
Synthesis of nanoporous poly-melamine-formaldehyde (PMF) based on Schiff base chemistry as a highly efficient adsorbent†
Shuaihua
Zhang‡
 ab,
Qian
Yang‡
c,
Xin
Zhou
a,
Zhi
Li
a,
Wenjin
Wang
a,
Xiaohuan
Zang
a,
Chun
Wang
a,
Muhammad J. A.
Shiddiky
d,
Adharvana Chari
Murugulla
e,
Nam-Trung
Nguyen
d,
Zhi
Wang
*a and
Yusuke
Yamauchi
*fg
ab,
Qian
Yang‡
c,
Xin
Zhou
a,
Zhi
Li
a,
Wenjin
Wang
a,
Xiaohuan
Zang
a,
Chun
Wang
a,
Muhammad J. A.
Shiddiky
d,
Adharvana Chari
Murugulla
e,
Nam-Trung
Nguyen
d,
Zhi
Wang
*a and
Yusuke
Yamauchi
*fg
aDepartment of Chemistry, College of Science, Hebei Agricultural University, Baoding 071001, China. E-mail: wangzhi@hebau.edu.cn
bInternational Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: y.yamauchi@uq.edu.au
cCollege of Science and Technology, Hebei Agricultural University, Cangzhou 061100, China
dSchool of Environment and Science and Queensland Micro- and Nanotechnology Centre, Griffith University (Nathan Campus), QLD 4111, Australia
eDr. MACS Bio-Pharma Private Limited, Plot no.79/B & C, EPIP Pashamyalaram, Patancheru Mandal, Sangareddy Dist., Telangana 502307, India
fSchool of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia
gDepartment of Plant & Environmental New Resources, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 446-701, South Korea
First published on 6th November 2018
Abstract
This study proposes the construction of nanoporous poly-melamine-formaldehyde (PMF) through the Schiff base condensation reaction of paraformaldehyde and melamine. The PMF nanoparticles showed a good adsorption capability to some benzene-ring-containing dyes including acid fuchsine, nigrosine, and methyl orange. Moreover, the as-prepared PMF nanoparticles were employed as the coating adsorbent for the solid-phase microextraction (SPME) of seven volatile fatty acids (VFAs) with high enrichment factors. A PMF-assisted SPME method was established for the enrichment of VFAs from environmental water samples with satisfactory recoveries (88.5%–102.0%) and acceptable precisions (relative standard deviations <10.9%). This contribution might furnish an advanced benchmark for the exploitation of new porous organic polymers as the effective adsorbents for SPME or other fields of utilization.
Introduction
There has been a growing tendency for the exploitation of novel highly efficient adsorbent materials for sample pretreatment processes including sampling, clean-up, preconcentration and separation, because the characteristics of adsorbents are very important to the selectivity, sensitivity and precision of the specific analytical methods.1 Porous materials play crucial roles in the sample pretreatment techniques such as solid-phase extraction, magnetic solid-phase extraction and solid-phase microextraction (SPME).2 Traditional inorganic porous materials, such as activated carbon3 and zeolites,4 have exhibited good enrichment capacity for diverse analytes. Nevertheless, the selectivity and practicability of these inorganic materials are still confronted with some issues since their limited synthetic diversifications and wide pore size distributions.2a The inorganic–organic hybrid porous materials such as metal–organic frameworks (MOFs),5 featured by tunable nanoscale porosity, high adsorption ability along with good “molecular sieving effect”1,6 towards target analytes, are crystalline porous coordination polymers by the self-assembly of organic ligands with metal ions (or metal clusters).5 MOFs have been demonstrated with numerous merits and diverse applications in analytical chemistry,6b however, the insufficient chemical stability of some MOFs in moisture or solvents due to their weak coordination bonds7 still limits their further practical application in sample pretreatment techniques.As a new generation of porous materials, porous organic polymers (POPs) latterly attracted a significant interest owing to their remarkable characteristics such as large surface area, well-defined porosities, good chemical resistance and thermostability.8 By adopting different categories of synthetic reactions, POPs can be subdivided into hypercrosslinked polymers (HCPs),9 polymers intrinsic microporosity (PIMs),10 conjugated microporous polymers (CMPs),11 covalent-organic frameworks (COFs),12 and porous aromatic frameworks (PAFs),13etc. Recently, Tan and co-workers14 reported the construction of a novel melamine-based POP, termed as poly-melamine–formaldehyde (PMF), based on the Schiff base condensation of low-cost and abundant melamine and paraformaldehyde. The PMF nanoparticles featured with a good micro- and meso-porosity, high percentage of aminal (–NH–CH2–NH–) and triazine functional groups, and has demonstrated high activities in catalysis14a and adsorption (e.g., heavy metal14b and CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14c). However, in sharp contrast to the excellent properties of PMF, the applications of this promising material as the adsorbents in the sample pretreatment techniques still remain unexploited.
14c). However, in sharp contrast to the excellent properties of PMF, the applications of this promising material as the adsorbents in the sample pretreatment techniques still remain unexploited.
In this contribution, the PMF nanoparticles were prepared through a solvothermal synthesis method based on Schiff base chemistry. The as-synthesized PMF nanoparticles emerged a fast absorptivity towards some phenyl-dyes such as acid fuchsine, nigrosine, and methyl orange. For the first time, the PMF nanoparticles have been utilized as SPME coating for the enrichment of volatile fatty acids (VFAs) with the carboxyl groups. VFAs are important malodorous compounds, which assumed to act as carbon sources for anaerobic bacterial fermentation in food15 or water samples.16 Furthermore, a PMF-assisted SPME method combined with gas chromatography-mass spectrometry (GC-MS) detection was developed for the analysis of VFAs from the environmental water samples.
Experimental
Materials and reagents
Melamine (99.0%) and paraformaldehyde (95.0%) were obtained from Makclin Biochemical Co., Ltd (Shanghai, China) for the synthesis of PMF nanoparticles without further purification. Dyes including acid fuchsine (>70%), nigrosine (water soluble, >98.0%), and methyl orange (96.0%) were bought from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). The seven VFAs standards: acetic acid (ACA, 99.8%), propionic acid (PPA, 99.5%), isobutyric acid (IBA, 99%), butyric acid (BTA, 99%), isovaleric acid (IVA, 99%), valeric acid (VLA, 99%) and hexanoic acid (HAA, 99%) were purchased from Makclin Biochemical Co., Ltd (Shanghai, China). The standard mixture stock solution of VFAs was configured in acetone (concentration, 100 μg L−1) and maintained at 4 °C in the darkness. For the method optimization and establishment, the solutions at various concentrations of the VFAs were produced by the appropriate dilutions of their homologous stock solutions with double-distilled water. Other chemical reagents including sodium chloride (NaCl, 99.5%), anhydrous dimethyl sulfoxide (DMSO, >99.9%), dichloromethane (CH2Cl2, 99.5%) and tetrahydrofuran (THF, >99.0%), all of analytical grade, were from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Neutral multifunctional silicone sealant was obtained in the local building materials market (Baoding, China). The stainless steel wire substrates were purchased from High Pigeon Industry & Trade Co., Ltd (Shanghai, China).Instrumentation
The scanning electron microscopy (SEM) images of the PMF nanoparticles and PMF-coated SPME fibers were obtained on a Hitachi S4800 instrument at an accelerating voltage of 3.0 kV (Tokyo, Japan). Transmission electron microscopy (TEM) characterization was conducted on a JEM-2010HR TEM instrument operated at 200 kV (JEOL, Tokyo, Japan). Fourier transform infrared spectra (FT-IR) was measured on a Bruker Alpha FT-IR spectrometer (Ettlingen, Germany). The nitrogen adsorption–desorption isotherm was obtained by a Quantachrome Autosorb-iQ Automated Gas Sorption System at 77 K (Quantachrome Instruments, Florida, USA) and the surface area of the PMF nanoparticles was estimated based on the Brunauer–Emmett–Teller (BET) method. Thermogravimetric analyser (Henven HCT-2, Beijing, China) was applied to evaluate the thermal stability of the PMF nanoparticles coating from ambient temperature to 600 °C under N2 atmosphere. A UV/Vis spectrometer (UV-1780, Shimadzu, Japan) was utilized for the quantitative analysis of dye adsorption experiments.The SPME performance of the PMF-coated fiber was executed with an Agilent 7820A-5977E GC-MS (Santa Clara, CA, USA) equipped with a DB-WAX capillary column (30 m × 0.25 mm i.d. × 0.25 μm film thickness, Agilent J&W Scientific, CA, USA). The Helium with the purity higher than 99.999% was utilized as the carrier gas with the flow rate at 1.2 mL min−1. The instrumental conditions for VFAs were as follows: splitless mode; injector temperature, 280 °C; the GC programmed temperature, 50 °C maintained for 1 min, then raised to 160 °C at the heating rate of 5 °C min−1, finally increased at 50 °C min−1 to 260 °C. The total running time was 25.0 min. The mass spectrometer was operated in the electron ionization mode (70 eV). The ion source, quadruple and GC-MS interface temperatures were set at 230 °C, 150 °C and 250 °C, respectively. The MS was performed in the full-scan mode with the m/z ranging from 50 to 300 for the analysis of VFAs while the data acquisition was conducted in the selected ion monitoring (SIM) mode to achieve a higher selectivity and sensitivity.
Synthesis of PMF nanoparticles
The PMF nanoparticles were constructed via a previously reported one-pot catalyst-free method14a with some modifications. As the synthetic scheme illustrated in Fig. 1A, the monomers of paraformaldehyde (0.648 g, 21.6 mmol) and industrially accessible melamine (1.512 g, 12 mmol) were dissolved in a 15 mL of DMSO under magnetic stirring. Then, the above solution of monomers together with the magnetic stir bar was transferred into a 50 mL Teflon-lined autoclave for solvothermal reaction at 120 °C for one hour. The Teflon-lined autoclave was then gingerly moved to a magnetic plate for further stirring for another one hour, followed by heated to 170 °C for 10 h. After dropped to ambient temperature, the white PMF product was filtered and washed with DMSO, acetone, THF and CH2Cl2 sequentially, and dried overnight under vacuum at 80 °C.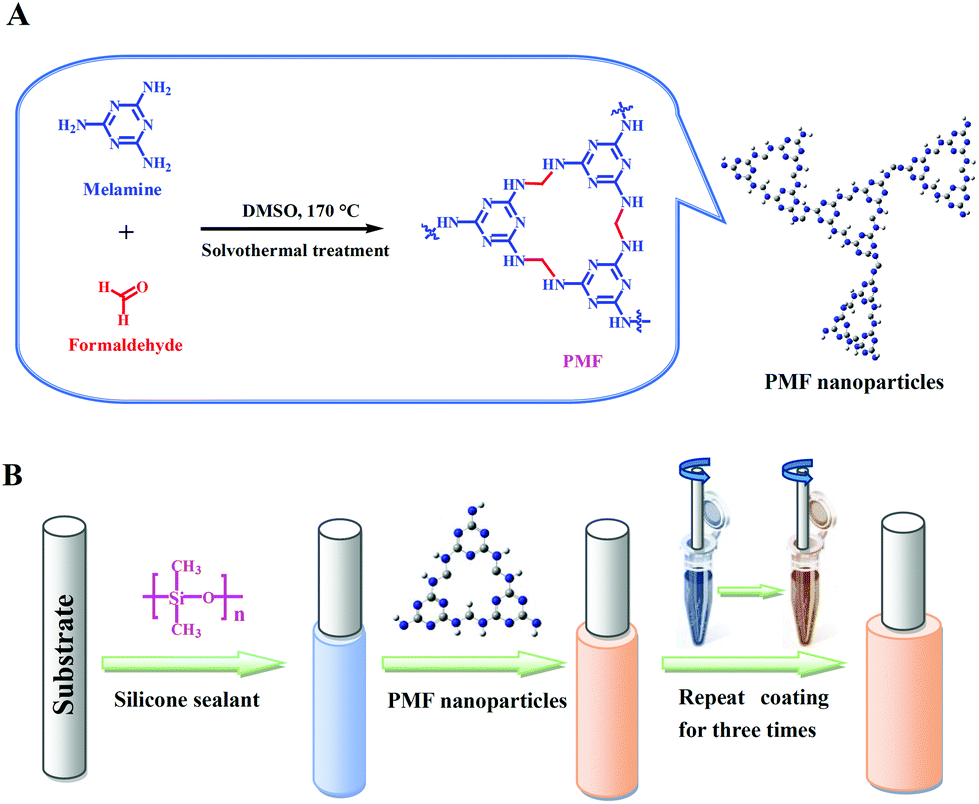 | ||
| Fig. 1 (A) Schematic illustration for the synthesis of the PMF nanoparticles and (B) fabrication of the PMF-coated SPME fiber (B). | ||
Dye adsorption experiments
Twenty milligram of the PMF nanoparticles were dispersed into the dye solutions of acid fuchsine, nigrosine, or methyl orange at the concentration of 20 ppm. After strenuous oscillating for 15 min, the mixture solution was kept for another 5 min and then contrasted with the untreated dye solution to observe the appearance of dye-fading. The mixture solution was subsequently filtered and the collection of filter liquor was used to compare with the untreated dye solution again.Headspace-SPME procedure
The PMF nanoparticles were physically coated onto the stainless steel fiber substrate with the assistance of silicone sealant17 for the construction of the PMF coated fiber (Fig. 1B). Prior to use, the PMF coated fiber was conditioned in the GC-MS injector port (280 °C) for 60 min under Helium atmosphere for the avoidance of carryover.The pond water samples were collected from our campus pond (Baoding, China) and river water samples were obtained from the YiShui River (Baoding, China). Two environmental waters were filtered through a 0.45 μm filter membrane before the following SPME procedure. For headspace (HS)-SPME, 15.0 mL of water sample was dissolved with saturated NaCl in a 25.0 mL sealed-vial and then added with 15 μL of the VFAs standard solution at an appropriate concentration to achieve a spiked water sample for the optimization experiments. The PMF coated fiber was exposed to the headspace of water samples at 30 °C for 30 min of HS-SPME. After the extraction, the fiber was promptly inserted into the injector port of GC at 280 °C for thermal desorption.
Results and discussion
Structural characterization
The PMF nanoparticles were synthesized through a solvothermal method.14a By mixing paraformaldehyde and melamine in hot DMSO, the poly-condensation reaction enables the generation of aminal-based polymer structures (–NH–CH2–NH–). The foamy PMF materials demonstrated an interconnected porous architecture in Fig. S1A,† while the high-magnification SEM (Fig. S1B and C†) and TEM (Fig. S1D–F†) images further showed that the PMF materials were composed of spherical nanoparticles with nanoscale-sizes. Furthermore, with the assistance of silicone sealant, the PMF nanoparticles were uniformly coated on the stainless steel wire substrate (Fig. 2A). The PMF nanospheres in Fig. 2B, with the sizes ranging from 20 to 50 nm, are aggregated and interconnected together, which allows the construction of a loosely spumescent framework with porous architecture.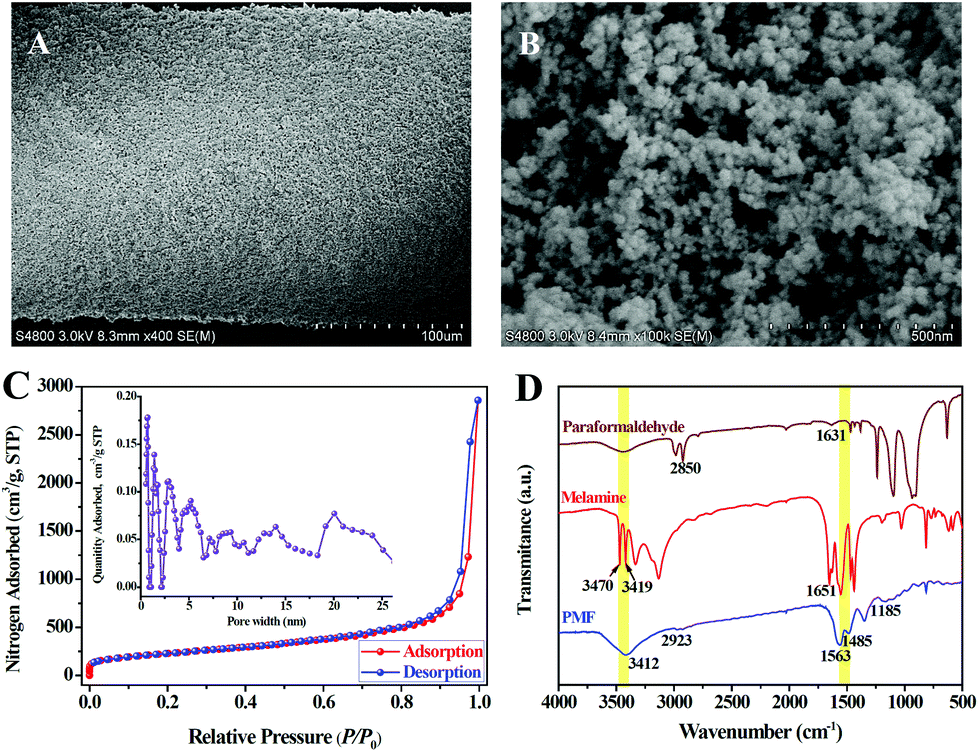 | ||
| Fig. 2 (A, B) Scanning electron microscopy images of the PMF-coated fiber at different magnifications, (C) the nitrogen adsorption–desorption isotherm of the PMF nanoparticles (the inset picture is the pore size distribution calculated using the NLDFT method), and (D) FT-IR spectrums of paraformaldehyde, melamine and PMF nanoparticles. | ||
The nitrogen adsorption–desorption isotherms were further used to investigate the porous structure of PMF nanoparticles. As revealed in Fig. 2C, the sharp raise of N2 uptake at low relative pressures (P/P0) displays the existence of permanent micropores in PMF nanoparticles. Gradual increase of N2 adsorbed amount in the high relative pressure region indicates the presence of mesopores with broadly distributed pore sizes which originated from the interconnected polymer networks between the PMF nanoparticles.18 Pore size distribution (PSD) of the PMF nanoparticles estimated by non-local density functional theory (NLDFT) method was shown in the internal image of Fig. 2C. This PSD pattern suggested that both micropores and mesopores exist in the PMF nanoparticles. The measured BET surface area and total pore volume was 934.6 m2 g−1 and 2.3 cm3 g−1, respectively, which indicates the potential advantages of the PMF nanoparticles as a high-performance adsorbent.
The FT-IR spectra of paraformaldehyde, melamine and PMF are compared and illustrated in Fig. 2D. For the spectrum of melamine, the characteristic bands of –NH2 stretching at 3470, 3419 cm−1 and –NH2 deformation at 1651 cm−1 are attributed to three –NH2 groups of melamine. While, the characteristic bands of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1631 cm−1 and C–H stretching at 2850 cm−1 represent the aldehyde groups (–CHO) of paraformaldehyde.18 The disappearance or significant reduction in the intensity of these bands shows a successful Schiff base condensation between –NH2 and –CHO groups.18,19 On the contrary, the appearance of a broad and strong band of free N–H stretching at 3412 cm−1, together with the –CH2 stretching band at 2923 cm−1 and the C–N stretching band from secondary aminal –NH– at 1185 cm−1, indicating the presence of free –NH2 and –NH–CH2–NH– groups in PMF.18 In addition, the bands at 1563 and 1485 cm−1 in the spectrum of PMF suggest that the triazine units of melamine monomer are retained in the formation of PMF.18,19 Furthermore, the thermogravimetric (TG) analysis of the PMF nanoparticles coating after conditioned at 280 °C for 60 min indicated that this SPME coating can be stabilized to 350 °C with a weight loss of approximately 2.2% (Fig. S2†).
O stretching at 1631 cm−1 and C–H stretching at 2850 cm−1 represent the aldehyde groups (–CHO) of paraformaldehyde.18 The disappearance or significant reduction in the intensity of these bands shows a successful Schiff base condensation between –NH2 and –CHO groups.18,19 On the contrary, the appearance of a broad and strong band of free N–H stretching at 3412 cm−1, together with the –CH2 stretching band at 2923 cm−1 and the C–N stretching band from secondary aminal –NH– at 1185 cm−1, indicating the presence of free –NH2 and –NH–CH2–NH– groups in PMF.18 In addition, the bands at 1563 and 1485 cm−1 in the spectrum of PMF suggest that the triazine units of melamine monomer are retained in the formation of PMF.18,19 Furthermore, the thermogravimetric (TG) analysis of the PMF nanoparticles coating after conditioned at 280 °C for 60 min indicated that this SPME coating can be stabilized to 350 °C with a weight loss of approximately 2.2% (Fig. S2†).
Rapid dye adsorption
Emerging the remarkable characteristics such as high surface area, good porosity as well as aromatic conjugated architecture, the PMF nanoparticles were demonstrated to have a good and quick dye adsorption capacity. After the dye solution with the addition of the PMF nanoparticles was violently oscillated for 15 min and subsequently kept still for another 5 min, the color of the solution was observed to become shallower (Fig. 3A). Then, the dye solutions were filtered to examine the adsorption results clearly and the filtrates were gathered for comparison with the untreated counterparts. The filtrates became achromatic and clear in Fig. 3B, indicating that the adsorption of dyes were close to completely finish by the PMF nanoparticles. In addition, the absorption kinetics of the dyes was further studied by utilizing UV/vis spectrometry (Fig. S3†) and the removal ratios of the PMF nanoparticles to acid fuchsine, nigrosine, or methyl orange were 92%, 94% and 90%, respectively. As the structures of the dyes illustrated in Fig. S3,† all the three dyes, including acid fuchsine, nigrosine, and methyl orange contained benzene rings, –CC–, or –NN– in their architectures, it was reasonable that the PMF nanoparticles possessing many aromatic triazine-rings showed a rapid adsorption capability towards these benzene-ring-containing dyes. The satisfactory dye removal ratios might be attributed to the π–π stacking effect between these dyes and the aromatic conjugated structure from the PMF nanoparticles.2a
 | ||
| Fig. 3 Dye adsorption images of the PMF nanoparticles before (A) and after (B) filtration. Dye solutions from left to right are acid fuchsine, nigrosine and methyl orange. | ||
SPME applications
In HS-SPME, the extraction temperature has an enormously essential effect on the extraction efficiency of analytes. Elevating temperature could enhance the diffusion coefficients of analytes from aqueous phase to headspace. On the other side, the adsorption onto the SPME coating is regarded as an exothermic process and the extraction efficiency will be decreased under a higher temperature. Herein, the effect of extraction temperature was examined in the range of 25–60 °C with other conditions unchanged. Fig. S4A† exhibited that the peak areas of the VFAs excluding HAA firstly increased as the extraction temperature rose from 25 to 30 °C with the maximum values achieved at 30 °C while HAA was obtained at 35 °C. However, their peak areas gradually dropped when the extraction temperature further increased beyond 30 °C (except for HAA). Consequently, an extraction temperature of 30 °C was adopted for the further experiments.
SPME process takes an effusion of time to achieve the extraction equilibrium between analytes and coating adsorbent. The influence of extraction time on the peak areas of VFAs was investigated in the range of 5–50 min. As illustrated in Fig. S4B,† the peak areas of these VFAs were gradually incremental with the extension of extraction time from 5 min to 30 min but their peak areas no longer increased with a prolonged extraction time after 30 min. Therefore, 30 min of extraction time was selected in subsequent experiments.
Salt concentration is another crucial factor for HS-SPME, which can enhance the ionic strength in aqueous solution, and thus conducive to motivate the analytes from aqueous phase to the headspace. The influence of salt concentration for the extraction efficiency of VFAs was examined by increasing the NaCl concentration from 0 to saturation. The experimental data demonstrated that the increase of NaCl concentration resulted in a favorable positive contribution to the extraction efficiency of all VFAs and the NaCl saturated solution achieved the greatest peak areas. As a result, saturated NaCl solution was used for the subsequent studies.
Both the desorption temperature and time was optimized to ensure rapid and maximum transfer of VFAs from the PMF coating into the GC-MS injection port. During the optimizations of the desorption conditions, the temperature of desorption was examined by ranging from 230 °C to 290 °C while the desorption time varied between 1 and 10 min. According to the experimental results, the response for the VFAs have enhanced with increasing desorption temperature from 230 °C to 280 °C, but no longer increased as the further raise to 290 °C. As shown in Fig. S4C,† the peak areas of VFAs increased with the desorption time from 1 to 5 min, indicating the VFAs were completely desorbed in 5 min. Therefore, the best desorption conditions were 5 min at 280 °C. Besides, the PMF nanoparticles coated fiber could be utilized in more than 120 of adsorption/desorption cycles without remarkable reduction in their extraction efficiencies (Fig. S4D†). Compared with some commercial fibers (40–100 times),20 this PMF nanoparticles coating showed a relatively long lifetime with higher stability and ruggedness.
:1 and 10:1, were within the scope of 0.012–0.040 μg L−1 and 0.04–0.13 μg L−1, respectively. The repeatability of the method was examined by utilization of one single fiber for five replicated HS-SPMEs of VFAs, and the relative standard deviations (RSDs) were ranging from 6.1% to 9.4%. For the assessment of the fiber-to-fiber reproducibility, the RSDs of four different PMF nanoparticles fibers under the same fabricated conditions were in the range of 5.9%–11.5%, indicating that the PMF nanoparticles coating possessed a good repeatability and the coating fabrication procedure was reproducible. In addition, the enrichment factors (EFs) were calculated by the ratios from peak areas of the analytes after SPME to that gained by direct injection of standard solution,1,6a which ranging from 187 to 1089 (Table 1). These high EFs might be ascribed to the acid–base interaction between the carboxyl groups of VFAs and the amine groups of the PMF nanoparticles.1 The PMF nanoparticles provided a considerable electron donors (aminal-groups) as the binding sites and the acidic VFAs with carboxyl groups acted as the acceptors. Therefore, based on the π–π affinity, the nitrogen-rich feature of the PMF coating was expected to facilitate the highly efficient SPME of acidic VFAs.
| VFAs | Linear ranges (μg L−1) | r 2 | LODs (μg L−1) | LOQs (μg L−1) | One fiber repeatabilitya (n = 5, %) | Fiber to fiber reproducibilityb (n = 4, %) | EFs |
|---|---|---|---|---|---|---|---|
| a n = 5 means that one single fiber has been employed for five replicated HS-SPMEs of VFAs. b n = 4 means that four different fibers were prepared under the same batch and used for HS-SPMEs of VFAs. | |||||||
| ACA | 0.13–10.0 | 0.996 | 0.040 | 0.13 | 6.7 | 8.6 | 187 |
| PPA | 0.11–10.0 | 0.994 | 0.033 | 0.11 | 8.5 | 10.9 | 219 |
| IBA | 0.08–10.0 | 0.998 | 0.025 | 0.08 | 7.8 | 9.0 | 309 |
| BTA | 0.07–10.0 | 0.998 | 0.021 | 0.07 | 6.1 | 6.3 | 364 |
| IVA | 0.06–10.0 | 0.996 | 0.019 | 0.06 | 6.7 | 5.9 | 563 |
| VLA | 0.04–5.0 | 0.995 | 0.013 | 0.04 | 8.3 | 9.7 | 912 |
| HAA | 0.04–5.0 | 0.996 | 0.012 | 0.04 | 9.4 | 11.5 | 1089 |
To evaluate the practicality of the PMF-assisted SPME method in real sample analysis, two environmental samples such as pond and river water were analyzed. The results indicated that VFAs was undetected in these environmental water samples. The recoveries of the developed method were studied by spiking the standard VFAs into the waters at three concentration levels, 0.2, 1.0, and 5.0 μg L−1, respectively. As shown in Table 2, the recoveries of the VFAs were in the range of 89.4%–102.0% and 88.5%–101.0% for pond and river water samples, with the corresponding RSDs for five replicated extractions were 4.5%–10.9% and 4.7%–9.2%, respectively. Based on the above results, the PMF-coated SPME fiber exhibited good recoveries for the VFAs, and the established method could be, therefore, applicable for the enrichment of the VFAs in environmental water samples. The GC-MS chromatogram of the river water before and after spiking with the VFAs was illustrated in Fig. 4.
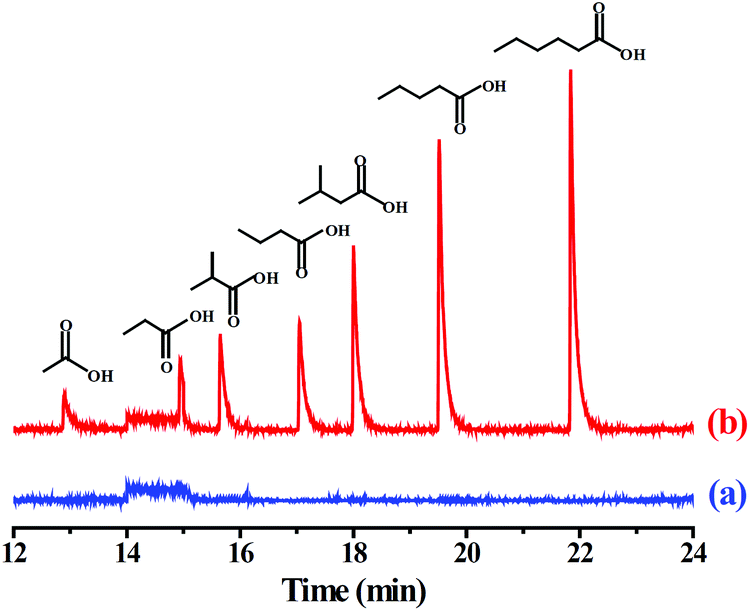 | ||
| Fig. 4 GC-MS chromatograms of the VFAs by the PMF-assisted HS-SPME-GC-MS method for (a) blank river water sample and (b) the river water sample spiked with each of the VFAs at 1.0 μg L−1. | ||
| VFAs | Spiked (μg L−1) | Pond water | River water | ||||
|---|---|---|---|---|---|---|---|
| Found (μg L−1) | Recovery (%) | RSDs (%) | Found (μg L−1) | Recovery (%) | RSDs (%) | ||
| ACA | 0.2 | 0.195 | 97.5 | 8.4 | 0.188 | 94.0 | 8.5 |
| 1.0 | 0.926 | 92.6 | 6.6 | 0.913 | 91.3 | 6.3 | |
| 5.0 | 4.91 | 98.2 | 5.1 | 4.90 | 98.0 | 4.7 | |
| PPA | 0.2 | 0.183 | 91.5 | 9.0 | 0.177 | 88.5 | 9.0 |
| 1.0 | 0.941 | 94.1 | 5.7 | 0.962 | 96.2 | 9.2 | |
| 5.0 | 4.47 | 89.4 | 4.5 | 4.93 | 98.6 | 8.1 | |
| IBA | 0.2 | 0.197 | 98.5 | 8.6 | 0.195 | 97.5 | 8.0 |
| 1.0 | 0.950 | 95.0 | 8.5 | 0.937 | 93.7 | 5.9 | |
| 5.0 | 4.88 | 97.6 | 6.2 | 4.79 | 95.8 | 6.3 | |
| BTA | 0.2 | 0.185 | 92.5 | 7.1 | 0.183 | 91.5 | 6.8 |
| 1.0 | 0.921 | 92.1 | 6.4 | 0.973 | 97.3 | 7.4 | |
| 5.0 | 4.93 | 98.6 | 6.9 | 4.99 | 99.8 | 5.7 | |
| IVA | 0.2 | 0.192 | 96.0 | 7.3 | 0.195 | 97.5 | 7.0 |
| 1.0 | 0.959 | 95.9 | 6.9 | 0.910 | 91.0 | 8.3 | |
| 5.0 | 5.06 | 101.2 | 6.0 | 4.71 | 94.2 | 4.9 | |
| VLA | 0.2 | 0.186 | 93.0 | 5.7 | 0.198 | 99.0 | 6.7 |
| 1.0 | 0.947 | 94.7 | 4.6 | 1.01 | 101.0 | 6.4 | |
| 5.0 | 4.76 | 95.2 | 5.0 | 5.03 | 100.6 | 5.3 | |
| HAA | 0.2 | 0.204 | 102.0 | 10.9 | 0.196 | 98.0 | 7.6 |
| 1.0 | 0.996 | 99.6 | 9.2 | 0.967 | 96.7 | 5.5 | |
| 5.0 | 4.95 | 99.0 | 7.6 | 4.91 | 98.2 | 4.9 | |
Furthermore, Table S1† summarizes the analytical characteristics of some reported SPME methods with different coatings21 such as commercial carboxen/polydimethylsiloxane (CAR/PDMS),21a–c polyacrylate (PA),21d,e divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS),21f or polymeric ionic liquids (PILs),21c SNW-1,1 along with those of the present method for the enrichment and determination of VFAs from the viewpoint of linear ranges, LODs as well as repeatability. As shown in Table S1,† the extraction performance of the PMF-assisted SPME method is comparable1 or superior21 to the other reported SPME methods combined with GC-MS or GC-FID for the determination of VFAs.
Conclusion
The PMF nanoparticles were synthesized through a facile Schiff base condensation and used as the SPME adsorbent for the first time. The as-synthesized PMF nanoparticles demonstrated a fast adsorption capacity to some benzene-ring-containing dyes due to the π–π stacking interaction. The extraction characteristics of the PMF-coated SPME fiber were evaluated by HS-SPME of VFAs as the target analytes. Coupled with GC-MS detection, the PMF-assisted SPME method showed low limits of detection (0.012–0.040 μg L−1) as well as high enrichment superiority (EFs, 187–1089) towards VFAs, which mainly resulted from the acid–base interaction of the sufficient aminal groups of PMF and the carboxyl groups of VFAs. Furthermore, this PMF-coated SPME fiber with a high thermal stability and long lifetime was successfully applied for the enrichment of VFAs from environmental waters with high recoveries and good sensitivities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Foundation of Hebei Agricultural University (ZD201703), the Youth Top-notch Talent Foundation of Hebei Provincial Universities (BJ2018024), the Natural Science Foundation of Hebei Province (C2017204019), and the National Natural Science Foundation of China (31571925 and 31671930). This work was also partly supported by the Australian Research Council (ARC) Future Fellow (FT150100479), JSPS KAKENHI (17H05393 and 17K19044), a collaborative research grant between Dr MACS Bio-Pharma Pvt. Ltd (India) and QMNC, Griffith University, and the research fund by the Suzuken Memorial Foundation.References
- J. Pan, S. Jia, G. Li and Y. Hu, Anal. Chem., 2015, 87, 3373–3381 CrossRef PubMed.
- (a) S. Liu, D. Chen, J. Zheng, L. Zeng, J. Jiang, R. Jiang, F. Zhu, Y. Shen, D. Wu and G. Ouyang, Nanoscale, 2015, 7, 16943–16951 RSC; (b) M. Wu, G. Chen, P. Liu, W. Zhou and Q. Jia, Analyst, 2016, 141, 243–250 RSC; (c) L. Chen and X. Huang, Analyst, 2017, 142, 4039–4047 RSC; (d) S. Yu, Z. Liu, H. Li, J. Zhang, X. Yuan, X. Jia and Y. Wu, Analyst, 2018, 143, 883–890 RSC; (e) H. L. Qian, C. X. Yang, W. L. Wang, C. Yang and X. P. Yan, J. Chromatogr. A, 2018, 1542, 1–18 CrossRef CAS PubMed.
- (a) T. Sun, J. Jia, N. Fang and Y. Wang, Anal. Chim. Acta, 2005, 530, 33–40 CrossRef CAS; (b) S. Lirio, C. W. Fu, J. Y. Lin, M. J. Hsu and H. Y. Huang, Anal. Chim. Acta, 2016, 927, 55–63 CrossRef CAS PubMed.
- (a) J. Zhao, T. Luo, X. Zhang, Y. Lei, K. Gong and Y. Yan, Anal. Chem., 2012, 84, 6303–6307 CrossRef CAS PubMed; (b) E. Fernández, L. Vidal and A. Canals, J. Chromatogr. A, 2016, 1458, 18–24 CrossRef PubMed.
- (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- (a) N. Chang, Z. Y. Gu, H. F. Wang and X. P. Yan, Anal. Chem., 2011, 83, 7094–7101 CrossRef CAS PubMed; (b) Z. Y. Gu, C. X. Yang, N. Chang and X. P. Yan, Acc. Chem. Res., 2012, 45, 734–745 CrossRef CAS PubMed.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F. Coudert, Nat. Chem., 2016, 9, 11–16 CrossRef PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- (a) Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed; (b) L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- (a) N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC; (b) M. Carta and N. B. Mckeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- (a) J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky and Y. Z. Khimyak, Angew. Chem., Int. Ed., 2007, 119, 8574–8578 CrossRef PubMed; (b) A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS; (c) Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed; (b) H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed; (c) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC; (d) J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC; (e) C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed; (f) A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed; (g) T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- (a) T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed; (b) T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC; (c) C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11–21 RSC.
- (a) M. X. Tan, L. Gu, N. Li, J. Y. Ying and Y. Zhang, Green Chem., 2013, 15, 1127 RSC; (b) M. X. Tan, Y. N. Sum, J. Y. Ying and Y. Zhang, Energy Environ. Sci., 2013, 6, 3254 RSC; (c) M. X. Tan, Y. Zhang and J. Y. Ying, ChemSusChem, 2013, 6, 1186 CrossRef CAS PubMed.
- M. J. Trujillo-Rodríguez, H. Yu, W. T. S. Cole, T. D. Ho, V. Pino, J. L. Anderson and A. M. Afonso, Talanta, 2014, 121, 153–162 CrossRef PubMed.
- (a) M. A. Ullah, K. H. Kim, J. E. Szulejko and J. Cho, Anal. Chim. Acta, 2014, 820, 159–167 CrossRef CAS PubMed; (b) M. A. A. Rocha, S. Raeissi, P. Hage, W. M. A. Weggemans, J. van Spronsen, C. J. Peters and M. C. Kroon, Chem. Eng. Sci., 2017, 165, 74–80 CrossRef CAS.
- (a) L. Xie, S. Liu, Z. Han, R. Jiang, H. Liu, F. Zhu, F. Zeng, C. Su and G. Ouyang, Anal. Chim. Acta, 2015, 853, 303–310 CrossRef CAS PubMed; (b) S. Zhang, Q. Yang, Z. Li, W. Wang, C. Wang and Z. Wang, Analyst, 2016, 141, 1127–1135 RSC.
- Y. Wang, Y. Xie, Y. Zhang, S. Tang, C. Guo, J. Wu and R. Lau, Chem. Eng. Res. Des., 2016, 114, 258–267 CrossRef CAS.
- G. Yang, H. Han, C. Du, Z. Luo and Y. Wang, Polymer, 2010, 51, 6193–6202 CrossRef CAS.
- M. Wu, C. Gang, J. Ma, L. Ping and Q. Jia, Talanta, 2016, 161, 350–358 CrossRef CAS PubMed.
- (a) M. Abalos and J. M. Bayona, J. Chromatogr. A, 2000, 891, 287–294 CrossRef CAS PubMed; (b) M. Ábalos, J. M. Bayona and J. Pawliszyn, J. Chromatogr. A, 2000, 873, 107–115 CrossRef; (c) M. J. Trujillo-Rodriguez, H. Yu, W. T. Cole, T. D. Ho, V. Pino, J. L. Anderson and A. M. Afonso, Talanta, 2014, 121, 153–162 CrossRef CAS PubMed; (d) S. J. Olivero and J. P. Trujillo, Anal. Chim. Acta, 2011, 696, 59–66 CrossRef PubMed; (e) A. F. González-Córdova and B. Vallejo-Cordoba, J. Agric. Food Chem., 2001, 49, 4603–4608 CrossRef; (f) D. Fiorini, M. C. Boarelli, R. Gabbianelli, R. Ballini and D. Pacetti, Anal. Biochem., 2016, 508, 12–14 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01623b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |