DOI:
10.1039/C8AN01390J
(Paper)
Analyst, 2019,
144, 331-341
AIE active piperazine appended naphthalimide-BODIPYs: photophysical properties and applications in live cell lysosomal tracking†
Received
24th July 2018
, Accepted 7th October 2018
First published on 15th October 2018
Abstract
Piperazine appended naphthalimide-BODIPYs (NPB1–NPB4) exhibiting solvatochromism and aggregation-induced emission with a large Stokes shift (up to 146 nm) have been described. Separation of naphthalimide and BODIPY fluorophores by piperazine in these conjugates creates a donor–acceptor system and induces twisted intramolecular charge transfer, in addition to photoinduced electron transfer. The crucial role of naphthalimide, the alkyl chain length, the piperazine ring, and the solid-state packing on AIE has been extensively investigated by various studies. Superior cell permeability coupled with bio-compatibility of these conjugates offers a unique opportunity for their potential applications in live cell lysosomal tracking.
Introduction
Boron dipyrromethene (BODIPY) conjugates have attracted enormous current interest due to their excellent spectral features like their strong absorption of visible light, intense emission in longer wavelength regions, and good photostability as well as their wide applications in diverse areas.1 Creation of systems showing outstanding photophysical properties and superior biological activity has been challenging. Recent studies have shown that it can be achieved by a judicious choice of fluorophores, auxochromes, and substituents.2 Notably the majority of fluorophores are highly emissive in solution; however, these fluoresce weakly in the aggregated/solid-state due to aggregation-caused quenching (ACQ) which hampers their realistic values.3 In this context, aggregation-induced emission (AIE) has fascinated the scientific community as it enhances emission in the solid-state and overcomes ACQ.4 The explanation of AIE can be related to several processes like restricted intramolecular rotation (RIR), reduced face-to-face intermolecular π–π interactions, twisted intramolecular charge transfer (TICT), and photoinduced electron transfer (PET).4a Furthermore, naphthalimides (NPIs) are widely encountered organic fluorophores due to their interesting optical properties, biological activity and significance in developing fluorescent materials.5 The inclusion of NPIs into a BODIPY core may improve photophysical and biological properties, and donor–acceptor (D–A) systems involving NPI-BODIPY conjugates may offer an in-depth understanding of AIE mechanisms.2b,6 In addition, AIE active NPI-BODIPY conjugates with a D–A construct are rather scarce.6a It is presumed that systems involving NPI connected to BODIPY via flexible piperazine may be interesting as piperazine can donate the charge density to fluorophores (NPI and BODIPYs) via the push–pull mechanism creating a D–A construct.6d–f Moreover, the D–A construct has attracted great interest due to the sensitivity of optical behaviours towards surrounding media (solvents/pH/chemical ions), dual emission, emissions with a large Stokes shift and their applications in various fields.6f
Thus, developing piperazine-appended NPI-BODIPY conjugates having alkyl groups of diverse chain lengths (–CH3, –C2H5, –C6H13, and –C8H17) will be highly attractive. The alkyl groups may offer flexibility and hydrophobicity apart from directing the packing pattern and influencing the photophysical properties of conjugates. Furthermore, photoinduced electron transfer (PET) is strongly influenced by structural variations viz. orientation of the D–A moiety, conformational changes, separation length, etc.7 Structural modulations extending flexibility via conformational changes influence PET and may account for the mechanisms of AIE, TICT, and solvatochromism. Also, PET active systems are vital for pH-dependent optical responses.7f,g It may provide valuable information about biological systems as intracellular pH is related to cell processes and abnormalities.8 Usually, the pH of acidic cell organelles in a typical mammalian cell such as endosomes and lysosomes varies from 6.3 to 4.7. It is a daunting task to develop selective probes for this particular pH range and track acidic cell organelles, particularly lysosomes.2b,9
With an objective of developing AIE active piperazine-appended NPI-BODIPYs, four new conjugates (NPB1–NPB4) have been synthesized and thoroughly characterized. These exhibit prominent solvatochromism, AIE, solid-state fluorescence, and a pH sensing ability. Through this contribution, we describe the influence of TICT and PET on AIE in BODIPY conjugates and their potential applications in lysosomal tracking.
Results and discussion
Synthesis and characterization
A simple synthetic route adopted for the preparation of the conjugates NPB1–NPB4 is illustrated in Scheme 1. Compound 2 has been synthesized using 1,2-dihydroacenaphthylene (1) following literature procedures.10a Condensation of this compound with respective amines yielded isoquinoline derivatives 3A–3D in reasonably good yield.10b
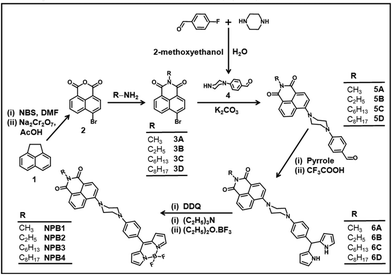 |
| | Scheme 1 Synthetic route to NPB1–NPB4 (NBS = N-bromosuccinimide, DMF = N,N-dimethylformamide, and DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone). | |
Piperazinylbenzaldehyde (4) was prepared by reacting 4-fluorobenzaldehyde with piperazine adopting literature procedures.10c The isoquinoline derivatives 3A–3D reacted with 4 in the presence of K2CO3 in dry N,N-dimethylformamide (DMF) to afford the aldehydes 5A–5D in good yield.6a,g Condensation of 5A–5D with an excess of pyrrole and catalytic amounts of trifluoroacetic acid (TFA) gave dipyrromethanes 6A–6D. These dipyrromethanes upon oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) followed by treatment with BF3·Et2O in the presence of Et3N afforded the desired conjugates NPB1–NPB4 in good yield.6a,10d Elemental analyses, IR, ESI-MS, NMR (1H, 13C, 11B, and 19F), absorption and emission spectral studies confirmed their formation (Fig. S1–S24†). The structures of NPB1, NPB3, and NPB4 have been unequivocally determined by X-ray single crystal analyses.
Electronic absorption and fluorescence spectroscopy
The absorption spectra of NPB1–NPB4 (THF, room temperature) showed a characteristic strong band at ∼498 nm along with a shoulder peak at ∼460 nm due to the meso-aryl BODIPY core and a relatively broader one at ∼381 nm due to both BODIPY and naphthalimide units (Fig. 1).11 Furthermore, because of an analogous chromophore, the conjugation length and an auxochrome, these showed absorption maxima at almost the same position. The emission spectra of these conjugates upon excitation at 498 nm exhibited bands at ∼525 nm [NPB1, 520; NPB2, 522; NPB3, 526; NPB4, 527 nm] caused by typical locally excited (LE) state emission and at ∼627 nm [NPB1, 630; NPB2, 629; NPB3, 626; NPB4, 626 nm] due to TICT transitions (Fig. 1).1b,6c
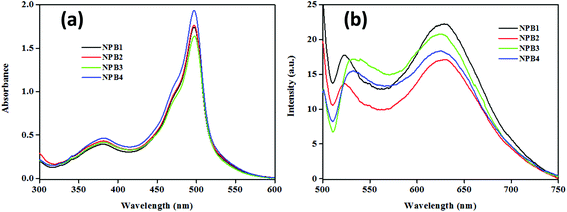 |
| | Fig. 1 Absorption (a) and emission spectra (b) of NPB1–NPB4 (c, 50 μM, THF; λex, 498 nm). | |
Solvatochromism
The presence of piperazinyl nitrogen in NPB1–NPB4 in conjunction with acceptors (BODIPY and NPI) enables them to behave as ideal D–A constructs capable of exhibiting the push–pull mechanism and thus solvent dependent optical properties.6c–e Solvatochromism for these conjugates has been investigated by performing absorption and emission studies.
The resulting data are collected in Table S1† while spectra are depicted in Fig. 2 and Fig. S25–S27.† Expectedly, with an increase in solvent polarity, they showed significant lowering of the emission intensity followed by a red shift. Emission with the highest intensity has been observed in hexane at ∼516 nm [NPBl, 516; NPB2, 522; NPB3, 516; NPB4, 521 nm] while the least intense red-shifted band in methanol at ∼641 nm [NPBl, 641; NPB2, 639; NPB3, 640; NPB4, 640 nm]. On the other hand, the absorption spectra showed insignificant changes with variation in the solvent polarity. Furthermore, the emission color for these conjugates has been observed with the naked eye in solvents of varying polarities under UV illumination (λex, 365 nm). In a non-polar solvent like hexane they strongly emitted green, in benzene and toluene they emitted yellow, and orange/red in polar solvents such as chloroform, dichloromethane (DCM), 1,4-dioxane, and tetrahydrofuran (THF). On the other hand, in highly polar solvents such as acetonitrile (MeCN), methanol, N,N-methylformamide (DMF), and dimethyl sulfoxide (DMSO), they emitted faint red light (Fig. 2 and Fig. S25–S27†). Calculated quantum yields offered further insights into the emission profile for NPB1–NPB4 in different solvents (Table S2†).
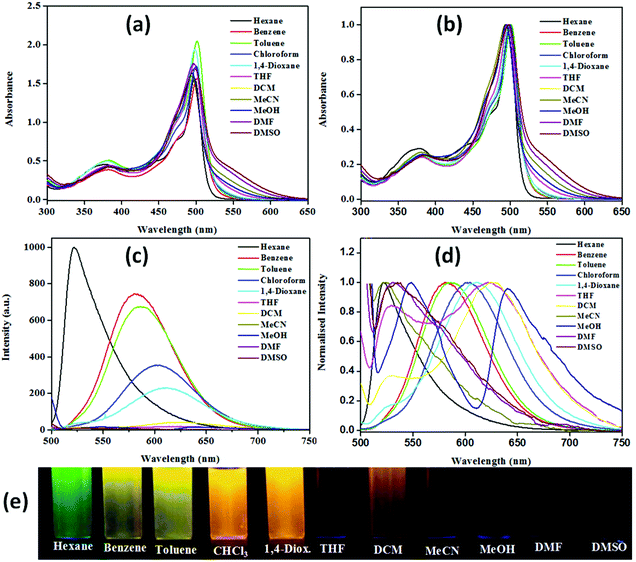 |
| | Fig. 2 Spectra showing the variation of intensity with solvent polarity in different solvents: absorption spectra (a) and its normalized representation (b), emission spectra (c) and its normalized representation (d) and a photograph showing emission color changes in diverse solvents under UV irradiation (λex, 365 nm) (e) for NPB4 (c, 50 μM, THF; λex, 498 nm) [1,4-diox. = 1,4-dioxane, THF = tetrahydrofuran, DCM = dichloromethane, MeCN = acetonitrile, MeOH = methanol, DMF = N,N-methylformamide, DMSO = dimethylsulfoxide]. | |
The plot of emission intensity vs. solvent polarity parameter (Δf) showed a downward curve, indicating the probability of non-radiative decay and charge transfer states in polar solvents (Fig. 3a and Fig. S28†).1b In addition, the excitation spectra monitored at LE emission did not match with the absorption spectra, which also reveals the existence of charge transfer states (Fig. 3b and Fig. S29†).12a
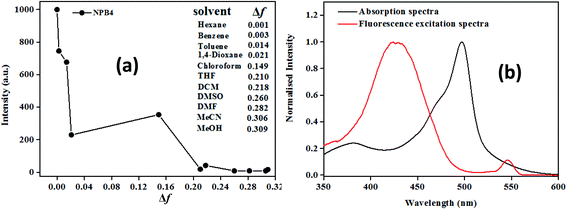 |
| | Fig. 3 Plot of emission intensity vs. solvent polarity parameter (a) and fluorescence excitation spectra at λem, 527 nm (b) for NPB4 (c, 50 μM, THF; λex, 498 nm). | |
The substantial fluorescence solvatochromism (∼125 nm) shown by the conjugates indicated the possibility of a polar excited state.6c–e Piperazinyl nitrogen in NPB1–NPB4 upon interaction with light may induce charge transfer via delocalization of the electron density onto the adjacent acceptor units.6d–e The resulting excited species may stabilize itself via interaction with solvent molecules and single bond rotation causing a TICT state in polar solvents. Thus, intramolecular rotation may bring LE to the TICT state enforcing luminogens to show a large Stokes shift with a low intensity.6c–e,1b To confirm the occurrence of the TICT, the hydrophobicity effect has been examined using a THF/hexane mixture with varying hexane fractions (fh). Increasing fh from 0 to 90% led to a persistent blue shift for the TICT band with an increase in emission intensity while LE emission showed insignificant changes. Furthermore, at fh of 99%, the emission band associated with LE prevailed while the band due to TICT completely vanished (Fig. 4a, Fig. S30 and Table S3†). This observation is in agreement with the occurrence of the TICT state.12a,6f Temperature-dependent studies on emission behaviour for NPB1–NPB4 also offered valuable information about the hydrophobic effect and TICT. As depicted in Fig. 4b, and Fig. S31–S32 in the ESI,† at ∼0 °C the emission spectra showed a single band at ∼525 nm [NPB1, 520; NPB2, 522; NPB3, 530; NPB4, 530 nm] due to LE emission. Upon raising the temperature to 10 °C, a new band emerged at ∼640 nm [NPB1, 644; NPB2, 640; NPB3, 635; NPB4, 635 nm] due to TICT. A further increase in temperature (10 to 60 °C) led to a continual intensity enhancement for the TICT band and emission maxima blue shifted (Table S4†). This type of behaviour in polar solvents due to the temperature effect is usually observed in systems exhibiting TICT and strongly supported the existence of the TICT state.6c,13
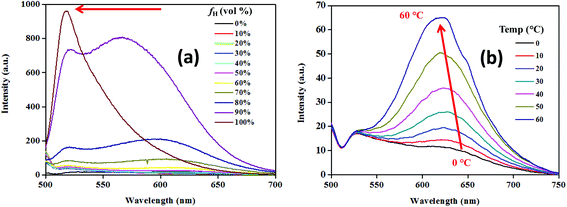 |
| | Fig. 4 Emission spectra with increasing hexane volume fraction (a) and temperatures (b) for NPB4 (c, 50 μM, THF; λex, 498 nm). | |
Aggregation-induced emission
Solvent-dependent emissive behaviour for NPB1–NPB4 encouraged us to investigate their emission properties due to aggregation. In this direction, fluorescence experiments have been performed in a THF/water mixture. These conjugates emit weakly in pure THF solution (c, 50 μM). Gradual addition of water (fw, up to 70%) does not cause any apparent changes; however, at a high water fraction (fw >70 to 99%) the emission intensity radically enhanced with a large red shift which is a typical feature of AIE (Fig. 5b, Fig. S34–S35 and Table S5†).4a,b,12 The quantum yield at the maximum water fraction (99%) provided a quantitative picture of AIE and fell in the range of 7–15.5% [ΦF = 7%, NPB1; 9.7%, NPB2; 13.1%, NPB3; 15.5%, NPB4], much higher relative to those in THF (∼0.8%) (Table S2†). In addition, emission maxima showed a red shift of ∼125 nm [NPB1, 131; NPB2, 123; NPB3, 116; NPB4, 113 nm] due to the aggregate formation (Table S5†).4a The absorption spectra of these conjugates do not show major changes up to a fw of 80% but at higher fw (≥90%) a levelling-off tail emerged in the visible region most likely due to Mie scattering usually observed in nanoaggregate suspension.12 The changes in the absorption spectrum at fw ≥ 90% are consistent with the observations made in emission spectral studies and supported the formation of nanoaggregates (Fig. 5a, Fig. S33, and Table S5†).4b,12 The size of the aggregates has been confirmed by dynamic light scattering (DLS) measurements which revealed average sized nanoaggregates [NPB1, 352.6; NPB2, 273.2; NPB3, 151.5 and NPB4, 166.7 nm] at a fw of 99% (Fig. S36†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) studies further unveiled the formation of nanoaggregates in an aqueous mixture (Fig. S37 and S38†).6a
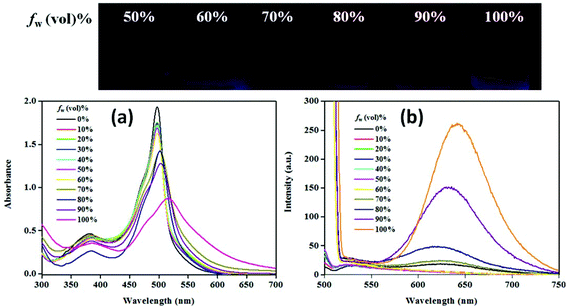 |
| | Fig. 5 Absorption (a) and emission (b) spectra for NPB4 in a THF/water mixture with increasing water volume fractions (c, 50 μM, THF; λex, 498 nm). | |
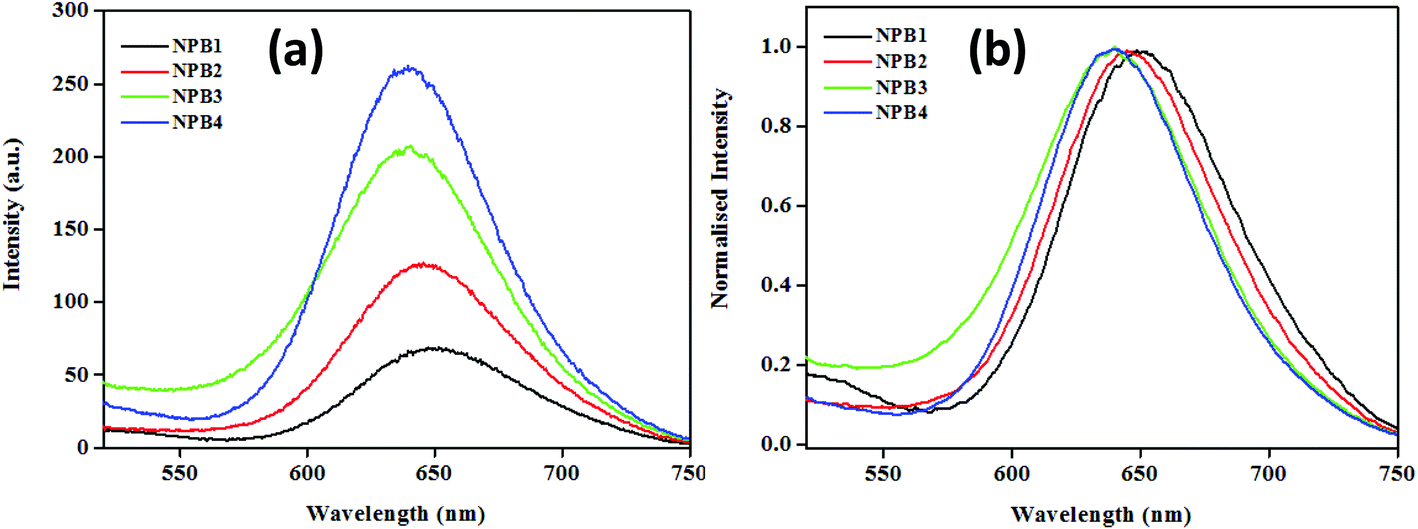
The conjugates under study upon aggregation exhibited AIE to varying extents (NPB1, 3.25; NPB2, 9; NPB3, 11.39; and NPB4, 16.25 times) as evidenced by fluorescence enhancements. Among these, NPB4 showed superior AIE relative to other derivatives probably due to the presence of a long alkyl chain as it may cause an increase in hydrophobicity to a greater extent, thereby promoting aqueous aggregation.14 A comparison of AIE for these conjugates revealed that upon complete aggregation (∼99%, fw) emission maxima for the methyl derivative red shifted to a greater extent and the intensity was lower, while the octyl derivative showed a small red shift and enhanced intensity (Fig. 6 and Table S5†). As AIE and TICT compete with each other and the methyl (NPB1) derivative is less hydrophobic relative to octyl (NPB4), TICT may dominate in NPB1 compared to NPB4 and may cause large red-shifted emission. These offer a reasonable correlation between AIE, TICT and alkyl chain lengths.
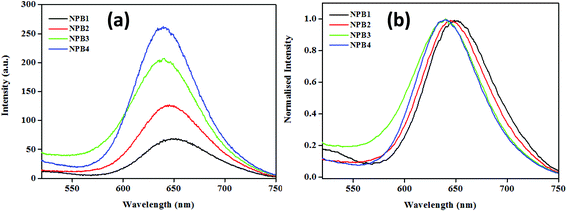 |
| | Fig. 6 Comparison of emission spectra (a) and its normalized representation (b) for NPB1–NPB4 in the aggregated state at a fw of 99% (c, 50 μM, THF; λex, 498 nm). | |
Mechanism of aggregation-induced emission
After scrutinizing AIE, an attempt has been made to investigate its mechanism. According to the literature, restriction of the intramolecular bond rotation (i.e., RIR) of units in any system causes significant emission enhancements due to the physical restraint on the nonradiative relaxation channel and thereby makes ample contributions towards AIE.4a To confirm the role of RIR in AIE, viscosity experiments have been performed using a methanol/glycerol mixture (c, 50 μM, 0.5 vol% THF) with a varying glycerol fraction (fg) (Fig. 7 and Fig. S39 and Table S6†).4b The emission intensity and wavelength showed negligible changes up to a fg of 60%; however, a further increase in fg led to a continual intensity enhancement and ultimately a dramatic increase in emission intensity at a fg of 90%. NPB1–NPB4 showed fluorescence enhancements in the red region (∼646 nm) with a large Stokes shift of up to 147 nm [NPB1, 146; NPB2, 145; NPB3, 147 and NPB4, 147 nm] at a fg of 90% (Table S6†). These enhancements may be related to RIR that makes ample contribution towards AIE.6c,12 Besides, emission enhancement may also be significantly influenced by PET and piperazine conformational changes. In a medium with lower viscosity, piperazine freely undergoes changes (boat/chair form) and can adopt a conformation wherein the nitrogen atoms are close to each other and favour the highest PET and effective fluorescence quenching. But in a medium with high viscosity, aggregated or crystalline state, such changes are restricted and piperazine may adopt a more stable chair conformation wherein the nitrogen atoms lie away from each other (as evident from the crystal structure, vide-infra) and may not favour PET and ultimately enhance the emission intensity.7d,e The PET and TICT operate together in the same direction and in a low viscous medium; the former brings down the emission intensity in a manner analogous to the TICT state.7e
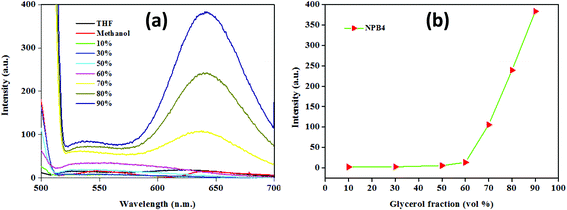 |
| | Fig. 7 Emission spectra in a methanol/glycerol mixture (a) and the plot of emission intensity changes (b) with increasing glycerol volume fractions for NPB4 (c, 50 μM, THF; λex, 498 nm). | |
The above results suggested that an increase in the viscosity of the solvent system inhibits intramolecular rotation and conformational flipping and thereby suppresses non-radiative pathways and causes an increase in the fluorescence intensity.4b,12
Time-resolved fluorescence spectroscopy
To gain deep insight into the AIE behaviour of NPB1–NPB4, the time-resolved emission decay profile has been acquired in THF/water. The conjugates under study showed a weak fluorescence in THF and a THF/water mixture up to a fw of 70% and their lifetimes (τ) came out to be small and beyond the detection limit of the instrument. However, in the aggregated state (fw, 80–99%) they exhibited decay via two relaxation pathways with a continual fluorescence lifetime enhancement as fw increased from 80 to 99% (Fig. 8, Fig. S40 and Table S7†).1b,4b An enhanced lifetime at high water fraction indicated the lowering of the non-radiative channels and high emission intensity in THF/water due to the restriction of the intramolecular rotation via aggregate formation supporting AIE.1b,12 Conversely, the diminished lifetime at a low water fraction (up to 70%) indicated rapid non-radiative decay favouring the TICT process. Increasing the water fraction raises the polarity of the media and enhances TICT causing quenching due to fast non-radiative decay and a lowered fluorescence lifetime.1b,12a
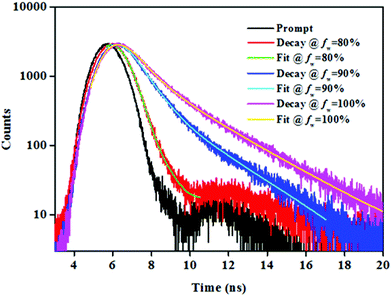 |
| | Fig. 8 Time-resolved emission decay profiles at 639 nm with varying fw for NPB4 (c, 50 μM, THF). | |
Solid-state fluorescence
As shown in Fig. 9, the solid-state emission spectra of NPB1–NPB4 showed a relatively broad band at ∼650 nm [NPB1, 661; NPB2, 640; NPB3, 640; NPB4, 630 nm] with a good quantum yield [NPB1, 9.25%; NPB2, 10.21%; NPB3, 12.76%; NPB4, 13.39% (Table S2†)]. On going from solution to the solid-state, the emission maxima showed an appreciable red shift [NPB1, 141; NPB2, 118; NPB3, 114; NPB4, 104 nm] and followed the trends observed with AIE.6a In the solid-state, the intramolecular rotation and conformational flexibility of the compounds get completely restricted causing fluorescence revival.15
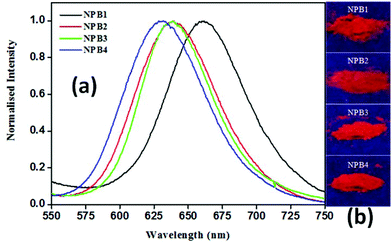 |
| | Fig. 9 Normalised solid-state emission spectra (a) and photographs (b) of powder samples under UV irradiation (λex, 365 nm) for NPB1–NPB4. | |
Crystal structure and packing
To gain deep insight into the emission enhancement in the aggregated and solid-states of these conjugates, the structures of NPB1, NPB3, and NPB4 have been determined by X-ray single crystal analyses. Suitable crystals were obtained by slow evaporation of a DCM–methanol solution of the respective compounds (Fig. 10). The resulting crystallographic data and refinement parameters are summarized in Tables S8–S11.† Quality crystals of NPB3 could not be obtained; however, an attempt has been made to solve and refine the structure from the data acquired on this system too. The structure has been refined to a good approximation signifying intermediate π-interactions in this molecule too, relative to NPB1 and NPB4. The unit cell of NPB1 contains two independent molecules (A and B) with different dihedral angles while the unit cells of NPB3 and NPB4 possess single molecules.
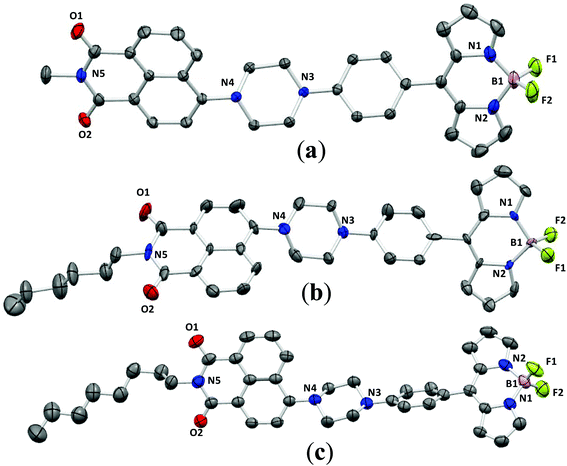 |
| | Fig. 10 ORTEP views of NPB1 (a), NPB3 (b) and NPB4 (c) at 40% ellipsoidal probability (hydrogen atoms have been omitted for clarity). | |
An examination of the dihedral angles (Table S11†) revealed that they adopted a highly twisted non-planar conformation which can prevent intermolecular π–π stacking and creation of adverse excimers/exciplexes responsible for ACQ.6c It is assumed that the twisted conformations of NPB1–NPB4 favor active intramolecular rotation in solution and strengthen non-radiative channels making them non-emissive in solution. But in the aggregated state, intramolecular rotations are strictly restricted and cause fluorescence recovery.6c
Crystal packing analyses for NPB1, NPB3, and NPB4 revealed that amongst these, NPB1 possesses a maximum number (six) of relatively short π–π interactions. Therefore, the molecules of NPB1 are forced to pack closely in a head-to-tail fashion creating a dimeric structure responsible for the lowest AIE and most red-shifted emission (Fig. 11). Only one π–π interaction between the molecules of NPB3 due to relatively greater crowding of the hexyl relative to the methyl chain causes this molecule to show superior AIE relative to NPB1. Conversely, NPB4 lacked any π–π interaction due to a specific orientation and steric hindrance from a long octyl chain. These factors collectively kept NPB4 away from close packing and provided the maximum AIE. Other intermolecular interactions like C–H⋯π, C–H⋯H–C, C–H⋯O, C–H⋯F, and B–F⋯C are believed to be responsible for exerting physical restraints on intramolecular rotation and lead to ordered aggregates making the luminogens more emissive in the aggregated state (Fig. S52–S60†).3c,16
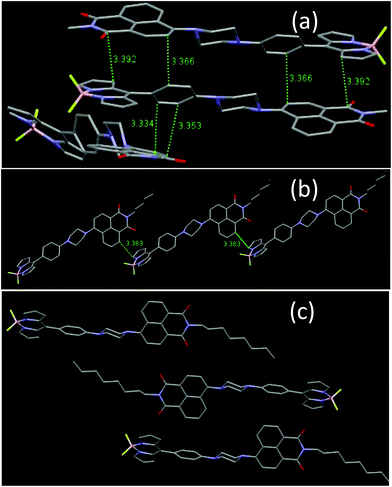 |
| | Fig. 11 Comparison of intermolecular π–π stacking in NPB1, NPB3 and NPB4. | |
Theoretical considerations
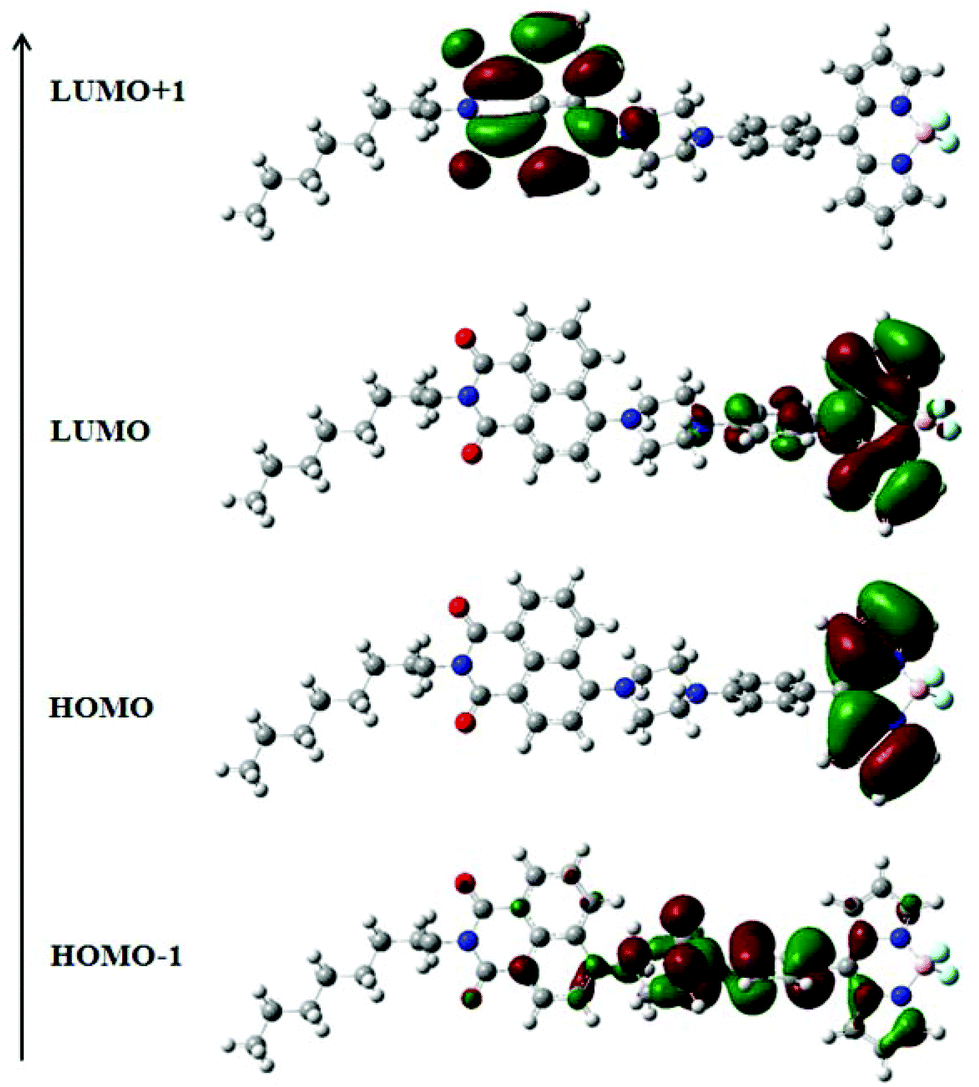
The electronic structures of NPB1–NPB4 have been explained by DFT using the (B3LYP 6-31G**) method. The resulting data are gathered in Fig. 12, Fig. S41–S42 and Tables S9, S10 and S12 in the ESI.† Computational studies on these molecules clearly indicated that the HOMO is mainly concentrated on the BODIPY core and the LUMO over the aryl-BODIPY moiety (Fig. 12 and Fig. S42†). Considering the poor information available from the HOMO and LUMO itself, the results have further been scrutinized using the distribution of HOMO−1 and LUMO+1. It can be seen that HOMO−1 is mainly distributed over the phenyl piperazine unit which serves as an actual donor, whereas LUMO+1 is associated with the naphthalimide core. Hence, the LUMO and LUMO+1 are spread over BODIPY and the naphthalimide core, respectively, and support the acceptor nature of these units.12a In this type of D–A construct, one can categorically say that charge-transfer transitions are not only contributed by the HOMO–LUMO transitions alone but also other possible transitions arising from HOMO−1 and LUMO+1 make important contributions.12a Time-dependent (TD) DFT calculations have also been performed on NPB1–NPB4 and TD-DFT simulated UV/vis spectra are comparable to the experimental results (Fig. S43 and S44, and Table S13†).12a
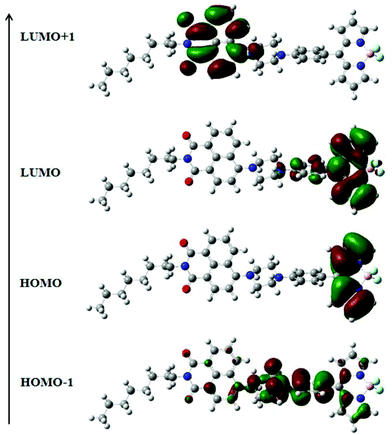 |
| | Fig. 12 Frontier molecular orbitals of NPB4 obtained from DFT calculations. | |
pH sensing and selective lysosome tracking
Conjugates NPB1–NPB4 fulfill a typical molecular format “fluorophore–spacer–receptor (one or more N/O containing functional groups)” for PET; thus they may find potential applications in the detection of protons in chemical and biological systems.2b
As shown in Fig. 13 and Fig. S45 in the ESI,† the pH sensitivity of the conjugates has been examined under aqueous conditions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, ACN/buffer, v/v). At basic and neutral pH, these fluoresce weakly due to PET from the strong donor piperazinyl nitrogen to the fluorophore unit, but at acidic pH, the fluorescence intensity enhanced significantly. A large emission enhancement at acidic pH may be attributed to the gradual protonation of piperazinyl nitrogen inhibiting PET, leading to the recovery of the quenched fluorescence.2b,8 Notably, they showed a significant increase in fluorescence intensity [NPB1, 32; NPB2, 56.4; NPB3, 32; NPB4, 58-fold] in the pH range of 7.0–3.0, indicating their ability to serve as a selective sensor for acidic pH.9 However, at a low pH (<3.0) only a small increase in emission intensity was observed; therefore, it is concluded that they are more sensitive probes in the pH range of 6–3. The sensitivity of the probes NPB1–NPB4 toward pH has also been investigated by UV/vis spectral studies which did not show significant changes (Fig. 13 and S45†). Moreover, the photostability of the probes (NPB1–NPB4) was also investigated by UV/vis spectral measurements in DMSO (c, 50 μM). The absorption spectra of the probes were monitored under the irradiation of visible light (400–700 nm) at different time intervals up to 6 h.17 As shown in Fig. S46 in the ESI,† there is no appreciable change in the intensity of the band observed after long-term irradiation of up to 6 h, indicating that these probes are photostable under irradiation with visible light.17
:1, ACN/buffer, v/v). At basic and neutral pH, these fluoresce weakly due to PET from the strong donor piperazinyl nitrogen to the fluorophore unit, but at acidic pH, the fluorescence intensity enhanced significantly. A large emission enhancement at acidic pH may be attributed to the gradual protonation of piperazinyl nitrogen inhibiting PET, leading to the recovery of the quenched fluorescence.2b,8 Notably, they showed a significant increase in fluorescence intensity [NPB1, 32; NPB2, 56.4; NPB3, 32; NPB4, 58-fold] in the pH range of 7.0–3.0, indicating their ability to serve as a selective sensor for acidic pH.9 However, at a low pH (<3.0) only a small increase in emission intensity was observed; therefore, it is concluded that they are more sensitive probes in the pH range of 6–3. The sensitivity of the probes NPB1–NPB4 toward pH has also been investigated by UV/vis spectral studies which did not show significant changes (Fig. 13 and S45†). Moreover, the photostability of the probes (NPB1–NPB4) was also investigated by UV/vis spectral measurements in DMSO (c, 50 μM). The absorption spectra of the probes were monitored under the irradiation of visible light (400–700 nm) at different time intervals up to 6 h.17 As shown in Fig. S46 in the ESI,† there is no appreciable change in the intensity of the band observed after long-term irradiation of up to 6 h, indicating that these probes are photostable under irradiation with visible light.17
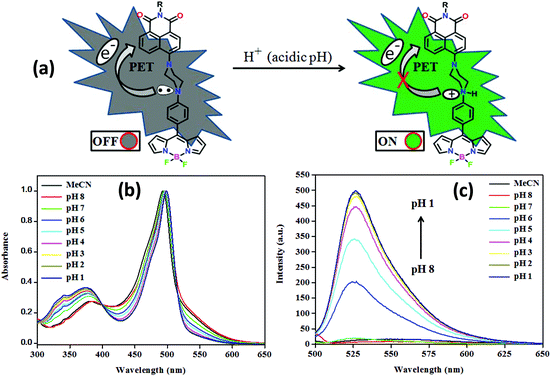 |
| | Fig. 13 A graphical representation of PET ON/OFF mechanism (a), normalised absorption (b), and emission (c) spectra of NPB4 at different pH (PET = photoinduced electron transfer). | |
As this range of pH sensitivity matches well with the lysosomal pH and NPB1–NPB4 shows good photostability in solution, it was presumed that NPB1–NPB4 may find potential applications in bioimaging and lysotracking.9 To realize the efficacy of these systems for bioimaging and lysosomal tracking, live cell imaging experiments have been performed on a human cervical cancer cell line (HeLa). The cell line was procured from the National Center for Cell Science (NCCS) Pune, India. Firstly, the cytotoxicity (cell viability) of NPB1–NPB4 has been examined on HeLa cells by the standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assay over 16 h and 48 h employing concentrations of up to 50 μM.2b,18 As shown in Fig. S47 and S48 in the ESI,† they exhibited marginal toxicity at a rather high concentration (50 μM) and in a long incubation period (48 h). The IC50 was not obtained up to the studied concentration and time period. NPB4 shows the minimum cell viability with approximately 60% of live cells after 48 h at 50 μM while approx. 80% of cells were alive in the case of NPB1. Considering the cell viability results even at high concentrations and in a long incubation time, it is concluded that these conjugates can be safely employed for bio-imaging purpose.2b,8,18 Furthermore, to investigate the bioimaging potential and cellular uptake of NPB1–NPB4, HeLa cells were treated with a 10 μM solution of these compounds for 30 min and imaged under a confocal microscope. The fluorescence signal in Fig. S49† (λex, 488; λem, 500–550 nm) shows good cellular uptake for NPB1–NPB4 and the generality of the cell staining over the whole population highlighting the potential of these compounds as bioimaging agents. A strong green fluorescence signal concentrated in the extranuclear region indicated selective lysosome staining. A strong green signal in the lysosomal region may be related to the restriction of PET in the unaggregated monomeric species of the probe in the acidic environment of lysosomes.2b,8
To confirm the selective staining ability of NPB1–NPB4 for lysosomes, their staining patterns have been compared with commercial LysoTracker stain “LysoTracker™ Red DND-99”.2b As shown in Fig. 14, green fluorescence from NPB1–NPB4 matches well with a red signal of LysoTracker™ Red and both the signals are concentrated in the perinuclear region. Colocalization of the probes NPB1–NPB4 is clearly seen with yellow spots in the merged image. The cellular co-localization was also calculated using Pearson's colocalization coefficient (NPB1, 0.60; NPB2, 0.80; NPB1, 0.88 and NPB4, 0.67) with respect to LysoTracker™ Red, which further confirmed their localization in the same cellular region. To verify their compatibility with other nucleus-specific probes and perinuclear localization, the nucleus-specific stain “Hoechst33342” was used as a counter-stain with lysosome specific NPB1–NPB4.2b As shown in Fig. S50 in the ESI,† they showed superior compatibility with Hoechst and the overlap showed perinuclear distribution which further validated specific lysosomal staining without nuclear uptake. These results indicated that NPB1–NPB4 can be used as pH probes for lysotracking in living cells. To verify the generality of cell staining, 50000 events were recorded using a flow cytometer and the relative intracellular fluorescence intensity of NPB1–NPB4 was compared with that of the control. As shown in Fig. S51 in the ESI,† a clear shift from the fluorescence signal of the control over the entire population verifies the generality of the cell staining using NPB1–NPB4.
 |
| | Fig. 14 Confocal images of HeLa cells after treatment with NPB1–NPB4 and counter staining with “LysoTracker™ Red DND-99”. | |
Conclusion
In summary, through this work, four new piperazine appended BODIPYs NPB1–NPB4 with diverse alkyl chain lengths have been synthesized and thoroughly characterized. The D–A skeleton of the conjugates enabled them to exhibit TICT and PET which has further been explored by AIE and solvatochromism with a large Stokes shift of up to 146 nm. Variable AIE and solid-state emission have been related to crystal packing patterns especially diverse π–π interactions in these conjugates arising from the flexibility of the compounds due to piperazine and alkyl chain lengths. PET induced by the “fluorophore–spacer–receptor” format for NPB1–NPB4 has been exploited toward pH sensitivity and selective lysosome tracking in living cells. These probes show good biocompatibility and potential for long-term monitoring of the lysosomal morphology and can track physiological and pathological changes during various cellular processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from the Science and Engineering Research Board (SERB), New Delhi, through the scheme EMR/2015/001535. B. K. D. acknowledges the University Grants Commission, New Delhi, India, for a Senior Research Fellowship (22/6/2014(i)EU-V).
References
-
(a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed;
(b) R. Hu, C. F. A. Gómez-Durán, J. W. Y. Lam, J. L. Belmonte-Vázquez, C. Deng, S. Chen, R. Ye, E. Peña-Cabrera, Y. Zhong, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 10099–10101 RSC;
(c) T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC;
(d) C. Zhang, J. Zhao, S. Wu, Z. Wang, W. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566–10578 CrossRef CAS PubMed;
(e) T. Bura, N. Leclerc, S. Fall, P. Lévêque, T. Heiser, P. Retailleau, S. Rihn, A. Mirloup and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 17404–17407 CrossRef CAS PubMed.
-
(a) K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC;
(b) Y. Bao, H. De Keersmaecker, S. Corneillie, F. Yu, H. Mizuno, G. Zhang, J. Hofkens, B. Mendrek, A. Kowalczuk and M. Smet, Chem. Mater., 2015, 27, 3450–3455 CrossRef CAS;
(c) H. A. Meylemans, C.-F. Lei and N. H. Damrauer, Inorg. Chem., 2008, 47, 4060–4076 CrossRef CAS PubMed;
(d) C. F. A. Gomez-Duran, R. Hu, G. Feng, T. Li, F. Bu, M. Arseneault, B. Liu, E. Peña-Cabrera and B. Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 15168–15176 CrossRef CAS PubMed;
(e) D. C. Magri, Analyst, 2015, 140, 7487–7495 RSC;
(f) X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
-
(a) M. T. F. Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef PubMed;
(b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC;
(c) M. Yang, D. Xu, W. Xi, L. Wang, J. Zheng, J. Huang, J. Zhang, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2013, 78, 10344–10359 CrossRef CAS PubMed.
-
(a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC;
(c) R. Jiang, M. Liu, C. Li, Q. Huang, H. Huang, Q. Wan, Y. Wen, Q.-Y. Cao, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 708–714 CrossRef CAS PubMed;
(d) V. Kachwal, I. S. V. Krishna, L. Fageria, J. Chaudhary, R. K. Roy, R. Chowdhury and I. R. Laskar, Analyst, 2018, 143, 3741–3748 RSC;
(e) G. Balamurugan, S. Velmathi, N. Thirumalaivasan and S. P. Wu, Analyst, 2017, 142, 4721–4726 RSC;
(f) D. Wei, Y. Xue, H. Huang, M. Liu, G. Zeng, Q. Wan, L. Liu, J. Yu, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 81, 120–126 CrossRef CAS PubMed.
-
(a) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC;
(b) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC;
(c) D. Kolosov, V. Adamovich, P. Djurovich, M. E. Thompson and C. Adachi, J. Am. Chem. Soc., 2002, 124, 9945–9954 CrossRef CAS PubMed;
(d) M. L. Giuffrida, G. T. Sfrazzetto, C. Satriano, S. Zimbone, G. A. Tomaselli, A. Copani and E. Rizzarelli, Inorg. Chem., 2018, 57, 2365–2368 CrossRef CAS PubMed;
(e) K. Xu, L. He, X. Yang, Y. Yang and W. Lin, Analyst, 2018, 143, 3555–3559 RSC.
-
(a) S. Mukherjee and P. Thilagar, Chem. – Eur. J., 2014, 20, 9052–9062 CAS;
(b) P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS PubMed;
(c) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS;
(d) J. Do, J. Huh and E. Kim, Langmuir, 2009, 25, 9405–9412 CrossRef CAS PubMed;
(e) G. Haberhauer, Chem. – Eur. J., 2017, 23, 9288–9296 CrossRef CAS PubMed;
(f) S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC;
(g) S. Mukherjee and P. Thilagar, Chem. – Eur. J., 2014, 20, 8012–8023 CrossRef CAS PubMed.
-
(a) J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS PubMed;
(b) S. Madhu and M. Ravikanth, Inorg. Chem., 2014, 53, 1646–1653 CrossRef CAS PubMed;
(c) S. Doose, H. Neuweiler and M. Sauer, ChemPhysChem, 2009, 10, 1389–1398 CrossRef CAS PubMed;
(d) F. D. Lewis and E. L. Burch, J. Am. Chem. Soc., 1994, 116, 1159–1160 CrossRef CAS;
(e) T. Liu, X. Liu, D. R. Spring, X. Qian, J. Cui and Z. Xu, Sci. Rep., 2014, 4, 5418 CrossRef CAS PubMed;
(f) A. P. d. Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC;
(g) X.-L. Wang, X.-J. Li, R. Sun, Y.-J. Xu and J.-F. Ge, Analyst, 2016, 141, 2962–2969 RSC.
-
(a) K. Zhou, H. Liu, S. Zhang, X. Huang, Y. Wang, G. Huang, B. D. Sumer and J. Gao, J. Am. Chem. Soc., 2012, 134, 7803–7811 CrossRef CAS PubMed;
(b) M. H. Lee, N. Park, C. Yi, J. H. Han, J. H. Hong, K. P. Kim, D. H. Kang, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 14136–14142 CrossRef CAS PubMed.
- S.-L. Shen, X.-P. Chen, X.-F. Zhang, J.-Y. Miao and B. X. Zhao, J. Mater. Chem. B, 2015, 3, 919–925 RSC.
-
(a) C. Zhang, Z. Liu, Y. Li, W. He, X. Gao and Z. Guo, Chem. Commun., 2013, 49, 11430–11432 RSC;
(b) C. Peebles, P. M. Alvey, V. Lynch and B. L. Iverson, Cryst. Growth Des., 2014, 14, 290–299 CrossRef CAS PubMed;
(c) X. Feng, T. Zhang, J.-T. Liu, J.-Y. Miao and B.-X. Zhao, Chem. Commun., 2016, 52, 3131–3134 RSC;
(d) C.-H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427–11440 CrossRef CAS.
-
(a) S. Madhu, M. R. Rao, M. S. Shaikh and M. Ravikanth, Inorg. Chem., 2011, 50, 4392–4400 CrossRef CAS PubMed;
(b) Y. Tian, F. Su, W. Weber, V. Nandakumar, B. R. Shumway, Y. Jin, X. Zhou, M. R. Holl, R. H. Johnson and D. R. Meldrum, Biomaterials, 2010, 31, 7411–7422 CrossRef CAS PubMed.
-
(a) R. S. Singh, S. Mukhopadhyay, A. Biswas and D. S. Pandey, Chem. – Eur. J., 2016, 22, 753–763 CrossRef CAS PubMed;
(b) B. K. Dwivedi, V. D. Singh, R. P. Paitandi and D. S. Pandey, ChemPhysChem, 2018, 19, 1–12 CrossRef PubMed.
- A. Kawski, B. Kukliński and P. Bojarski, Chem. Phys. Lett., 2008, 455, 52–54 CrossRef CAS.
- C. Carayon, A. Ghodbane, L. Gibot, R. Dumur, J. Wang, N. Saffon, M.-P. Rols, K. M. Solntsev and S. Fery-Forgues, Small, 2016, 12, 6602–6612 CrossRef CAS PubMed.
-
(a) T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS PubMed;
(b) Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
-
(a) H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H.-Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287–2294 CrossRef CAS;
(b) X. Zhang, Z. Chi, B. Xu, C. Chen, X. Zhou, Y. Zhang, S. Liua and J. Xu, J. Mater. Chem., 2012, 22, 18505–18513 RSC.
-
(a) M. K. Raza, S. G. A. Garai, K. Mitra, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2017, 56, 11019–11029 CrossRef CAS PubMed;
(b) Q.-Y. Cao, R. Jiang, M. Liu, Q. Wan, D. Xu, J. Tian, H. Huang, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 578–583 CrossRef CAS PubMed;
(c) R. Jiang, H. Liu, M. Liu, J. Tian, Q. Huang, H. Huang, Y. Wen, Q.-Y. Cao, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 81, 416–421 CrossRef CAS PubMed.
-
(a) Q.-Y. Cao, R. Jiang, M. Liu, Q. Wan, D. Xu, J. Tian, H. Huang, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 411–416 CrossRef CAS PubMed;
(b) H. Huang, D. Xu, M. Liu, R. Jiang, L. Mao, Q. Huang, Q. Wan, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 78, 862–867 CrossRef CAS PubMed;
(c) S. Yu, D. Xu, Q. Wan, M. Liu, J. Tian, Q. Huang, F. Deng, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 78, 191–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01390j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Daya Shankar
Pandey
b and
Daya Shankar
Pandey