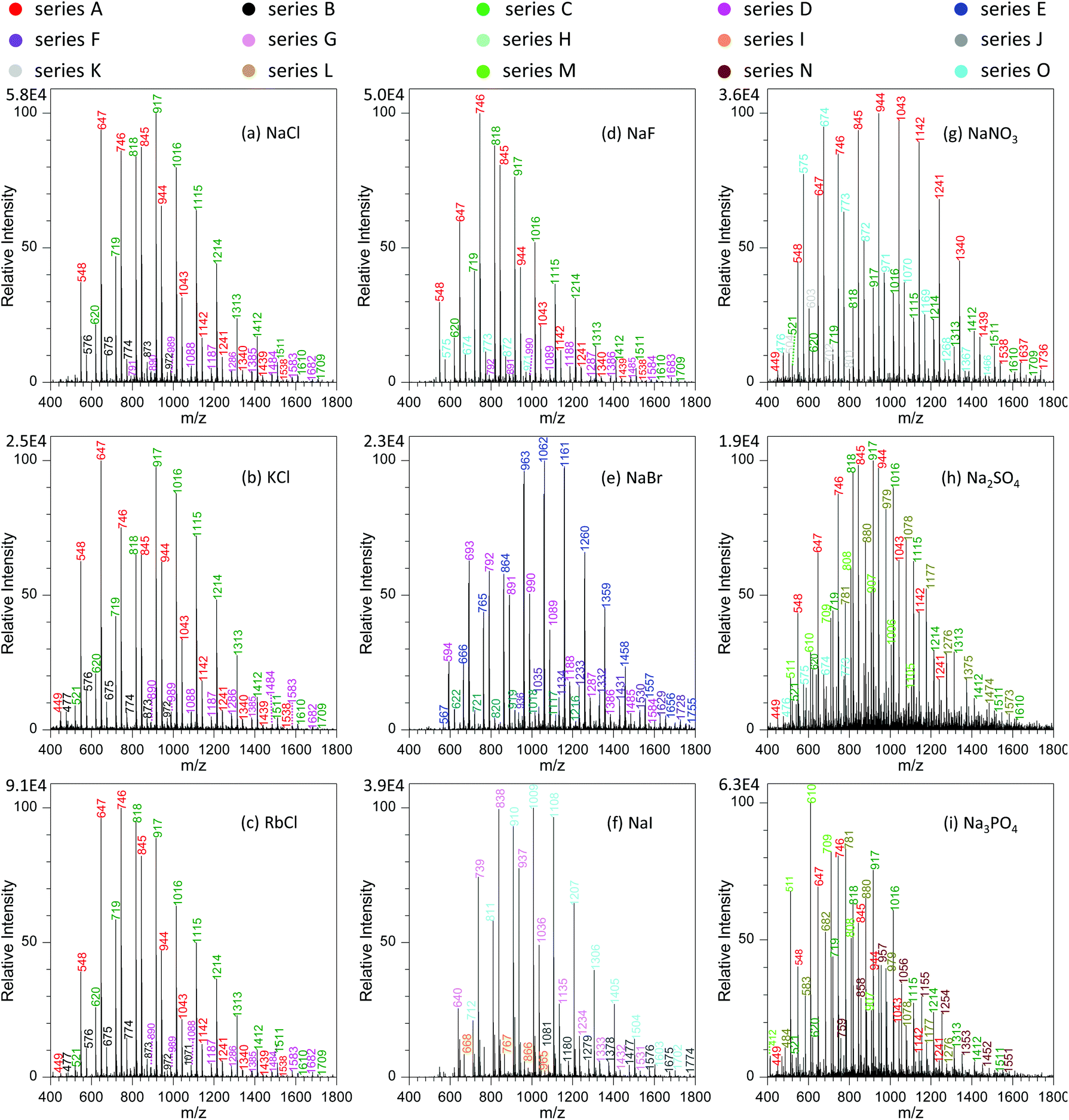 | ||
| Fig. 5 Effect of different types of metal salts on the analysis of 100 μg mL−1 GAP #1: (a) NaCl, (b) KCl, (c) RbCl, (d) NaF, (e) NaBr, (f) NaI, (g) NaNO3, (h) Na2SO4, and (i) Na3PO4 (sample volume: 25 μL for each spray; applied voltage: −3.5 kV; concentration of each metal salt in GAP #1 solution: 1.0 mmol L−1). | ||
| Chloride | NaNO3 or not | Series | Composition | Observed peaks | M n | M w | PDI | |
|---|---|---|---|---|---|---|---|---|
| “P-without” means “positive ion mode and without NaNO3”, “P-with” means “positive ion mode and with NaNO3”, “N-without” means “negative ion mode and without NaNO3”, and “N-with” means “negative ion mode and with NaNO3”. | ||||||||
| Basic unit | M1 | 486, 585, 684, 783, 882, 981, 1080, 1179, 1278, 1377 | ||||||
| NaCl | N-with | C | [M1 + Cl]− | m/z | 620, 719, 818, 917, 1016, 1115, 1214, 1313, 1412, 1511, 1610, 1709 | 987.2 | 1031.8 | 1.045 |
| KCl | N-with | C | [M1 + Cl]− | m/z | 521, 620, 719, 818, 917, 1016, 1115, 1214, 1313, 1412, 1511, 1610, 1709 | 987.9 | 1032.6 | 1.045 |
| RbCl | N-with | C | [M1 + Cl]− | m/z | 521, 620, 719, 818, 917, 1016, 1115, 1214, 1313, 1412, 1511, 1610, 1709 | 949.3 | 996.1 | 1.049 |
| basic unit | M2 | m/z | 414, 513, 612, 711, 810, 909, 1008, 1107, 1206 | |||||
| NaCl | N-with | A | [M2 + Cl]− | m/z | 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538 | 816.6 | 858.0 | 1.051 |
| KCl | N-with | A | [M2 + Cl]− | m/z | 449, 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538 | 796.8 | 843.5 | 1.059 |
| RbCl | N-with | A | [M2 + Cl]− | m/z | 449, 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538 | 789.5 | 825.3 | 1.045 |
| basic unit | M3 | 442, 541, 640, 739 | ||||||
| NaCl | N-with | B | [M3 + Cl]− | m/z | 576, 675, 774, 873, 972 | 736.6 | 759.2 | 1.031 |
| KCl | N-with | B | [M3 + Cl]− | m/z | 477, 576, 675, 774, 873, 972 | 695.9 | 736.2 | 1.058 |
| RbCl | N-with | B | [M3 + Cl]− | m/z | 477, 576, 675, 774, 873, 972, 1071 | 730.5 | 754.7 | 1.033 |
| basic unit | M4 | 756, 855, 954, 1053, 1152, 1251 | ||||||
| NaCl | N-with | D | [M4 + Cl]− | m/z | 791, 890, 989, 1088, 1187, 1286, 1385, 1484, 1583, 1682 | 1206.8 | 1249.5 | 1.035 |
| KCl | N-with | D | [M4 + Cl]− | m/z | 890, 989, 1088, 1187, 1286, 1385, 1484, 1583, 1682 | 1204.2 | 1239.5 | 1.029 |
| RbCl | N-with | D | [M4 + Cl]− | m/z | 890, 989, 1088, 1187, 1286, 1385, 1484, 1583, 1682 | 1256.2 | 1301.2 | 1.036 |
Furthermore, careful observation of the mass spectra with the involvement of alkali metal chlorides revealed that for the dominant series A and C, the peaks shifted steadily from higher mass-to-charge ratios to lower ones (Fig. 5a–c). For example, with variation in the alkali metal chlorides from NaCl to KCl, and then to RbCl, the Mn value for series A changed from 816.6 to 796.8, and then to 789.5, and the weight-average molecular weight (Mw) from 858.0 to 843.5, and then to 825.3 as listed in Table S1.† To our knowledge, this case could be presumably due to the decreasing trend in the solubility or dissociation energy from NaCl to CsCl in the employed solution (Fig. S3†), which should have an impact on the concentration of free Cl− available for GAP adduction. Therefore, with the decrease in the radius of alkali metal in used chlorides, a softer ionization was observed for the system with NaCl, whereas a contrary case occurred for RbCl.
Based on the above discussion, the possible mechanisms for the ionization of GAP oligomers are proposed as shown in Fig. 6. When no metal salt was involved in the spray solution, the cations such as H+ in spray solution would move to the negative pole of the applied potential (left side), and GAP− moved to the positive pole (right side). Due to the small radius and high charge density of H+, its moving speed was much faster than the flow rate of the spray solution, and a good separation between H+ and GAP− occurred before they were sprayed out. When GAP− was in the spray plume, the electric field force would be applied directly on the structure of GAP− (Fig. 6a), which induced its fragmentation. So various series of peaks with low mass-to-charge ratios were observed for GAP polymers (Fig. 2d). When NaCl was involved in the spray solution, similar to the system without the metal salt, the cations such as H+ and Na+ would move quickly to the negative pole of the applied potential due to their relatively high charge density on the surfaces, and the anions such as [GAP + Cl]− and Cl− moved to the positive pole and were sprayed out upon the applied high voltage (−3.5 kV). As [GAP + Cl]− and Cl− were involved in the spray plume, the bound Cl− in the structure of [GAP + Cl]− adducts could shield partially the electric field induced force applied to the GAP structure (Fig. 6b). As a result, a softer ionization event occurred when NaCl was added into the GAP solution compared to that without NaCl (Fig. 6a). It should be mentioned that this way did not allow to produce intact GAP species in the gas phase. As listed in Table 2, the maximum Mn value for GAP #1 was no more than 1300 using the MSAESI-MS method, which was less than the value (Mn = 1477) from gel permeation chromatography (GPC). This indicated that the current way could not thoroughly prevent GAP species from fragileness during ionization. With the increase in the radius of alkali metal ions from Na+ to Rb+, the dissolution ability of the alkali salt in methanol demonstrated a decreasing trend, and the content of Cl− in the sample solution gradually decreased, which would limit the complexation of Cl− with GAP oligomers. Therefore, the amount of formed [GAP + Cl]− showed a decreasing trend. After paper spray, a small amount of GAP− existed in the spray plume, which made a majority of the electric field force to directly apply on GAP− with little shielding from bound CI− and the fragmentation of GAP− occurred because of its fragileness (Fig. 6c). The peaks in the mass spectrum shifted to lower mass-to-charge ratios (Fig. 5c).
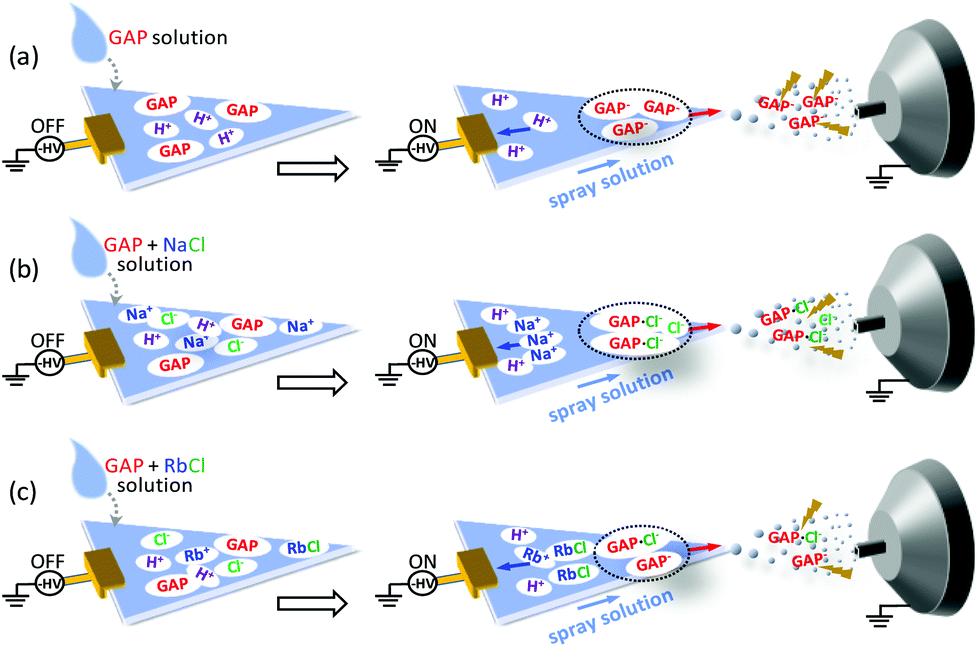 | ||
| Fig. 6 Proposed electrospray mechanisms of MSAESI-MS for the analysis of 100 μg mL−1 GAP #1 (a) without and with the presence of different metal salts: (b) NaCl, and (c) RbCl. | ||
In the current study, we also found that the type of anions in added metal salts had a great effect on the analysis of GAP oligomers. Fig. 5a and d–f show the mass spectra of GAP #1 solution with the addition of 1.0 mmol L−1 NaF, NaCl, NaBr, and NaI, respectively. It is obvious that when NaF was involved in the GAP #1 solution (Fig. 5d), the peaks from series A, B, C and D were available in the mass spectra, which were very similar to those in the presence of NaCl, KCl and RbCl (Fig. 5a–c). According to the above discussion, the peaks in those series were the adducts between Cl− and the basic units of GAP oligomers (Table 2). After further calculation, no formation of [M + F]− adducts was found in the mass spectrum when NaF was involved (Table S4†). These results illustrated that the observed peaks in Fig. 5d could be assigned to the cross-contamination from the previous experiments, and F− had very weak complexing interaction with GAP oligomers by the formation of [GAP + F]−. As NaBr was introduced into the sample solution (Fig. 5e), two new series of peaks (i.e., series E and F) appeared in the mass spectrum, whereas the mass-to-charge ratios for the peaks in series C and D were same as those with the involvement of NaCl (Fig. 5a) although both had different patterns. According to the calculation, the results demonstrated that for series E and F, they had compositions of [M1 + Br]− and [M4 + Br]−, and for series C and D, their compositions were [M3 + Br]− and [M2 + Br]− as listed in Table S4.† The isotopic ratio between the intensity of 79Br and 81Br involved peaks close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S2b†) also suggested the formation of [M + Br]−. For the system with NaI, the formation of [GAP + I]− adducts was also observed (Fig. 5f and Table S4†). From the above, it is apparent that except for F−, other halides (i.e., Cl−, Br− and I−) could form adducts with GAP oligomers, attributable to the relative higher dissolution of the latter in methanol than that of the former (Fig. S4†). In addition, careful observation on the variation in the mass spectra revealed that with the increase in the radius of halide ions, the mass-to-charge ratios of obtained adducts shifted first to higher values, and then to relatively lower values. For example, for the basic unit M1 (namely series C for Cl− as Fig. 5a, series E for Br− as Fig. 5e, and series H for I− as Fig. 5f), the Mn value changed from 986.6 to 1082.1, and then to 1065.6, and the Mw value varied from 1029.2 to 1126.4, and then to 1103.5. For other units such as M2, M3 and M4, a similar trend was observed as listed in Table S4.† This phenomenon was presumably due to the fact that with variation in the employed halides from NaCI to NaBr, then to NaI, their dissolution ability into used solvent presented a gradually increasing trend, whereas the complexing interaction between GAP and halide ions illustrated a decreasing one resulting from surface charge density or polarity. As a compromise, NaBr gave different series of [M + Br]− adducts with relatively higher mass-to-charge ratios.
:1 (Fig. S2b†) also suggested the formation of [M + Br]−. For the system with NaI, the formation of [GAP + I]− adducts was also observed (Fig. 5f and Table S4†). From the above, it is apparent that except for F−, other halides (i.e., Cl−, Br− and I−) could form adducts with GAP oligomers, attributable to the relative higher dissolution of the latter in methanol than that of the former (Fig. S4†). In addition, careful observation on the variation in the mass spectra revealed that with the increase in the radius of halide ions, the mass-to-charge ratios of obtained adducts shifted first to higher values, and then to relatively lower values. For example, for the basic unit M1 (namely series C for Cl− as Fig. 5a, series E for Br− as Fig. 5e, and series H for I− as Fig. 5f), the Mn value changed from 986.6 to 1082.1, and then to 1065.6, and the Mw value varied from 1029.2 to 1126.4, and then to 1103.5. For other units such as M2, M3 and M4, a similar trend was observed as listed in Table S4.† This phenomenon was presumably due to the fact that with variation in the employed halides from NaCI to NaBr, then to NaI, their dissolution ability into used solvent presented a gradually increasing trend, whereas the complexing interaction between GAP and halide ions illustrated a decreasing one resulting from surface charge density or polarity. As a compromise, NaBr gave different series of [M + Br]− adducts with relatively higher mass-to-charge ratios.
To further investigate the effect of anions with multiple valences on the analysis of GAP oligomers, we investigated the ionization performance of GAP #1 with the addition of NaNO3, Na2SO4 and Na3PO3 in the spray solution, and Fig. 5g–i show their corresponding mass spectra. It was obvious that when NaNO3 was introduced into the spray solution (Fig. 5g), abundant peaks in series A, C and O from GAP #1 presented in the mass spectrum as well as relatively weak peaks in series K, and they could be assigned to the adducts of [M1 + NO3]−, [M2 + NO3]−, [M4 + NO3]−, and [M3 + NO3]− as listed in Table S4,† respectively. The dominant peaks were centered on the one at m/z 944. Also, careful observation revealed that the mass-to-charge ratios of the peaks in series O (Fig. 5g) had only an 1 Da difference from series B shown in Fig. 5a–c. Such a difference was due to the various complexes ([M3 + Cl]− for series B and [M2 + NO3]− for series O as Tables 2 and S4†). As Na2SO4 was involved in the GAP solution #1 (Fig. 5h), the mass spectrum became complex, and, more importantly, the peaks shifted to lower mass-to-charge ratios. Careful observation revealed that there were five series of peaks in the mass spectrum, namely series A, B, C, L and M. After analysis based on the basic units of GAP oligomers, series A, B and C were from cross-contamination with Cl− or NO3−, and series L and M were the adducts of [M1 + HSO4]− and [M2 + HSO4]− as listed in Table S4.† When Na3PO3 was added into the sample solution (Fig. 5i), the obtained peaks shifted more to lower mass-to-charge ratios than that with Na2SO4, and five series of peaks (i.e., series A, C, L, M, and N) appeared in the mass spectrum, in which series A and C were from the cross-contamination of either Cl− or NO3−, and series L, M and N were the adducts of [M1 + H2PO3]−, [M2 + H2PO3]− and [M3 + NaHPO3]−, respectively. From the mass spectra as shown in Fig. 5g–i, it is obvious that the complexing ability between GAP and NO3−, SO42− or PO43− demonstrated a steadily decreasing trend, and the mass-to-charge ratios of the generated peaks shifted gradually to low values. As is well-known, the dissolution ability of NaNO3, Na2SO4 and Na3PO4 into methanol illustrates a decreasing trend (Fig. S5†), whereas their average charge at the surfaces [NO3−: −0.542, SO42−: −1.005, and PO43−: −1.289] demonstrates an increasing trend.55 Based on both parameters, it can be seen that the dissolution ability of those salts overwhelmed their average charge at the surfaces. This also suggests that for the purpose of obtaining a soft ionization of [GAP + anion]− adducts, the dissolution ability of selected salts is more important than other parameters if the complexing interaction between GAP molecules and corresponding anions is strong enough.
Application of MSAESI-MS to other GAP samples and nanoelectrospray systems
To demonstrate the performance of the developed MSAESI-MS, several GAP samples including GAP #2, #3 and #4 from different synthesis parameters were analyzed. Fig. 7a shows the mass spectrum of GAP #2 solution without the addition of NaNO3. Apparently, there were three series of peaks in the mass spectrum, namely series A (m/z 449, 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, 1538, 1637, 1736, 1835, and 1934), series B (m/z 575, 674, 773, 872, 971, 1070, 1169, 1268, 1367, 1466, 1565, 1664, 1763, and 1862), and series C (m/z 603, 702, 801, 900, 999, 1098, 1197, and 1296). Among these series, only the peaks from series A were dominant and centered on the one at m/z 647. After NaNO3 was added into GAP #2 solution, the mass spectrum greatly changed (Fig. 7a′). On the one hand, the peaks in series B became the leading ones in the spectrum, and the intensity of the peaks in series C turned into more abundant ones than those without NaNO3. However, the intensity of the peaks in series A was weaker than those in the absence of NaNO3. On the other hand, the peaks in different series shifted to higher mass-to-charge ratios. For example, for series A with a component of [M1 + NO3]−, the Mn and Mw values increased from 789.2 and 881.7 to 934.2 and 1033.0, respectively, as listed in Table S5.† For series B with a component of [M2 + NO3]−, the corresponding values increased from 924.9 and 1028.6 to 957.2 and 1068.7, and for series C with a component of [M3 + NO3]−, they increased from 785.9 and 831.3 to 1036.4 and 1116.2. Besides GAP #2, the developed MSAESI-MS also worked for the soft ionization of other GAP samples such as GAP #3 (Fig. 7b and b′) and GAP #4 (Fig. 7c and c′). For the sample of GAP #3, the Mn and Mw values for series B increased from 758.0 and 790.6 to 1093.0 and 1163.9, respectively. For the sample of GAP #4, the Mn and Mw values for series B increased from 962.4 and 1046.1 to 1154.1 and 1241.3 (Table S5†). From these results, it is obvious that the present reported method is a soft and robust ionization source for the analysis of different GAP samples.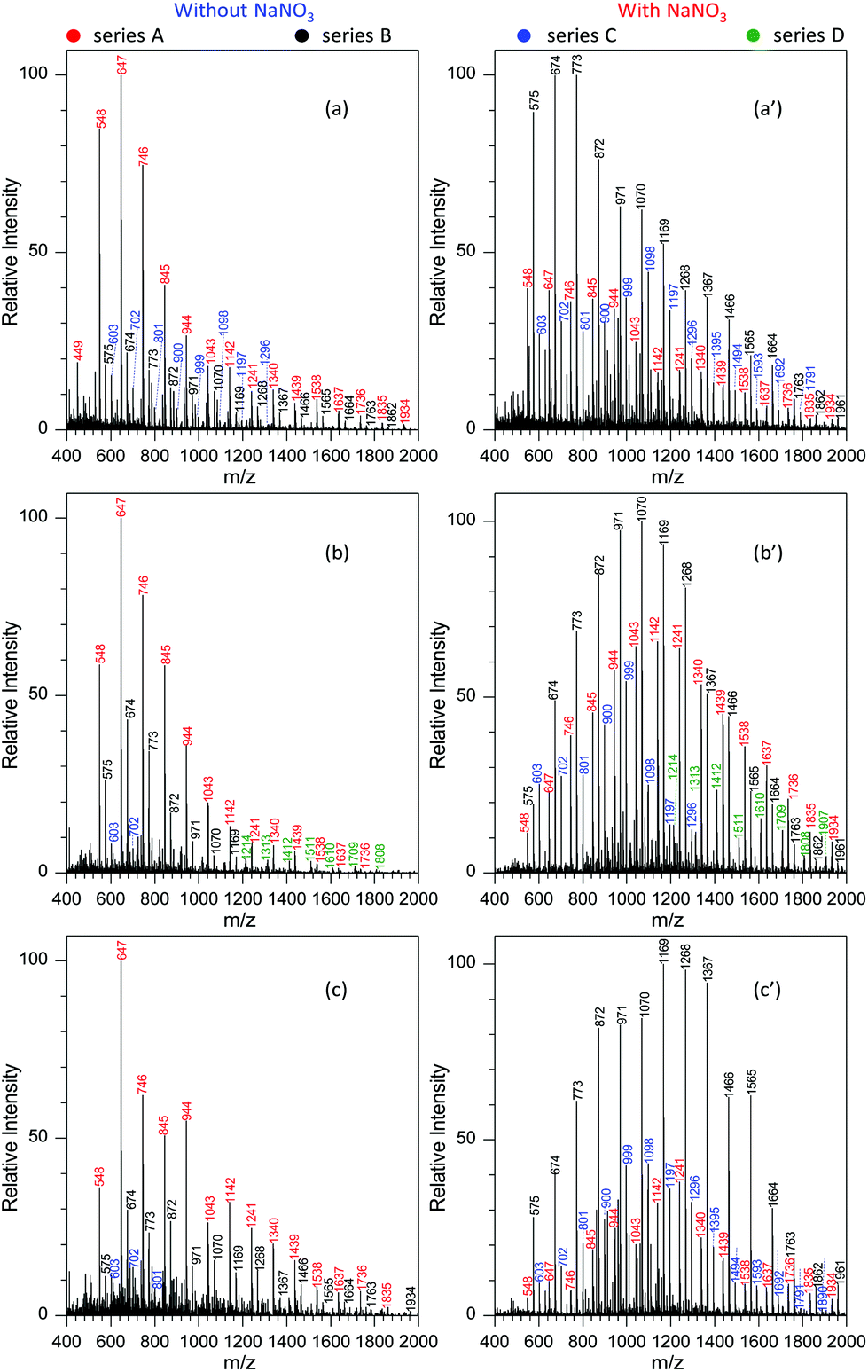 | ||
| Fig. 7 Comparison of the mass spectra of different GAP samples before and after the addition of 1.0 mmol L−1 NaNO3 into the sample solution: (a) and (a′) GAP #2, (b) and (b′) GAP #3, and (c) and (c′) GAP #4 (applied voltage: −3.5 kV; sample volume: 25 μL). | ||
Similar to paper spray ionization, nanoelectrospray ionization (nanoESI) is also a soft technique to ionize compounds prior to MS analysis.33 In order to promote the ionization performance of various target analytes, many attempts have been made to suppress the influence of metal salts in sample matrices during nanoESI in previous studies. For example, Wei et al.56 reported a step-voltage nanoESI method to remove the interference of salt cations and anions in small-volume biological samples. Huang and co-workers57 reported an induced nanoESI ionization to analyze raw serum and whole urine with a high concentration of salts. Gong and colleagues58 made use of a polarity-reversing high voltage strategy for the generation of nanoESI, which showed excellent desalting effect to the analysis of different biological samples. Contrary to the above studies, herein we introduced metal salts such as NaNO3 into the sample solution during nanoESI for the purpose of improving the soft ionization of GAP samples. Analogous to the electrospray mechanism of paper spray with the involvement of metal salts (Fig. 6), Fig. 8a shows the procedure for the analysis of the GAP sample in the presence of NaNO3 with nanoESI. Upon the action of a negative electric field, H+/Na+ and [GAP + NO3]− ions in the sample solution would migrate rapidly in opposite directions in the tip zone. After the anions such as [GAP + NO3]− are sprayed out from the capillary, the fragmentation of [GAP + NO3]− would be greatly prohibited due to the complexing interaction between GAP and NO3− or the shielding effect from bound NO3−. As a result, a soft ionization is observed for the system with the involvement of NaNO3. Fig. 8b–e and b′–e′ compare the mass spectra of different GAP samples without and with the presence of NaNO3. For sample GAP #1, when there was no NaNO3 in the sample solution (Fig. 8b), different series of peaks, including series A ([M1 + NO3]−, m/z 449, 548, 647, 746, 845, 944, 1043, 1142, 1241, 1340, 1439, and 1538), series B ([M2 + NO3]−, m/z 674, 773, 872, 971, and 1070), series C ([M4 + NO3]−, m/z 620, 719, 818, 917, 1016, 1115, 1214, 1313, 1412, and 1511), series D ([M2 + HSO4]− or [M2 + H2PO4]−, m/z 412, 511, 610, 709, and 808), and series E ([M1 + HSO4]− or [M1 + H2PO4]−, m/z 484, 583, 682, 781, 880, 979, 1078, and 1177), appeared in the mass spectrum, in which series D was the dominant one. The leading peaks were focused on the one at m/z 610. When NaNO3 was involved in the sample solution, much difference was observed (Fig. 8b′). The peaks in series A, B, and C appeared in the mass spectrum. More importance was that the above peaks shifted to higher mass-to-charge ratios in contrast to those without NaNO3. For example, the Mn and Mw values for series A increased from 839.6 and 891.4 to 1079.5 and 1111.5, respectively. For series C, those values increased from 941.1 and 976.8 to 1225.2 and 1260.5. With other samples such as GAP #2 (Fig. 8c and c′), GAP #3 (Fig. 8d and d′) and GAP #4 (Fig. 8e and e′), a similar phenomenon was observed for different series of peaks as listed in Table S6.† The above results suggest that the addition of metal salts such as NaNO3 is also an effective and facile avenue to improve the performance of nanoESI in GAP analysis, which suppresses greatly the fragmentation of various GAP samples.
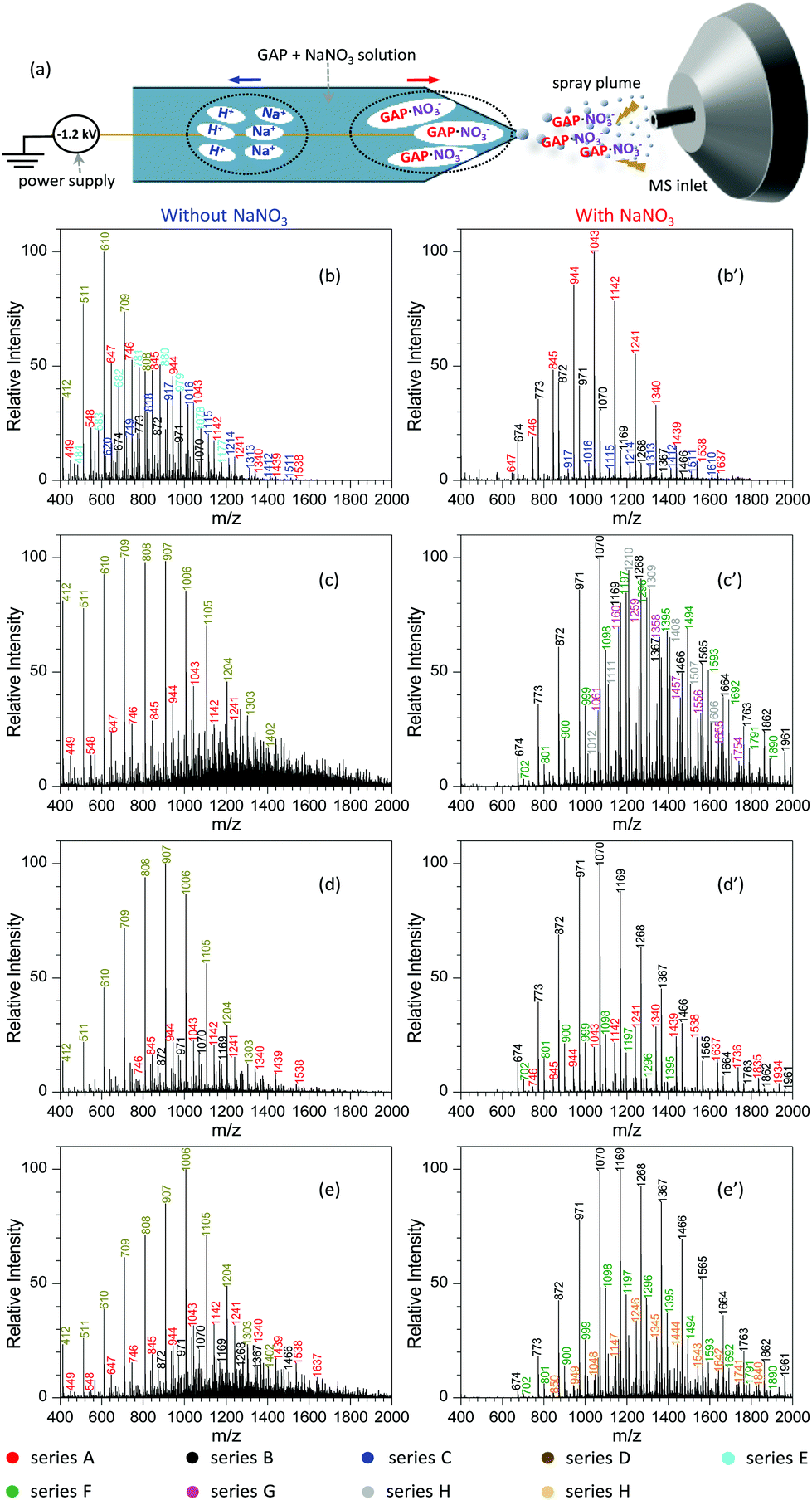 | ||
| Fig. 8 (a) Schematic representation of the procedure for analyzing the GAP sample containing 1.0 mmol L−1 NaNO3 using nanoESI-MS; comparison of the mass spectra of different GAP samples before and after the addition of 1.0 mmol L−1 NaNO3 into the sample solution: (b) and (b′) GAP #1, (c) and (c′) GAP #2, (d) and (d′) GAP #3, and (e) and (e′) GAP #4 (applied voltage: −1.2 kV). | ||
Conclusions
In summary, a novel MSAESI-MS method has been developed for the first time to softly ionize fragile GAP samples, which is promising for applications in the rapid analysis of energetic polymers. We showed that the ion mode, applied voltage, and the concentration and type of metal salt were crucial in determining the performance of the developed MSAESI-MS. Because of the formation of [GAP + NO3]− adducts, the negative ion mode was more advantageous to the soft ionization of GAP samples with metal salts such as NaNO3 than the positive ion mode. In the process of GAP analysis, for cations with equal charge, a smaller radius of cations would be more favorable for soft ionization due to the variation in dissolution ability (e.g., NaCl > KCl > RbCl), and, on the contrary, a larger radius of anions allows the observation of the peaks with higher mass-to-charge ratios in the mass spectrum (e.g., NaF < NaCl < NaBr < NaI). According to the experimental observation and evidence from MS, possible mechanisms were proposed to elucidate the electrospray ionization mechanisms of MSAESI-MS for GAP analysis in the presence of metal salts. The developed method has also been applied to the successful analysis of various GAP samples. More importantly, the present protocol is not limited to paper spray ionization for the soft ionization of energetic polymers such as GAP samples, but also works for other types of ionization sources such as nanoESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for funding from the National Natural Science Foundation of China (No. 21575112, 21777128 and 21705125) and the Natural Science Basic Research Program of Shaanxi Province of China (Grant No. 2019JC-33).Notes and references
- D. M. Badgujar, M. B. Talawar, V. E. Zarko and P. P. Mahulikar, Combust., Explos. Shock Waves, 2017, 53, 371–387 CrossRef.
- E. N. Volkov, A. A. Paletsky, A. G. Tereshchenko and O. P. Korobeinichev, Combust., Explos. Shock Waves, 2006, 42, 663–671 CrossRef.
- A. N. Nazare, S. N. Asthana and H. Singh, J. Energ. Mater., 1992, 10, 43–63 CrossRef CAS.
- S. M. Pedreira, J. R. A. Pinto, E. A. Campos, E. D. C. Mattos, M. S. D. Oliveira Junior, J. I. S. D. Oliveira and R. D. C. L. Dutra, J. Aerosp. Technol. Manage., 2016, 8, 18–25 CrossRef.
- M. Xu, Z. Ge, X. Lu, H. Mo, Y. Ji and H. Hu, RSC Adv., 2017, 7, 47271–47278 RSC.
- E. J. Vandenburg, Polymers Containing Azidomethyl Side Chains, US Patent3645917, 1972 Search PubMed.
- X. Xu, M. Liu, Y. Yin, C. Zheng, P. Deng and D. Xue, Green Chem., 2016, 18, 1364–1367 RSC.
- H. Fazlıoğlu and J. Hacaloğlu, J. Anal. Appl. Pyrolysis, 2002, 63, 327–338 CrossRef.
- T. Wang, S. Li, B. Yang, C. Huang and Y. Li, J. Phys. Chem. B, 2007, 111, 2449–2455 CrossRef CAS.
- Y. Wu, Z. Ge and Y. Luo, J. Therm. Anal. Calorim., 2016, 124, 107–115 CrossRef CAS.
- C.-J. Tang, Y. Lee and T. A. Litzinger, Combust. Flame, 1999, 117, 244–256 CrossRef CAS.
- D. S. Moore and S. D. McGrane, J. Mol. Struct., 2003, 661–662, 561–566 CrossRef CAS.
- J. F. Arenas, J. C. Otero and J. Soto, J. Mol. Struct., 1993, 294, 45–48 CrossRef CAS.
- T. Parr and D. Hanson-Parr, Combust. Flame, 2004, 137, 38–49 CrossRef CAS.
- Y. Sun and S. Li, J. Hazard. Mater., 2008, 154, 112–117 CrossRef CAS.
- S. P. Panda, S. G. Kulkarni, S. K. Sahu, V. N. Bhoraskar and P. A. Dokhale, Bull. Mater. Sci., 1996, 19, 1125–1132 CrossRef CAS.
- Y. He, Y. Liang and D. Wang, Chem. Commun., 2015, 51, 12092–12094 RSC.
- S. Brochu and G. Ampleman, Macromolecules, 1996, 29, 5539–5545 CrossRef CAS.
- V. T. Bui, E. Ahad, D. Rheaume and R. Whitehead, Ind. Eng. Chem. Res., 1997, 36, 2219–2224 CrossRef CAS.
- A. Pelster, M. Körsgen, T. Kurosawa, H. Morita and H. F. Arlinghaus, Anal. Chem., 2016, 88, 9638–9646 CrossRef CAS.
- K. Krueger, C. Terne, C. Werner, U. Freudenberg, V. Jankowski, W. Zidek and J. Jankowski, Anal. Chem., 2013, 85, 4998–5004 CrossRef CAS PubMed.
- K. Fouyer, O. Lavastre and D. Rondeau, Anal. Chem., 2012, 84, 8642–8649 CrossRef CAS PubMed.
- N. Zhang, Y. Zhou, C. Zhen, Y. Li, C. Xiong, J. Wang, H. Li and Z. Nie, Analyst, 2012, 137, 5051–5056 RSC.
- M. Nefliu, A. Venter and R. G. Cooks, Chem. Commun., 2006, 888–890 RSC.
- S. D. Hanton, Chem. Rev., 2001, 101, 527–570 CrossRef CAS.
- P. M. Peacock and C. N. McEwen, Anal. Chem., 2004, 76, 3417–3428 CrossRef CAS.
- S. M. Weidner and S. Trimpin, Anal. Chem., 2010, 82, 4811–4829 CrossRef CAS.
- X. Li, L. Guo, M. Casiano-Maldonado, D. Zhang and C. Wesdemiotis, Macromolecules, 2011, 44, 4555–4564 CrossRef CAS.
- M. Farber, S. P. Harris and R. D. Srivastava, Combust. Flame, 1984, 55, 203–211 CrossRef CAS.
- H. Fazhoğlu and J. Hacaloğlu, J. Macromol. Sci., Part A: Pure Appl.Chem., 2002, 39, 759–768 CrossRef.
- L. V. Kuibida, O. P. Korobeinichev, A. G. Shmakov, E. N. Volkov and A. A. Paletsky, Combust. Flame, 2001, 126, 1655–1661 CrossRef CAS.
- J. Liu, H. Wang, N. E. Manicke, J.-M. Lin, R. G. Cooks and Z. Ouyang, Anal. Chem., 2010, 82, 2463–2471 CrossRef CAS PubMed.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS PubMed.
- Z. Zhang, W. Xu, N. E. Manicke, R. G. Cooks and Z. Ouyang, Anal. Chem., 2012, 84, 931–938 CrossRef CAS PubMed.
- T. Wang, Y. Zheng, X. Wang, D. E. Austin and Z. Zhang, Anal. Chem., 2017, 89, 7988–7995 CrossRef CAS PubMed.
- X. Wang, Y. Zheng, J. Shi, X. Gong, Y. Ji, W. Han, Y. Jiang, D. E. Austin, X. Fang and Z. Zhang, Anal. Chem., 2018, 90, 11138–11145 CrossRef CAS.
- X. Lu, H. Mo, M. Xu, N. Liu and X. Wang, Chin. J. Chem. Analysis Meterage (Huaxue Fenxi Jiliang), 2018, 27, 1–5 Search PubMed.
- N. C. Hughes, E. Y. K. Wong, J. Fan and N. Bajaj, AAPS J., 2007, 9, E353–E360 CrossRef CAS.
- A. Medvedovici, F. Albu and V. David, J. Liq. Chromatogr. Relat. Technol., 2010, 33, 1255–1286 CrossRef CAS.
- T. M. Crescentini, J. C. May, J. A. McLean and D. M. Hercules, Polymer, 2019, 173, 58–65 CrossRef CAS.
- D. Fati, V. Leeman, Y. V. Vasil'ev, T. Drewello, B. Leyh and H. Hungerbuhler, J. Am. Soc. Mass Spectrom., 2002, 13, 1448–1458 CrossRef CAS.
- P. Hurtado, A. R. Hortal, F. Gamez, S. Hamad and B. Martinez-Haya, Phys. Chem. Chem. Phys., 2010, 12, 13752–13758 RSC.
- J. Wei, A. W. T. Bristow and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2015, 26, 166–173 CrossRef CAS.
- Y. Chen, S. D. M. Chinthaka and M. T. Rodgers, J. Phys. Chem. A, 2013, 117, 8274–8284 CrossRef CAS.
- R. Chen and L. Li, J. Am. Soc. Mass Spectrom., 2001, 12, 832–839 CrossRef CAS.
- G. Hart-Smith and C. Barner-Kowollik, Polymer, 2009, 50, 5175–5180 CrossRef CAS.
- A. R. Hortal, P. Hurtado, B. Martinez-Haya, A. Arregui and L. Banares, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 859–863 CrossRef CAS.
- R. Knochenmuss, E. Lehmann and R. Zenobi, Eur. Mass Spectrom., 1998, 4, 421–426 CrossRef CAS.
- S. Lin-Gibson, L. Brunner, D. L. Vanderhart, B. J. Bauer, B. M. Fanconi, C. M. Guttman and W. E. Wallace, Macromolecules, 2002, 35, 7149–7156 CrossRef CAS.
- M. C. Bridoux, A. Schwarzenberg, S. Schramm and R. B. Cole, Anal. Bioanal. Chem., 2016, 408, 5677–5687 CrossRef CAS PubMed.
- M. V. Kosevich, V. G. Zobnina, V. V. Chagovets and O. A. Boryak, Rapid Commun. Mass Spectrom., 2011, 25, 713–718 CrossRef CAS PubMed.
- M. A. A. Rodrigues, D. C. Mendes, V. Ramamurthy and J. P. Da Silva, J. Am. Soc. Mass Spectrom., 2017, 28, 2508–2514 CrossRef CAS PubMed.
- J. Steinkoenig, M. M. Cecchini, S. Reale, A. S. Goldmann and C. Barner-Kowollik, Macromolecules, 2017, 50, 8033–8041 CrossRef CAS.
- V. G. Zobnina, M. V. Kosevich, V. V. Chagovets, O. A. Boryak, A. N. Kulik and A. Gomory, J. Anal. Chem., 2011, 66, 1341–1347 CrossRef CAS.
- M. F. C. Ladd, Theor. Chim. Acta, 1980, 54, 157–164 CrossRef CAS.
- Z. Wei, S. Han, X. Gong, Y. Zhao, C. Yang, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11025–11028 CrossRef CAS PubMed.
- G. Huang, G. Li and R. G. Cooks, Angew. Chem., Int. Ed., 2011, 50, 9907–9910 CrossRef CAS PubMed.
- X. Gong, X. Xiong, Y. Zhao, S. Ye and X. Fang, Anal. Chem., 2017, 89, 7009–7016 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an01887e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |