
A computational study of photonic materials based on Ni bis(dithiolene) fused with benzene, possessing gigantic second hyperpolarizabilities†
Aggelos
Avramopoulos
 *ab,
Nicolás
Otero
cd,
Heribert
Reis
a,
Panaghiotis
Karamanis
c and
Manthos G.
Papadopoulos
*a
*ab,
Nicolás
Otero
cd,
Heribert
Reis
a,
Panaghiotis
Karamanis
c and
Manthos G.
Papadopoulos
*a
aInstitute of Biology, Pharmaceutical Chemistry and Biotechnology, National Hellenic Research Foundation, 48 Vas. Constantinou Ave., Athens 11635, Greece. E-mail: aavram@eie.gr; mpapad@eie.gr
bDepartment of Computer Engineering, Technological Education Institute (TEI) of Sterea Ellada, Lamia 35100, Greece
cCNRS/UNIV Pau & Pays Adour, Institut Des Sciences Analytiques et de Physico-Chimie pour
l'environnement et les Materiaux MIRA*, UMR5254, 64000, Pau and Helioparc Pau Pyrnes 2 avenue du President Angot, 64053 PAU Cedex 09, France
dDepartamento de Química Física, Universidade de Vigo, Vigo, Galicia 36310, Spain
First published on 24th November 2017
Abstract
We propose a family of photonic materials based on nickel bis(dithiolene) [NiBDT] and benzene (B). Linear and planar oligomers of 1D and 2D polymers, together with several rich π-electron linkers (e.g. C14H8, C24H8, and C4O2H4), have been employed in order to demonstrate the superior performance of the proposed materials. A key property for the design of efficient photonic materials is the second hyperpolarizability. We have, thus, computed it together with several other properties, such as the dipole moment, polarizability, first excitation energy, etc. We have also used two indices to estimate the degree of the diradical character of the considered derivatives. The calculations have been performed by employing density functional theory. Considerable care has been taken to validate the computational procedure we have used. The main findings of the present work are as follows: (a) the 1D and 2D coordination polymers based on nickel bis(dithiolene) and benzene and in particular the linear ones, (B-NiBDT)n, are very likely to be efficient photonic materials, since they have gigantic second hyperpolarizability. (b) The second hyperpolarizability greatly depends on the organic linker with which NiBDT is fused and on the structure; for example, the linear oligomers have much higher second hyperpolarizability than the planar ones. The present work together with a recent article (A. Avramopoulos et al., J. Phys. Chem. C, 2016, 120, 9419–9435) demonstrates a mechanism for designing efficient photonic materials.
I. Introduction
There is an ever-increasing need for novel photonic materials for use in a large number of applications (e.g. laser protection,1 devices for all-optical control,2 optical communication, optical signal processing3). Here we propose a novel family of photonic materials: 1D and 2D coordination polymers (nanosheets), based on nickel bis(dithiolene) and benzene. We recently reported that the oligomers (RA-NiBDT)n, RA = radiaannulene, NiBDT = bis(ethylene-1,2-dithiolato)Ni, have giant second hyperpolarizabilities. We found in the literature that Kambe et al.4–7 synthesized 1D and 2D (nanosheets) coordination polymers, based on NiBDT. It was a natural extension of our previous work to compute the (hyper)polarizabilities of some of their models, employing density functional theory. Specifically, we got some fragments of the 1D and 2D polymers and using these linear and planar oligomers, we performed a systematic study of their linear and nonlinear optical properties. We found that some of these oligomers have extremely large nonlinearities, which is one of the prerequisites for an efficient photonic material.In order to demonstrate the superior non-linear optical (NLO) performance of the proposed materials, over related compounds, we have carried out extensive experimentation, which involved NiBDT, fused with a series of rich π-electron organic linkers:
(a) Benzene. This was fused with NiBDT to produce linear and planar oligomers (nanosheets). Extensive experimental work has been reported on the corresponding polymers.4–7 The linear oligomer, symbolically, is denoted as (B-NiBDT)n, where n = 1–6. The planar oligomers have been studied by employing 3 models (Fig. 1). This choice also allows studying the significant effect of the geometry on the properties of interest.
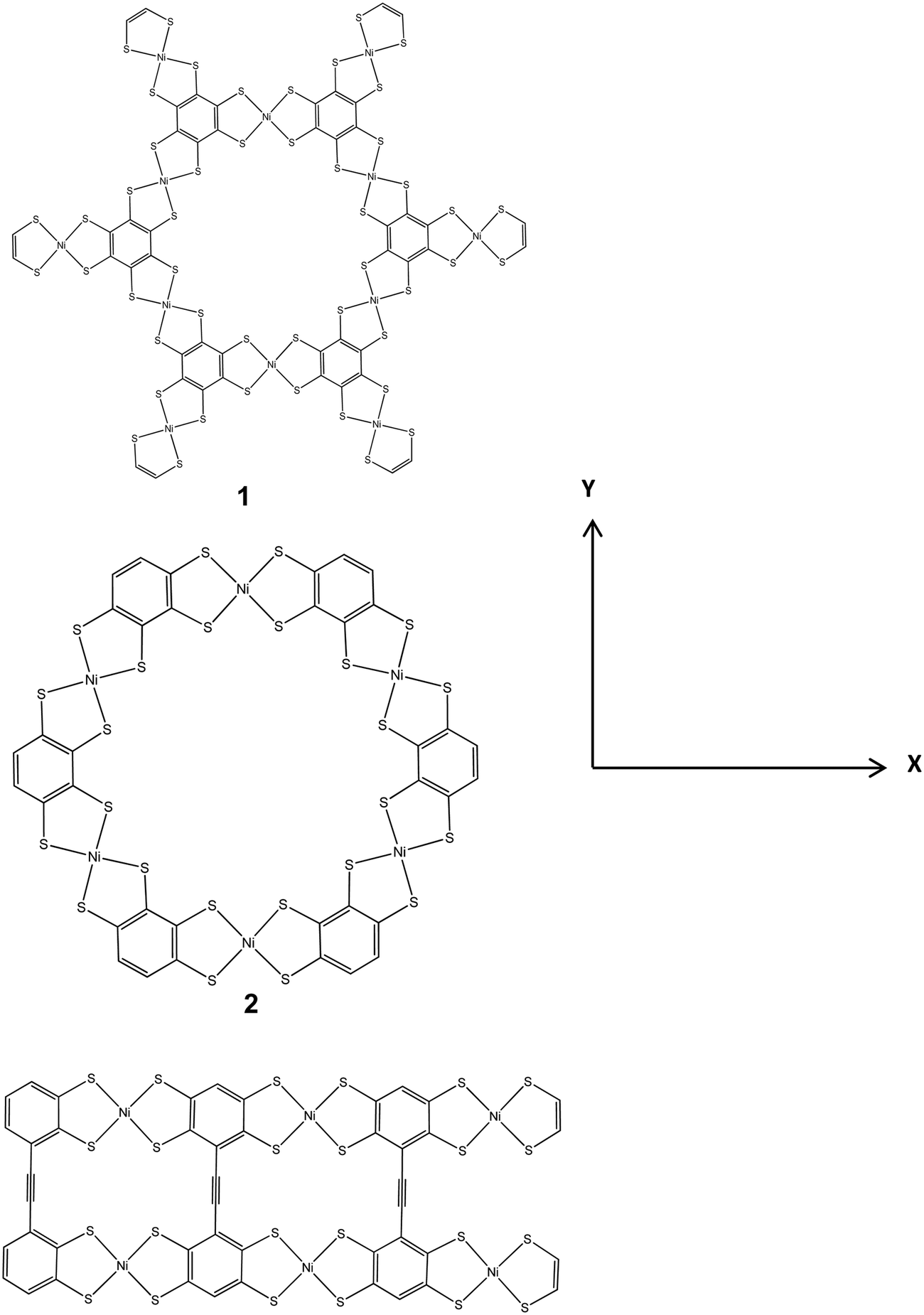 | ||
| Fig. 1 The structures (1–3) of several planar nickel bis(dithiolene) complexes, computed with B3LYP/6-31G*. | ||
(b) Linkers with extensive conjugation. We have used C14H8 (pyracyclene), C24H12 (dicyclopenta(cd,lm)perylene), and C24H8 (this is a radiaannulene, RA). These linkers allow studying the effect of the extensive π-electron network on the properties of interest.
(c) C 4 O 2 H 4 , C 4 S 2 H 4 , C 4 OH 4 , and C 4 SH 4 . We have used a series of small molecules to connect the NiBDT units, the effect of which has been compared with that of benzene. These are C4O2H4 (1,4-dioxin), C4S2H4 (1,4-dithiin), C4OH4 (furan), and C4SH4 (thiophene). Furan and thiophene are aromatic, 1,4-dioxin is non-aromatic and 1,4-dithiin is antiaromatic. It is known that thiophene is an important element of many photonic materials.8
Of major importance in this work are the (hyper)polarizabilities and in particular the second hyperpolarizability. For their rationalization we have used several other properties, such as EHOMO (energy of the highest occupied molecular orbital), ELUMO (energy of the lowest unoccupied molecular orbital), and the first excitation energy. A significant property of our studied compounds is their diradical character (DC). This is one of the major characteristics for understanding the intriguing properties of the NiBDT derivatives. Thus, we have used two indices which estimate, or they are related to, the strength of the DC. This work, in connection with our previous results, demonstrates a mechanism for designing photonic materials with exceptionally large nonlinearities. This mechanism includes a series of units consisting of NiBDT, π-electron rich linkers, Li substituents9 and the appropriate structure, for example a linear arrangement leads to a much larger second hyperpolarizability in comparison to the corresponding planar one.
This article is organized as follows: in Section II we briefly present the most relevant articles of the extensive literature, which is devoted to the various topics of this work; in Section III, we describe the methods we have used, in Section IV we analyze our results and finally in Section V, the concluding remarks are presented.
1. Literature survey
We briefly review some recent, relevant work, related to 2D nanosheets, polymers, NiBDT and thiophene.Intensive research is currently being carried out on 2D nanosheets (e.g. graphene,10 BN,11chalcogenides,12 silicene,13 and germanane14), due to the possibility of tuning their properties (e.g. photoluminescence15 and photoconductivity16) in order to optimize their performance in many applications (e.g. photonics, biomedicine, electronics, and energy storage).2,10,17,18 In particular various, π-conjugated (X = Ni, Pd, Pt, Co) bis(dithiolene) nanosheets have been studied.19–21 High conductivity was observed for Ni bis(dithiolene) π-nanosheets.4 Tang and Zhou22 studied the binding of ethylene to nickel bis(dithiolene). A 2D nickel bis(dithiolene) sheet is an organic topological insulator.4,16,23,24 Zhou found that this sheet behaves like a metal, when it interacts with graphene to form a heterobilayer material.25 Shojaei and Kang found that between two layers of a π-conjugated Ni bis(dithiolene) complex (NiS4C4), covalent bonds are formed. The bilayer is potentially useful for the design of electromechanical and optoelectronic devices.26 Shojaei et al.23 investigated the electronic and mechanical properties of a 2D π-conjugated Ni bis(dithiolene) sheet (NiS4C4) and its Pd analogue (PdS4C4). Liu et al.27 found, by using first principles calculations, that the π-conjugated Ni bis(dithiolene) complex with formula Ni3S12C12 can be used to detect poisonous gases (e.g. CO, NO). A change in the properties (from semiconducting to metallic) of the Ni3S12C12 nanosheet was induced by adsorbed molecules. Pal et al.20 reported the synthesis of palladium bis(dithiolene) nanosheets. Clough et al.21 used 2D polymer nanosheets containing Co-bis(dithiolene) units for efficient hydrogen evolution from water. It has also been reported that a cobalt dithiolene metal–organic framework exhibits band-like metallic conductivity.28 Other related materials have also been studied, for example, Ni3(HITP)2,29 Cu3(HITP)2 [22]30 and HTT-Pt,31 where HITP = 2,3,6,7,10,11-hexaiminotriphenylene and HTT = triphenylene hexathiol. Excellent NLO properties have been observed for low-dimensional materials, together with the possibility of developing new photonic devices based on them.1–3,32–35
Polymers possessing delocalized π-electrons have been investigated extensively, as a potential source of photonic materials.36–42
NiBDT. Many articles on Ni bis(dithiolene) related derivatives and
several of their properties have been reported (e.g. HOMO and LUMO
energy levels, third order NLO properties).43–52 Deplano et al.53 reviewed square-planar d8 metal mixed-ligand
dithiolene complexes as second order nonlinear optical chromophores. One of
their goals was to propose tools for optimizing the optical properties. Several
markers for the prediction of the optical properties have been proposed
(e.g. λmax, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)).53 Garreau-de Bonneval et al.54 reviewed the synthesis, electronic properties and
applications of neutral d8 metal bis-dithiolene complexes. They noted
that the NiBDT derivatives, due to their optical, electronic, magnetic and
conductive properties, provide an excellent basis for the development of many
devices (e.g. opto-electronic).54
C)).53 Garreau-de Bonneval et al.54 reviewed the synthesis, electronic properties and
applications of neutral d8 metal bis-dithiolene complexes. They noted
that the NiBDT derivatives, due to their optical, electronic, magnetic and
conductive properties, provide an excellent basis for the development of many
devices (e.g. opto-electronic).54
Thiophenes. Iyoda and Shimizu reviewed π-expanded oligothiophene macrocycles; they noted that their properties (e.g. optoelectronic) depend on the inserted π-systems. The authors discussed the remarkable optical and electronic properties of these systems.55 Li et al. employed a hybrid computational scheme, including MD, QM/MM and the Frenkel exciton theory of vibronic coupling, to study the optical spectra of α-sexithiophene nanoparticles.56 Gonzalez et al. discussed the self-assembly of fully conjugated π-expanded macrocyclic oligothiophenes, complexed with fullerenes. The role of non-covalent interactions in controlling their electronic properties was analysed.57 Oligothiophene linkers were used to create a series of structural scaffolds. A series of molecules were studied and this allowed for fine-tuning of emissions in the range of 412–540 nm.58
II. Methods
This section discusses the following: (i) a description and justification of the functionals we employed; (ii) definition of (hyper)polarizabilities; (iii) definition of the diradical character and the indices we used to compute it; (iv) description of the procedures we have employed to validate the methods we have used; (v) description of the methods we have employed to compute the molecular structures, (vi) calculation of the excitation spectrum; (vii) decomposition of the molecular polarizabilities of some selected systems into atomic contributions by using the fractional occupation Hirshfeld-iterative method and (viii) vibrational effects on the (hyper)polarizabilities.(a) Functionals. The molecular properties were computed by employing a series of functionals which include the following:
(a) CAM-B3LYP.59 This is the long range (LR) corrected version of the B3LYP functional; it has given satisfactory (hyper)polarizabilities for large and extended molecules.9,60,61
(b) LC-BLYP.62 This long-range corrected functional (BLYP) has been shown to provide reasonable second hyperpolarizability values for singlet diradical molecules63,64 and a series of fullerenes.65
(c) ωB97X.66 This LC functional gave satisfactory results for the second-order nonlinear optical responses of a series of ferrocene-tetrathiafulvalene hybrids67 and several properties (e.g. affinities, dissociation energies, and dipole moments) of ytterbium doped silicon clusters.68
(d) LC-PBE.69 This long-range corrected version of PBE was developed by Perdew et al. Lu used this functional to satisfactorily compute the dynamic quadratic polarizability of several organic molecules.70 Jacquemin et al. computed the longitudinal dipole moments and static electronic first hyperpolarizabilities of increasingly long polymethineimine oligomers.71 These authors noted the efficiency of long-range DFT, even in very pathological cases.
(e) BHandHLYP.72,73 This has been shown to provide satisfactory second order hyperpolarizability values for diradical molecules.74,75
(f) B2-PLYP.76 This functional provided reasonable second hyperpolarizabilities for NiBDT and several Ni-based derivatives, compared with those calculated with the more accurate CCSD(T) and CASPT2 methods.74
(b) Definitions. When a molecule is set in a uniform static electric field, F, its energy, E(F), is given by the following expansion:
| E(F) = E0 − μiFi − (1/2)αijFiFj − (1/6)βijkFiFjFk − (1/24)γijklFiFjFkFl − …, | (1) |
(c) Diradical character. Definitions of the diradical character may be found in the literature.74,82,83
It has been noted that in a singlet diradical, there are two singly occupied molecular orbitals of equal energy; these are weakly antiferromagnetically coupled.84 Singlet diradicals should be studied with a multi-configurational wavefunction, which can properly treat static correlation. However, such a method is computationally very demanding, in particular for molecules with the size of those considered in this work. It is known that the symmetry broken (BS) DFT can simulate the static correlation;84 this method has adequately described singlet diradicals.74,84–86
The diradical character may be estimated by using the following expressions:74
| (a) E1 = ERDFT(singlet) − EUDFT(singlet) |
| (b) E3 = (2(ET − E(U)DFT)/(2 − 〈S2〉BS)), |
(d) Validation. Most of our results have been calculated by employing the (U)CAM-B3LYP and (U)BHandHLYP functionals with the 6-31G* basis set for H, C, S, and O atoms.87,88 For Ni the quasi-relativistic, effective core potential, ECP28MWB(SDD), was used.89 Detailed studies on the performance of CAM-B3LYP and BHandHLYP, in connection with the 6-31G* basis set, for Ni derivatives, have been performed elsewhere.9 A comparison of the DFT results with the more rigorous CASSCF/CASPT2 [complete active space multiconfiguration SCF/CAS second order perturbation theory] and CCSD(T) [coupled-cluster with single, double and perturbative triple excitations (CCSD(T))] ones confirms the satisfactory performance of the employed DFT methods.74 It should also be noted that the adequacy of the 6-31G* basis set, for the selected compounds, has been confirmed by employing a series of larger basis sets, including more diffuse and polarization functions (Table S1, ESI†).
(e) Molecular structures. All the molecular structures have been computed by employing the B3LYP/6-31G* method. It has been demonstrated that this approach gives satisfactory structures for NiBDT derivatives having conjugated linkers.9 Furthermore, B3LYP was shown to give satisfactory geometries for diradical systems.90 It has been established that a broken symmetry method provides similar molecular geometries, compared with those computed with RDFT.84 Thus, RDFT was used for the geometry optimization. Vibrational analysis was performed for all structures employed in this study, to verify that a stationary point has been found.
(f) Excitation spectrum. Time dependent density functional theory (TD-DFT) was used to compute the transition energies and the transition dipole moments,91,92 with the CAM-B3LYP functional.59 This gave satisfactory estimates (in comparison with the corresponding experimental values) of transition energies of some organic dyes.93–96
(g) The fractional occupation Hirshfeld-iterative method. As in one of our recent studies9 on Ni-(bis)dithiolene derivatives, we decomposed the molecular polarizabilities of some selected systems of the current study into atomic contributions by means of the fractional occupation Hirshfeld-iterative method (FOHI).97,98 Through this well tested partitioning scheme, we obtained pure atomic polarizability values (hereafter intrinsic polarizabilities), free of size and origin dependent charge transfer interactions.99 In addition, we have computed and plotted contour maps of the mean intrinsic polarizability (MIP) densities in order to identify the most polarizable regions of some of the studied systems.99 The aim of these treatments is to obtain useful size independent chemical insights into the polarizability evolution of the systems considered by identifying the most polarizable atoms in the considered molecules and study effects which are expected to arise from the various structural changes that have been applied during our designing strategy. More information and some demonstrative examples on the application of the FOHI method in various study cases can be found in ref. 100–102 and references therein. All electron density matrices as well as their derivatives with respect to an electric field were computed analytically as implemented in GAUSSIAN 09,77 while atomic partitioning was carried out with the BRABO package,103 the STOCK program104 and homemade FORTRAN codes.
(h) Vibrational effects. The reliable calculation of the vibrational properties of molecules of the size investigated here is generally a rather difficult task. We computed the vibrational contributions of two relatively small models, related to the derivatives of interest. The perturbation theory, as pioneered by Bishop and Kirtman,105–107 was used for these calculations. Only the static contribution to the so-called nuclear relaxation (nr) was computed, which takes into account that the properties of the optimum geometry in an electric field differ from those of the field-free optimized geometry. The nr contribution was computed by means of the Bishop–Hasan–Kirtman approach, which is based on geometry optimizations in an applied electric field.108 The nr contribution is only the lowest order part to the total vibrational contribution. Further contributions, such as the zero-point average or the curvature contribution, are generally too costly to compute even for the smallest molecules considered here. This implies that no conclusions on the convergence behavior of the perturbational series can be drawn. The properties were computed by numerical differentiations from both energies and analytically computed dipole moments for several field strengths and applying the Romberg–Rutishauser differentiation method.80,81 The range of the applied field strengths was either 0.0001–0.0064, or, in some difficult cases, 0.000025–0.0064 a.u.
The dipole moments and (hyper)polarizabilities are expressed in atomic units; E1 and E2 are given in kcal mole−1, while the excitation energy is given in eV. Conversion factors to other systems of units are also provided.109
III. Results and discussion
The results presented in this section will demonstrate the extremely large second hyperpolarizabilities of 1D coordination polymers, based on Ni-bis(dithiolenes). This will be shown by employing a model involving (B-NiBDT)n, n = 1–6 (Table 1). Two important related questions that we deal with in this section are as follows: (a) how does the structure affect the properties of interest? In order to comment on this question we have employed several models, some of which involve planar fragments of the nanosheet Ni3S12C12 (Table 1, (B-NiBDT)n; Fig. 1). (b) We have found that an important element for the optimization of the L&NLO properties is the fused linkers with the NiBDT units. Thus, we have used several rich π-electron linkers to find their effect on the L&NLO properties (Fig. 2). It is well known that extended π-electron networks are of cardinal importance for the development of NLO materials and the rationalization of their properties.9 A molecular unit which is characterized as an important building block for the fabrication of photonic materials is thiophene and its related derivatives (Fig. 3). Thus, we consider its effect, as a linker, on the L&NLO properties of the resulting Ni-bis(dithiolene) compounds. This work mainly presents electronic contributions to (hyper)polarizabilities. However, for completeness, we will briefly comment on the vibrational properties, specifically on the nuclear relaxation contribution to (hyper)polarizabilities of two relatively small model derivatives, related to the compounds of interest.|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | P | |||||||||||
| μ x | α xx | α | β xxx × 10−3 | γ xxxx × 10−5 | (γxxxx × 10−5)/na | (γxxxx × 10−5)/Nb | E 1 | E HOMO | E LUMO | |H–L| | E exc | |
| E 2 | ||||||||||||
| a n = the number of benzene–NiBDT units*. b N: number of electrons. c E 1 = ER − EU, method: B3LYP/6-31G*. d E 2 = (2(ET − EU)/(2 − 〈S2〉BS)), method: B3LYP/6-31G*. e Method: (U)CAM-B3LYP/6-31G*. f Method: (U)BHandHLYP/6-31G. g Method: (U)B2PLYP/6-31G*. h (U)LC-ωPBE. i Method: (U)LC-PBEPBE/6-31G*. j Method: (U)ωB97X/6-31G*. k Method: (U)CCSD/6-31G*. l Method: (U)CCSD(T)/6-31G*. m Method: (U)LC-BLYP/6-31G*. | ||||||||||||
| 1 | −0.438f | 371f | 229f | 3.8f | 19f | 19.0f | 0.14f | 0.0 | −0.264f | −0.111f | 0.153f | 1.92 |
| −0.231e | 422e | 239e | 6.0e | 20e | 20.0e | 0.14e | 11.67 | −0.272e | −0.108e | 0.164e | ||
| −0.042g | 488g | 10.9g | 41g | 41.0g | 0.30g | −0.262g | −0.098g | 0.164 | ||||
| −0.302h | 396h | 5.4h | 23h | −0.334h | −0.074h | 0.260h | ||||||
| −0.380i | 369i | 387i | 2.70i | 14i | −0.293i | −0.089i | 0.204i | |||||
| −0.206j | 435j | 242j | 8.2j | 26j | ||||||||
| −0.163k | 421k | 2.1k | 13k | |||||||||
| −0.197l | 486l | 6.1l | 38l | |||||||||
| 2 | −0.670f | 1336f | 576f | 5.4f | 327f | 163.5f | 1.28f | 0.37 | −0.254f | −0.148f | 0.106f | 1.26 |
| −0.392e | 1684e | 766e | 17.1e | 570e | 285.0e | 2.23e | 7.05 | −0.263e | −0.139e | 0.124e | ||
| −0.282g | 2020g | 22.5g | 861g | 430.5g | 3.36g | −0.249g | −0.137g | 0.112g | ||||
| −0.569i | 1347i | 1275i | −16.1i | 640i | −0.315i | −0.117i | 0.198i | |||||
| −0.347j | 1779j | 782j | 25.2j | 899j | −0.286j | −0.161j | 0.165j | |||||
| 3 | −0.784f | 2732f | 1076f | −2.3f | 1693f | 564.3f | 4.50f | 1.14 | −0.256f | −0.157f | 0.099f | 1.13 |
| −0.497e | 3836e | 1617e | 13.7e | 5380e | 1793.3e | 14.30e | 5.72 | −0.256e | −0.148e | 0.118e | ||
| −0.455g | 4579g | 1.6g | 5670g | 1890.0g | 15.10g | −0.251g | −0.146g | 0.105g | ||||
| −0.584h | 3463h | −0.318h | −0.126i | 0.192i | ||||||||
| −0.670i | 2809i | 1525i | −178.0i | 3810i | −0.288j | −0.129j | 0.159j | |||||
| −0.441j | 4141j | 1667j | 23.3j | 9000j | ||||||||
| 4 | −0.846f | 4383f | 1667f | −16.6f | 4650f | 1162.5f | 9.38f | 2.01 | −0.257f | −0.161f | 0.096f | 1.04 |
| −0.571e | 6743e | 2764e | −29.6e | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600e 600e |
5900.0e | 47.60e | 2.55 | −0.266e | −0.151e | 0.115e | ||
| −0.509j | 7407j | 2857j | −40.2j | 42000j |
−0.289j | −0.133j | 0.156j | |||||
| 5 | −0.881f | 6173f | 2307f | −31.8f | 9160f | 1832.0f | 14.87f | 2.88 | −0.259f | −0.165f | 0.094f | 0.98 |
| −0.624e | 10212e |
4135e | −117.5e | 66000e |
13200.0e | 107.10e | 1.41 | −0.269e | −0.156e | 0.113e | ||
| −0.710h | 8800h | −110.0h | −0.321h | −0.134h | 0.187h | |||||||
| −0.775i | 6579i | 2892i | −36.7i | 10200i |
−0.290i | −0.137i | 0.153i | |||||
| −0.56j | 11372j |
4280j | −181.8j | 120000j |
−0.312j | −0.126j | 0.186j | |||||
| −0.829m | 6253m | 2390n | −30.7m | 7280m | ||||||||
| 6 | −0.901f | 8033f | 2971f | −44.7f | 14800f |
2466.7f | 20.11f | 3.76 | −0.259f | −0.168f | 0.091f | 0.94 |
| −0.660e | 14047e |
5606e | −233.8e | 139000e |
23166.7e | 188.86e | 1.01 | −0.270e | −0.158e | 0.112e | ||
| −0.743h | 11890h |
−180.0h | 86000h |
−0.291j | −0.139j | 0.152j | ||||||
| −0.597j | 15796j |
5858j | −380.1j | 250000j |
||||||||

 | ||
| Fig. 2 Structures of NiBDT-based (right column) and their corresponding non-NiBDT-based (left column) derivatives, optimized with the B3LYP/6-31G*/Ni(SDD) method, in the gas phase. | ||
 | ||
| Fig. 3 Structures of NiBDT-based derivatives, optimized with the B3LYP/6-31G* method, in the gas phase. | ||
The basic parameters using which we study the above issues are, besides the (hyper)polarizabilities, the dipole moment, the EHOMO, ELUMO, |EHOMO − ELUMO|(|H–L|) gap and the distribution of spin density. Of particular importance is the diradical character (DC); for its quantification we have employed two indices. The variety of systems we have studied and the properties we have computed highlight the comparative character of the present work.
1. NiBDT fused with benzene
E HOMO gets a maximum at n = 2 for BHandHLYP and B2PLYP, while for CAM-B3LYP, EHOMO gets a maximum at n = 3.
E LUMO decreases with n for BHandHLYP and CAMB3LYP.
|H–L|. It decreases with n for BHandHLYP, CAM-B3LYP and ωB97X.
Effect of a uniform external electric field on |HLG|↑ and |HLG|↓. It has been shown that upon application of an electric field the spin states on the two edges of a ribbon may be affected.110–112 We have considered the effect of a uniform electric field on |HLG| of (B-NiBDT)6, for spin α(↑) and spin β(↓) orbitals, |HLG|↑ and |HLG|↓, respectively. The main points of Fig. 4 are:
 | ||
| Fig. 4 The |EHOMO − ELUMO| gap (HLG) for α spin (↑) and β spin (↓) orbitals of the (B-NiBDT)6 derivative as a function of the field. All field values (F = Fx) were computed with the (U)B3LYP/6-31G* method at the B3LYP/6-31G* gas phase optimized geometry. | ||
(a) |HLG|↑ = |HLG|↓, for F = 0.
(b) If F > 0, then |HLG|↑ ≠ |HLG|↓; for example for 14 × 10−4 < F < 48 × 10−4 a.u., then |HLG|↑ > |HLG|↓; for F = 48 × 10−4 a.u., we observe |HLG|↑ = |HLG|↓, and for 48 × 10−4 < F ≤ 64 × 10−4 a.u. |HLG|↑ < |HLG|↓.
(c) The difference |HLG|↑ − |HLG|↓ is small/negligible.
E exc. This property decreases with increasing n and seems to reach a constant value (ca. 0.9 eV) (Table 1). In Fig. 5 the absorption spectrum of (B-NIBDT)n, n = 1–6, is shown. A strong absorption peak was computed, ranging from 646 to 1323 nm. For higher energies (shorter wavelengths) the observed absorptions are associated with small oscillator strengths (<0.1).
 | ||
| Fig. 5 Absorption spectra of several (B-NiBDT)n, n = 1–6, derivatives, computed with (U)CAM-B3LYP/6-31G* at the B3LYP/6-31G* gas phase optimized geometry. | ||
Dipole moments. Significant differences are observed between the dipole moment values calculated by some of the employed functionals for n = 1 (Table 1). A comparison of the dipole moment values, computed by the CCSD and CCSD(T) methods, shows the significant effect of triples (T). The dipole moment data and n of [(B-NiBDT)n] are fitted with the function: |μx| = −0.020n2 + 0.227n, R2 = 0.9874 (Fig. 6).
 | ||
| Fig. 6 The dependence of the dipole moment, polarizability and first hyperpolarizability on the number of (B-NiBDT) units (n). The values were computed with the (U)CAM-B3LYP/6-31G* method. The black line depicts the fitted curve. | ||
Polarizabilities. A comparison of the CCSD and CCSD(T) αxx values shows that the perturbative triples (T) have a significant effect (Table 1). The five employed functionals give values for αxx in the range 369–488 a.u. for n = 1. There is a remarkable dependence of αxx on the selected functional. For n = 1–3, the maximum αxx value is computed by B2PLYP. There is reasonable agreement for n = 1–3 for αxx, computed with CAM-B3LYP and B2PLYP. The polarizability data and n of [(B-NiBDT)n] have been connected with the function αxx = 429.83n1.9674, R2 = 0.9996 (Fig. 6).
The variation of αxx(−ω;ω) for (B-NiBDT)3 is shown in Fig. S1 (ESI†). This property gets a maximum value for λ = 1100 nm. The computed λ is in agreement with the corresponding value (1097 nm) of Table 1.
In Fig. 7 we present atomic intrinsic
polarizabilities (INPs) of (B-NiBDT)2 together with the computed
FOHI atomic charges and the corresponding MIP densities including those of
its constituent units. In this treatment, we used a triple ζ
quality basis set, namely 6-311G*, for all atoms. At the respective level of
theory, the computed total mean molecular polarizability of
(B-NiBDT)2, which amounts to 768.4 a.u., is decomposed into a
dominant charge transfer part (CT) of 679.8 a.u. (∼77%) and a size
independent intrinsic polarizability part (IN) of 106.6 a.u. (∼13%).
As in all prolate shaped molecules, the CT part, which defines the magnitude
of their dipole polarizability, is dominated by charge transfer interactions
along the longitudinal axis of the molecule. On the other hand, the
intrinsic polarizability of (B-NiBDT)2 is dictated by the
individual atomic INPs of Ni and S atoms. Their overall contribution
accounts for 48% of the total molecular INP of (B-NiBDT)2. By
comparing the illustrated integrated values on each atom (Fig. 7), we see that for this molecule: INP(S)
> INP(Ni) > INP(C) > INP(H). The respective ordering, also observed
in the isolated NiBDT unit (shown at the bottom of Fig. 7), follows only partially the number of electrons (or
size) of each atom, and thus their atomic polarizabilities, in their free
forms. Hence, while Ni and S (Ni:28e, S:18e)
are found to be more polarizable than all C and H atoms of this system, the
INP of S and Ni appears to follow the opposite trend. The relative INPs
loosely correlate with the charges on each of these atoms in the sense that
the more positively (negatively) charged an atom is in the molecule the less
(more) polarizable it becomes. This trend, though, does not apply to atoms
of the same type. For instance, the negatively charged S atoms attached to
the two carbon hexagons bear slightly smaller INPs than those linked to the
terminal HCCH unit which remains intrinsically as polarizable as in free
NiBDT. Nevertheless, the observed differences are small and are not expected
to play an important role in the behavior of the molecule. On the other
hand, the INP of Ni in (B-NiBDT)2 increases considerably in
comparison with free NiBDT. The opposite trend is observed in the case of S
atoms which lose some of their polarizable character. Additional information
about the character of (B-NiBDT)2 in terms of polarizability can
be retrieved by comparing its mean intrinsic polarizability densities (MIP)
with those of its constituent units, namely, benzene and NiBDT. As explained
elsewhere,99 positive MIP densities
imply easily polarizable regions of the molecules. On the other hand,
negative densities stand for “nonpolarizable” zones in which
the electron density is expected to reduce under an external field
perturbation. The illustrated contour maps suggest that the most affected
areas of (B-NiBDT)2, with respect to the free molecules, are
those in the vicinity of Ni atoms and on the central carbon hexagon. The
latter unit is expected to bear a considerably reduced aromatic character as
compared to the free molecule of benzene or aromatic hexagons of larger
graphene like molecular systems.102
 | ||
| Fig. 7 FOHI charges, intrinsic atomic polarizabilities (INPs) and MIP density contour maps of (B-NiBDT)2, C6H6 and NiBDT computed at the (U)CAM-B3LYP/6-311G* level. | ||
First hyperpolarizabilities. A comparison of CCSD(T) with the CCSD βxxx values shows that the triples (T) have a significant effect (Table 1). The five considered functionals give values for βxxx in the range 2.1–10.9 × 103 a.u. There is a remarkable change of sign for βxxx. Curve fitting (Fig. 6) has disclosed a rather complex function connecting βxxx with n[(B-NiBDT)n].
We have employed a two-state model (TSM)113 in order to rationalize the first hyperpolarizability results of (B-NiBDT)n. The geometries were computed with the B3LYP/6-31G* method, while the βxxx values were calculated with the CAM-B3LYP/6-31G* approach (Table 2). For this model we have computed the dipole moment of the ground and excited states, μg and μe, respectively, the transition dipole moment, μge, and the excitation energy, ΔE. For n = 1, we have employed several excited states; for n = 2, we have used 2 excited states and for the rest of the oligomers, one. Of course, as one would expect the value of βxxx may greatly depend on the selected excited state. Both the numerically computed and the TSM βxxx values follow the same trend; however, the first gets a maximum value for n = 2, while the second one approaches a maximum for n = 3. ΔE and (μg)x decrease with n, while |(μge)x| increases; (μe)x and (μg)x − (μe)x get a maximum value for n = 2, that is they follow a similar trend to βxxx (Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| P | N | HF | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| a This state is associated with the first allowed electronic transition. b State 5. c State 42. d State 49. e State 53. f State 11. g β xxx = [3(μe − μg)(μge)2]/(ΔE)2, ΔE = Eexc − Eg. h Numerical value. | |||||||
| ΔE | 0.070a | 0.046a | 0.041a | 0.038a | 0.036a | 0.034a | 0.558a |
| 0.053b | 0.050f | ||||||
| 0.169c | |||||||
| 0.188d | |||||||
| 0.197e | |||||||
| |(μge)x| | 1.491a | 3.924a | 7.047a | 9.629a | 11.911a | 13.936a | 0.935a |
| 1.279b | 2.366f | ||||||
| 0.955c | |||||||
| 2.206d | |||||||
| 1.152e | |||||||
| (μe)x | −0.907a | 3.028a | 0.983a | −0.092a | −0.650a | −0.965a | 0.555a |
| 2.137b | −4.032f | ||||||
| 0.432c | |||||||
| −0.090d | |||||||
| 0.050e | |||||||
| (μg)x | −0.231 | −0.392 | −0.497 | −0.571 | −0.624 | −0.661 | −0.747 |
| (μe)x − (μg)x | −0.676a | 3.420a | 1.480a | 0.479a | −0.026a | −0.304a | 1.302a |
| 2.368b | −3.640f | ||||||
| 0.663c | |||||||
| 0.141d | |||||||
| 0.281e | |||||||
| β xxx | −906a | 73320a
|
127580a |
91021a |
−8540a | −149540a |
10.2a |
| 4190b | −24222f |
||||||
| 63c | |||||||
| 58d | |||||||
| 28e | |||||||
| β xxx | 6047 | 17100 |
13700 |
−29560 |
−117460 |
−233850 |
16.9 |

For comparison we also report the first hyperpolarizability value of HF
(hydrogen fluoride). The |βxxx| value of
(B-NiBDT)6 is 13837 times larger than that of HF. How
can we interpret this large difference using the TSM? It appears that the
properties that could be used to rationalize the much larger
|βxxx| of the oligomer are
ΔE and
|(μge)x|; the
first(denominator) is much smaller and the second(numerator) is much larger
than the corresponding properties of HF (Table
2).
Second hyperpolarizabilities. Triples (T) have a significant effect (Table 1). The maximum γxxxx value for n = 1–3 is computed with B2PLYP. The value of γxxxx depends, remarkably, on the functional. For most of the considered cases the minimum γxxxx is given by BHandHLYP and the maximum by ωB97X. The ratio γxxxx (ωB97X)/γxxxx (CAM-B3LYP) equals 1.3 for n = 1, and, stabilizes at 1.8 for n ≥ 5. Both ωB97X and CAM-B3LYP are long-range corrected functionals. We observe that the ratio γxxxx (n + 1)/γxxxx (n) decreases as n increases. So, n2/n1 = 28.9, but n6/n5 = 2.1 (CAM-B3LYP).
We have considered two indices in order to quantify the change of γxxxx with the size of the derivative: (a) the first index is I1 = γxxxx/n, where n is the number of benzene (B)–NiBDT units. I1 demonstrates that CAM-B3LYP leads to much more polarizable units than BHandHLYP. (b) The second index is I2 = γxxxx/N, where N is the number of electrons of the derivative. Both I1 and I2 clearly demonstrate that the considered structures (i) are extremely polarizable and (ii) there is a rapid, nonlinear, increase of I1 and I2 with the size of the structure. One of the main points, emerging from Table 1, is that some of the oligomers (B-NiBDT)n have giant second hyperpolarizabilities. The hyperpolarizability data and n[(B-NiBDT)n] have been connected with the function γxxxx = 19.641n5.0248, R2 = 0.999 (Fig. 8).
 | ||
| Fig. 8 The dependence of the second hyperpolarizability on (a) the number of (B-NiBDT) units (n), (b) the excitation energy (Eexc), and (c) E2. The values were computed with the (U)CAM-B3LYP/6-31G* method, except those of E2, where the (U)B3LYP/6-31G* method was employed. | ||
Effect of benzene on the (hyper)polarizabilities. We have found that αxx and γxxxx of NiBDT are 221.9 and 558 × 103 a.u., respectively.74 The corresponding properties of NiBDT-B ((U)HBandHLYP) are 371 and 19 × 105 a.u., respectively. Thus, benzene has a very large effect on both αxx and γxxxx. Furthermore, one may consider two derivatives of NiBDT in which (i) one H has been substituted with CH3 and (ii) 2 hydrogen atoms have been substituted with the ligand CS3. These derivatives have the following γxxxx values: 739 × 103 a.u. and 1535 × 103 a.u., respectively. All these properties have been computed with the (U)BHandHLYP/6-31G*.74 The significant effect of benzene, as a substituent, is observed.
Diradical character (DC). We used two indices, E1 and E2, to estimate the DC of the considered derivatives (Table 1). These indices show that DC increases with increasing n. Moreover E2 is associated with the exchange interaction, J, (E2 = 2J), which can be experimentally measured by electron spin resonance (ESR).86 It has been found that E1 and E2, for NiBDT, are 0.05 and 9.91 kcal mol−1, respectively.74 Comparing E2 of NiBDT with that of B-NiBDT, we infer that the fusion of benzene with NiBDT leads to a small decrease of DC. However, for 2 ≤ n, all the oligomers have larger DC than NiBDT.
Effect of a uniform external electric field on DC. Su et al.114 reported that they controlled the singlet–triplet energy gap of a diradical in the solid state by temperature, demonstrating that an external stimulus can modify the DC. Here we shall comment on the effect of a uniform external electric field on DC. Most considered field values lead to an increase of ΔE(F) = E(F)(RDFT) − E(F)(UDFT), that is an increase of F leads to an increase of DC, in comparison to the value that is associated with F = 0 (Fig. 9).
 | ||
| Fig. 9 The dependence of ΔE = E(RDFT) − E(UDFT) on the field (F = Fx) for the (B-NiBDT)6 derivative. All values were computed with the (U)B3LYP/6-31G* method at the B3LYP/6-31G* gas phase optimized geometry. | ||
Spin density distribution (triplet state). It was suggested that, in a singlet diradical, the weakly coupled electrons, and in a triplet radical, the corresponding electrons are located at the same position of the molecule.84 We shall adopt the same hypothesis and comment on the Mulliken spin densities of the lowest triplet state of (B-NiBDT)n, where n = 1–3. For n = 1, we observe (Fig. 10) a large spin density at nickel and sulfur atoms; at the carbon atoms the spin density is smaller or negligible. For comparison, we have also computed the spin densities of NiBDT. We found that the spin density at Ni is very small, while at the sulfur atoms the spin density is, approximately, twice that of carbon atoms. For n = 2, the total spin densities at sulfur and nickel atoms are 1.004 and 0.68, respectively; one of the two nickel atoms has negligible spin density. At the carbon atoms the spin density is negligible. For n = 3, the total spin densities at sulfur and nickel are 1.046 and, 0.597, respectively. The spin density is concentrated at the middle Ni atom. At the carbon atoms the spin density is very small or negligible. Thus, these computations show that the unpaired electrons are localized at the sulfur and nickel atoms. However, our VB computations for NiBDT suggest that the unpaired electrons are localized at the carbon atoms.74 In Fig. S2 (ESI†) the spin density distribution for (B-NiBDT)n, n = 2, 6, is also depicted.
 | ||
| Fig. 10 Computed Mulliken triplet spin densities with the UB3LYP/6-31G* method. | ||
Substitution of Ni with Pd. The results of Table 3 present some properties of (B-MBDT)n, where M = Ni, Pd and n = 2, 3. An increase of the atomic number of the metal (going from Ni to Pd) decreases the values of μx, αxx, βxxx, and γxxxx (Table 3). However, the Pd derivatives are associated with much larger DC. This finding confirms our previous computations related to the DC and (hyper)polarizabilities of A-[(SC)2-M-(SC)2]-A, where A = SC3C2 and M = Ni, Pd.115 The results of Table 1 show a different trend, that is increase of γxxxx is connected with an increase of DC.
| n/M | P | |||||||
|---|---|---|---|---|---|---|---|---|
| μ x | α xx | β xxx × 10−3 | γ xxxx × 10−5 | E 1 | E HOMO | E LUMO | |H–L| | |
| a Method: (U)CAM-B3LYP/6-31G*. b E 1 (kcal mol−1) = E(RDFT) − E(UDFT). Method: B3LYP/6-31G*. | ||||||||
| 2 | ||||||||
| Ni | −0.392 | 1684 | 17.1 | 570 | 0.37 | −0.263 | −0.139 | 0.124 |
| Pd | −0.536 | 1496 | 7.1 | 332 | 1.63 | −0.264 | −0.140 | 0.124 |
| 3 | ||||||||
| Ni | −0.497 | 3836 | 13.7 | 5380 | 1.14 | −0.256 | −0.148 | 0.118 |
| Pd | −0.641 | 3162 | 3.7 | 1946 | 3.26 | −0.266 | −0.146 | 0.120 |
From the results of Table 3 it is also observed that (γxxxx(n = 3)/γxxxx(n = 2)) [Ni] = 9.43, (γxxxx(n = 3)/γxxxx(n = 2)) [Pd] = 5.86. Similarly for E1 it was found that (E1(n = 3)/E1(n = 2)) [Ni] = 3.08, (E1(n = 3)/E1(n = 2)) [Pd] = 2. The larger increase of γxxxx [Ni], compared with that of γxxxx [Pd], with the number of (B-MBDT) units, is associated with the larger increase of E1[Ni] compared with that of E1[Pd].
| Method/basis set | Property | |||||
|---|---|---|---|---|---|---|
| α xx | α yy | β xxx (×10−3) | γ xxxx (×10−5)a | E 1 | E exc | |
| a Value computed by taking the first derivative of βxxx/yyy with respect to the field. b E 1 (kcal mol−1) = E(RDFT) − E(UDFT), method: B3LYP/6-31G*. c Fig. 4. d β yyy component. e γ yyyy component. | ||||||
| Compound: 1c | ||||||
| (U)BHandHLYP/6-31G* | 5895 | 6455 | — | 3860 | 0.12 | 1.16 |
| 7350 | 7322 | 8005 | ||||
| (U)ωB97X/6-31G* | ||||||
| (U)CAM-B3LYP/6-31G* | ||||||
| Compound: 2c | ||||||
| (U)BHandHLYP/6-31G* | 2550 | 2550 | — | 204 | 0.63 | 1.57 |
| 2837 | 2837 | 288 | ||||
| (U)CAM-B3LYP/6-31G* | 2858 | 2858 | 343 | |||
| (U)ωB97X/6-31G* | ||||||
| Compound: 3c | ||||||
| (U)CAM-B3LYP/6-31G* | 6327 | 3959 | 99.3 | 5014 | 1.41 | 1.19 |
| (U)ωB97X/6-31G* | 6761 | 2477 | (−8.2)d | (−3500)e | ||
| 163.2 | 8200 | |||||
| (−17.3)d | (−1480)e | |||||
2. NiBDT fused with linkers having extensive conjugation
We have selected 4 different conjugated organic linkers (L): C6H6, C14H8, C24H12, and C24H8 (Table 5). We have then designed 8 oligomers involving, first, one of the above linkers, fused with two NiBDT units and, second, 3 NiBDT units fused with two linkers (Fig. 2). These computational experiments have been performed in order to show the effect of the linker, and the conjugation pattern in particular, on the (hyper)polarizabilities. For (NiBDT)2-L, we observe that γxxxx = 270 ± 46 × 105 a.u. (UCAM-B3LYP/6-31G*), while for (NiBDT)3-L2, three of the linkers (C14H8, C24H12, and C24H8) give rather similar γxxxx values; a significantly different and much larger second hyperpolarizability is associated with C6H6 (Table 4). An approximate estimate of the interaction induced γxxxx for NiBDT-C2H2-NiBDT is 25.4 × 106 a.u. (UCAM-B3LYP/6-31G*). The γxxxx values for NiBDT and C6H6 are 77200 a.u. and 1946 a.u.,
respectively. The largest αxx values for both
(NiBDT)2-L and (NiBDT)3-L2 are associated
with C26H12. An increase of n of the oligomer
(NiBDT)m-Ln leads to an increase
of DC and a decrease of the excitation energy (Table
5).
| Derivative | Propertyb | Derivative | Propertyb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α xx | γ xxxx × 10−5 | (γxxxx × 10−5)/Nc | E 1 | E exc | α xx | γ xxxx × 10−5 | (γxxxx × 10−5)/Nc | E exc | ||
| a Fig. 2. b Method: (U)CAM-B3LYP/6-31G*. c N: total number of electrons. d E 1 (kcal mol−1) = EDFT(singlet) − EUDFT(singlet); method: B3LYP/6-31G*. e RA: radiaannulene, NiBDT: bis(ethylene-1,2-dithiolato)nickel. f Values were taken from J. Phys. Chem. C, 2016, 120, 9419–9435. g z-Component. | ||||||||||
| NiBDT-C2H2-NiBDTe | 1333 | 256 | 1.11 | 0.50 | 1.28 | C6H6 | 71 | 0.019 | 0.00046 | 7.47 |
| NiBDT-C2H4-NiBDTe | 704 | 45 | 0.41 | 0.19 | 1.97 | |||||
| (NiBDT)3-(C2H2)2 | 3318 | 3480 | 9.94 | 1.34 | 1.12 | C10H8 | 150 | 0.180 | 0.00265 | 6.29 |
| (NiBDT)3-(C2H4)2 | 1215 | 125 | 0.35 | 1.91 | ||||||
| NiBDT-C10H4-NiBDT | 1297 | 280 | 1.00 | 1.26 | 1.99 | C14H8 | 179 | 0.354 | 0.00385 | 7.35 |
| (NiBDT)3-(C10H4)2 | 2755 | 1425 | 3.17 | 2.63 | 1.66 | C26H12 | 459 | 8.143 | 0.04847 | 6.40 |
| NiBDT-C20H8-NiBDT | 1775 | 316 | 0.92 | 0.63 | 2.04 | C24H12 | 465 | 6.567 | 0.04209 | 6.60 |
| (NiBDT)3-(C20H8)2 | 3834 | 1506 | 2.61 | 1.39 | 1.88 | C46H20 | 1364 | 168.438 | 0.56904 | 2.34 |
| NiBDT-RA-NiBDTe | 1592f | 224f | 0.66 | 0.50f | 1.52f | C24H8 | 339 | 0.952 | 0.00626 | 2.70 |
| (712)g | (15.440)g | |||||||||
| (NiBDT)3-(RA)2 | 3535f | 1517f | 2.66 | 0.63f | 1.43f | C46H12 | 845 | 19.137 | 0.06645 | 2.83 |
| (1212)g | (5.749)g | |||||||||
In order to show the effect of the delocalized π electrons – in
particular those connected with the linkers – on the properties of
interest, we fused NiBDT with C6H8 (Fig. S3, ESI†). This linker allows the disconnection of the π-systems
of NiBDT units. The results show that there is a very large effect on
γxxxx, for example:
| γxxxx[(NiBDT)3–(C2H2)2]/γxxxx[(NiBDT)3–(C2H4)2] = 28. |
This effect increases with n. The other properties (e.g. αxx) are associated with a significant, but less pronounced, effect.
In Fig. 11 we contrast the intrinsic atomic polarizabilities and the +MIP densities of NiANi and NiBNi (Fig. 11). In these planar polyaromatic systems, the CAM-B3LYP/6-311G* CT part of the dipole polarizabilities accounts for 84% (714.27 a.u.) and 82% (522.23 a.u.) of their total mean molecular polarizabilities, respectively. The stronger CT contribution obtained for NiANi is in accord with its larger size in comparison with NiBNi in both its molecular directions. As in (B-NiBDT)2, Ni and S are intrinsically the most polarizable atoms in both NiANi and NiBNi, with INP(S) > INP(Ni). In either molecule, out of the eight S atoms those which are bonded to the polyaromatic linkers appear to be the most polarizable ones. In terms of element contributions to the global molecular intrinsic polarizability of NiANi/NiBNi, FOHI analysis suggests that Ni and S atoms contribute 53%/62% of the total molecular intrinsic polarizability. In this case the contribution of the latter elements follows the inverse of the corresponding molecular sizes. In contrast to what we saw in (B-NiBDT)2, when NiBDT becomes a part of either DCPP or pyracyclene, there is a notable decrease in the intrinsic polarizability of the Ni atoms as compared to free NiBDT. On the other hand, while the inner S atoms show an increase in their intrinsic polarizabilities, the side S atoms retain most of their character in terms of INP. Considering the main fragments of either system, our results suggest that the global intrinsic polarizabilities of the carbon frames of the linkers in NiANi/NiBNi, calculated as the sum of all atomic INPs, slightly decrease after they are functionalized with NiBDT. The most affected C atoms are those connected to the S atoms which lose about 60% of their initial INPs. Turning our attention to Fig. 11, where the computed MIP density distributions are illustrated, we see that both linkers have similar effects on the attached NiBDT. In these plots we also see that the MIPs of the linkers exhibit small variations in NiANi and NiANi with respect to their free forms. The most affected areas are spotted in the vicinity of the terminal CC bonds as is also suggested by the INPs discussed above. Finally it would be interesting to note that when these densities are compared to those of the (B-NiBDT)2 compound we see that there is a change in the distribution of the positive and negative areas which should be related to the decreased Ni INPs in the NiANi and NiBNi compounds in comparison with (B-NiBDT)2.
 | ||
| Fig. 11 FOHI charges, intrinsic atomic polarizabilities (INPs) and MIP density contour maps of NiBDT-C20H8-NiBDT (NiANi), NiBDT-C10H4-NiBDT (NiBNi), DCPP and pyracyclene computed at the (U)CAM-B3LYP/6-311G* level. | ||
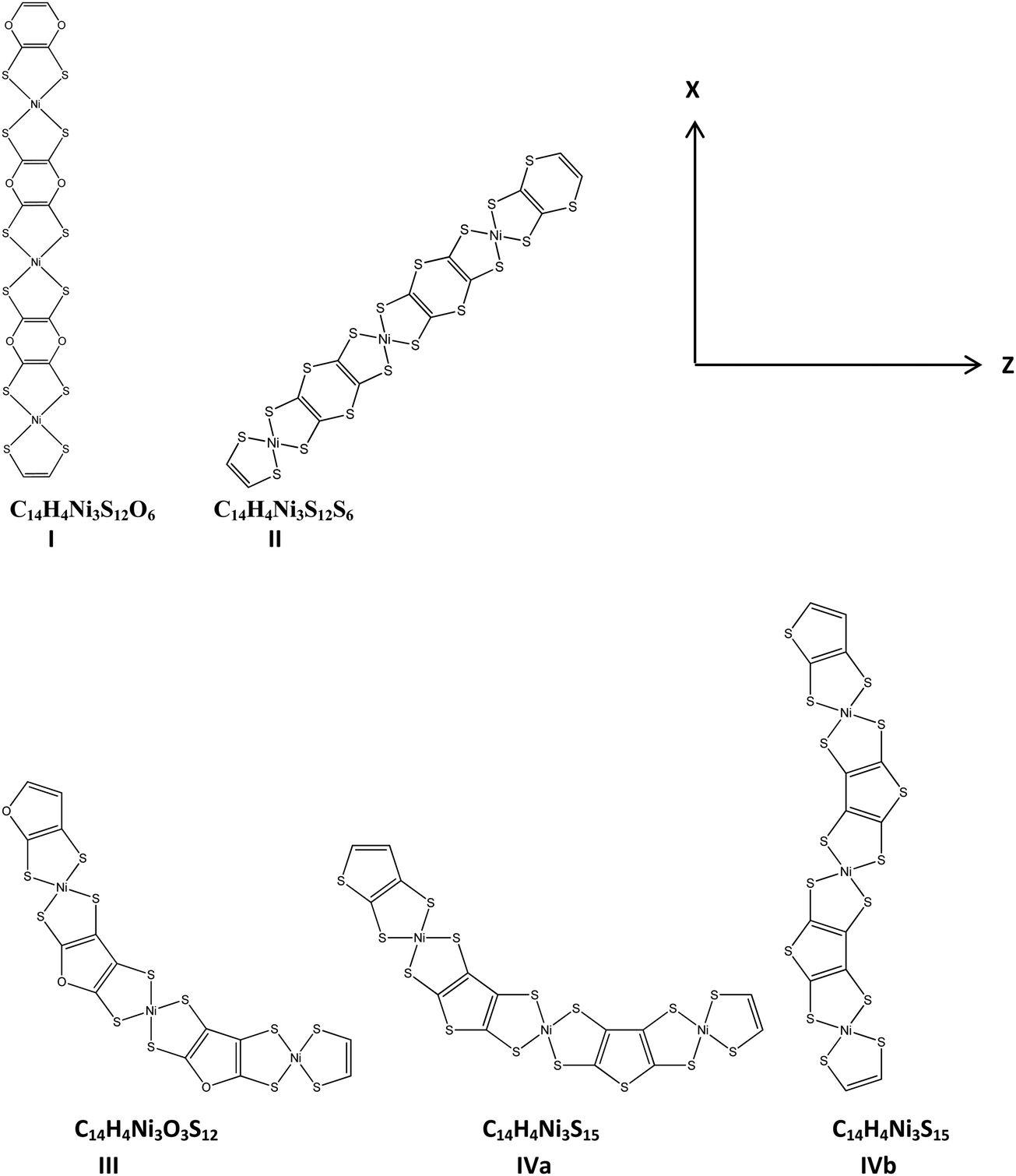
3. H4C4O2, H4C4S2, H4C4O and H4C4S fused with NiBDT
We have experimented with a further series of linkers: H4C4O2 (I), H4C4S2 (II), H4C4O (III) and H4C4S (IV). H4C4O2 and H4C4S2 were selected because we would like to find the effect of replacing CH (in benzene) with oxygen and sulfur (Table 6 and Fig. 3). H4C4O and H4C4S were selected in order to study the effect of replacing a six-membered ring with a five-membered one. All the molecules are planar, except those of II, which has a boat-shape (Fig. S4, ESI†). These derivatives have their dipole moment oriented along the x axis. For computation of all the molecular properties the CAM-B3LYP/6-31G* method has been employed. In addition, for the properties of IV(a,b), the BHandHLYP/6-31G* functional has been used. These functionals gave similar property values except for βzzz.| Method/basis set | Property | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| μ x | α zz | α xx | β xxx | β zzz | γ zzzz × 10−5 | γ xxxx × 10−5 | E 1 | E exc | |
| a Fig. 3. b The dipole moment is oriented along the x-axis (Fig. 3). c E 1 (kcal mol−1) = E(RDFT) − E(UDFT), method: B3LYP/6-31G*. | |||||||||
| (B-NiBDT)3 | |||||||||
| (U)CAM-B3LYP/6-31G* | 0.497 | 804 | 3836 | −13700 |
1 | −1100.0 | 5380.0 | 1.14 | 1.13 |
| C14H4Ni3S12O6 (I) | |||||||||
| (U)CAM-B3LYP/6-31G* | 1.242 | 463 | 1685 | 29880 |
0 | −0.4 | 645.8 | 5.65 | 1.47 |
| C14H4Ni3S12S6 (II) | |||||||||
| (U)CAM-B3LYP/6-31G* | 1.185 | 1259 | 1642 | 26079 |
18882 |
285.9 | 549.8 | 3.14 | 1.25 |
| C14H4Ni3O3S12 (III) | |||||||||
| (U)CAM-B3LYP/6-31G* | 1.004 | 1724 | 940 | 14935 |
−1010 | 574.9 | 74.2 | 3.14 | 1.06 |
| C14H4Ni3S15 (IVa) | |||||||||
| (U)BHandHLYP/6-31G* | 1.081 | 1665 | 864 | 8794 | −1260 | 591.0 | 43.4 | 1.25 | 1.88 |
| (U)CAM-B3LYP/6-31G* | 1.124 | 1912 | 923 | 6021 | −8811 | 889.0 | 59.5 | ||
| C14H4Ni3S15 (IVb) | |||||||||
| (U)BHandHLYP/6-31G* | 0.664 | 572 | 2009 | 18300 |
453 | −0.9 | 1013.0 | 1.57 | 1.82 |
| (U)CAM-B3LYP/6-31G* | 0.661 | 577 | 2366 | 22910 |
240 | −1.2 | 1100.0 | ||
I and II have similar properties along the x-direction, but present significant differences in their properties along the z-direction (αzz, βzzz, γzzzz). Their E1 values are also remarkably different. The above pattern of changes could be attributed (i) to the different linkers, that is H4C4O2 is the linker in I and H2C4S2 is the linker in II and (ii) the different structures of I and II. It was found that I has a linear structure, while II has a boat-shaped one (Fig. S4, ESI†). Benzene (C6H6) as a linker leads to much larger γxxxx values in comparison with 1,4-dioxin (H2C4O2) and 1,4-dithiin (H2C4S2). The same trend is observed for αxx.
In Fig. 12 we show the intrinsic atomic polarizabilities of compounds I and II (Fig. 3). Contrary to what was observed in the previous set of models the Ni atoms do not feature the largest intrinsic polarizability. Depending on their position in the framework of these chains, Ni loses large portions of its NPs. In terms of element contribution for either II/I, more than 50% of the molecular intrinsic mean polarizability originate from S atoms. In both cases, the atomic polarizabilities of Ni and S atoms notably vary depending on their position. The main trend holding for both systems is that the Ni and S atoms located at the outer chain sides are always more polarizable, and thus more sensitive to external field perturbations than those located close to the center of each chain. Also, there is a clear difference in the character of the various S atoms since FOHI analysis suggests that out of the 18 S atoms present in I, those which are directly bonded to Ni appear to be more polarizable than the S atoms of the embedded dithiin units. Finally, by comparing the average INP of O and S in free (1,4)-dithiine and in (1,4)-dioxine with the corresponding values in compounds I and II, it is revealed that there is a decrease of the average INP in all atoms of both fragments when they are present in I and II linear chains.
 | ||
| Fig. 12 FOHI intrinsic atomic polarizabilities and MIP density contour maps of C14H4Ni3S12S6 (II; Fig. 3) and C14H4Ni3S12O6 (I; Fig. 3) compounds computed at the UCAM-B3LYP/6-311G* level. Numbers in parentheses correspond to the INPs of (1,4)-dithiine and (1,4)-dioxine computed at the RCAM-B3LYP/6-311G* level. | ||
It is remarkable that II and III have, approximately, the same DC, as this is computed by E1, but several of their other properties have significant differences (e.g. βzzz, γxxxx). A similar trend has also been observed in other cases (Table 5; e.g. (NiBDT)2RA/(NiBDT)3(RA)2). These observations may be associated with the fact that the (hyper)polarizabilities are more sensitive probes, than the DC, for changes taking place in the structure of the molecule. A very small γxxxx value for III and IVa is observed. The (hyper)polarizabilities of IVa and IVb are quite different; these oligomers have a similar DC.
It is also noted that a comparison of the pairs I/II and III/IVa shows that the linkers with oxygen lead to oligomers with higher DC, in comparison to those with sulfur.
The employed linkers highlight the superiority of benzene for the production of oligomers with much larger second hyperpolarizability (γxxxx) in comparison with the other ones.
4. Vibrational contributions
We have chosen two relatively small models (Fig. S5, ESI†) to study both the electronic and vibrational contributions to αxx and γxxxx. The computed static nr contributions to the non-vanishing longitudinal diagonal component of the electrical properties computed for the spin-broken unrestricted DFT wavefunction are shown in Table 7, together with the purely electronic contributions. They were computed with the same basis set/pseudopotential combination as the electronic properties in the rest of this paper. To be consistent with the majority of the electronic results, the CAM-B3LYP functional was first tried for both molecules. It turned out, however, that the spin-broken solution of the C8O4NiS4H4 molecule (Fig. S4, ESI†) obtained with this functional showed no convergence for the property values obtained from the Romberg method. In order to have a full set of comparable values, the B3LYP functional was then applied for both molecules, where the problems observed with CAM-B3LYP did not occur. As shown in Table 7 for molecule C8S4NiS4H4 (Fig. S4, ESI†), the differences in the properties calculated with the two functionals are not very large (up to a factor of ∼2). While the electronic contributions to α and γ are of similar magnitude, the nr contributions are much larger for molecule C8S4NiS4H4 than for C8O4NiS4H4, especially for γ, which is about four orders of magnitude larger for C8S4NiS4H4 than for C8O4NiS4H4.In addition to the nr contribution to the electrical properties for the spin-broken unrestricted DFT wavefunction, we also computed the corresponding properties for the restricted DFT wavefunction, with CAM-B3LYP for molecule C8S4NiS4H4 and with B3LYP for molecule C8O4NiS4H4. It turns out that the two sets of properties differ substantially, both for the electronic as well as for the vibrational (nr) contributions (Table 7). While the polarizabilities are of similar order of magnitude, the second hyperpolarizabilities are about one order of magnitude larger with the unrestricted wavefunction.
IV. Conclusions
It is well known that there is a great need for novel photonic materials for use in a large number of applications (e.g. optical communications), which require an ever increasing improvement of their performance.In this work we propose a novel family of such materials: 1D and 2D coordination polymers, based on nickel bis(dithiolene) and benzene. Factors of cardinal importance for the photonic materials are their NLO properties; these are computed by employing density functional theory.
Extensive computational experimentation has been performed, by employing linear and planar oligomers of the 1D and 2D polymers, in order to document and demonstrate the superior NLO performance of the proposed materials. For comparison, we have also used several rich π-electron linkers (e.g. C6H6,C14H18, C24H12, and C4O2H4).
A property of key importance for this work is the second hyperpolarizability (γ). However, we computed several other properties (e.g. polarizabilities, EHOMO, ELUMO, and excitation energies), which have been correlated with γ and contributed to its rationalization. The diradical character, which was characterized by employing two indices, was also employed for the understanding of the properties of interest. This work mainly deals with the electronic contribution to (hyper)polarizabilities. However, we have also commented on the nuclear relaxation contribution to αxx and γxxxx of some relatively small model compounds. While the nr contribution for the O-containing molecule was modest, quite large contributions were found for the S-containing molecule. The electronic contributions for both molecules, on the other hand, were of similar magnitude. In addition, it was found that the spin-broken wavefunction yields quite different vibrational and electronic properties than the restricted wavefunction. Considerable care has been taken for the validation of the employed computational procedure. This involved several functionals and state-of-the-art techniques (e.g. UCCSD(T)).
The main findings of this work are the following:
(a) The 1D and 2D coordination polymers, and in particular the linear ones, (B-NiBDT)n, are likely to form the basis of a new class of efficient photonic materials with superior performance. There is a steep increase of γ with n and even for relatively small n, gigantic second hyperpolarizability values have been computed for (B-NiBDT)n.
(b) We have found a great dependence of γxxxx on (i) the linker with which the NiBDT units are fused (benzene, as a linker, leads to the maximum observed effect) and (ii) the structure, for example γ(linear; n = 6)/γ(3; planar) = 28 (Tables 1 and 3).
(c) This work, in connection with the results of our previous article,9 demonstrates a mechanism for designing photonic materials with exceptionally large nonlinearities. This mechanism involves a series of units consisting of NiBDT, π-electron rich linkers, Li substituents9 and the appropriate structure, for example a 1D arrangement leads to a much larger second hyperpolarizability in comparison to the corresponding 2D one.
Conflicts of interest
There are no conflicts to declare.References
- Y. Chen, T. Bai, N. N. Dong, F. Fan, S. F. Zhang, X. D. Zhuang, J. Sun, B. Zhang, X. Y. Zhang, J. Wang and W. J. Blau, Prog. Mater. Sci., 2016, 84, 118–157 CrossRef CAS.
- X. F. Liu, Q. B. Guo and J. R. Qiu, Adv. Mater., 2017, 29, 1605886 CrossRef PubMed.
- M. Feng, H. B. Zhan and Y. Chen, Appl. Phys. Lett., 2010, 96, 033107 CrossRef.
- T. Kambe, R. Sakamoto, T. Kusamoto, T. Pal, N. Fukui, K. Hoshiko, T. Shimojima, Z. F. Wang, T. Hirahara, K. Ishizaka, S. Hasegawa, F. Liu and H. Nishihara, J. Am. Chem. Soc., 2014, 136, 14357–14360 CrossRef CAS PubMed.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J. H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- K. Hoshiko, T. Kambe, R. Sakamoto, K. Takada and H. Nishihara, Chem. Lett., 2014, 43, 252–253 CrossRef CAS.
- R. Matsuoka, R. Sakamoto, T. Kambe, K. Takada, T. Kusamoto and H. Nishihara, Chem. Commun., 2014, 50, 8137–8139 RSC.
- Handbook of Thiophene-Based Materials: Applications in Organic Electronics and Photonics, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- A. Avramopoulos, H. Reis, N. Otero, P. Karamanis, C. Pouchan and M. G. Papadopoulos, J. Phys. Chem. C, 2016, 120, 9419–9435 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. Watanabe, T. Taniguchi and H. Kanda, Nat. Mater., 2004, 3, 404–409 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- E. Bianco, S. Butler, S. S. Jiang, O. D. Restrepo, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 4414–4421 CrossRef CAS PubMed.
- A. Splendiani, L. Sun, Y. B. Zhang, T. S. Li, J. Kim, C. Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- Y. M. Lin, C. Dimitrakopoulos, K. A. Jenkins, D. B. Farmer, H. Y. Chiu, A. Grill and P. Avouris, Science, 2010, 327, 662 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- T. Y. Lu, H. Feng, S. W. Yang and J. C. Zheng, Comput. Mater. Sci., 2017, 126, 170–175 CrossRef CAS.
- T. Pal, T. Kambe, T. Kusamoto, M. L. Foo, R. Matsuoka, R. Sakamoto and H. Nishihara, ChemPlusChem, 2015, 80, 1255–1258 CrossRef CAS.
- A. J. Clough, J. W. Yoo, M. H. Mecklenburg and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 118–121 CrossRef CAS PubMed.
- Q. Tang and Z. Zhou, J. Phys. Chem. C, 2013, 117, 14125–14129 CAS.
- F. Shojaei, J. R. Hahn and H. S. Kang, Chem. Mater., 2014, 26, 2967–2974 CrossRef CAS.
- Z. F. Wang, N. H. Su and F. Liu, Nano Lett., 2013, 13, 2842–2845 CrossRef CAS PubMed.
- J. Zhou, RSC Adv., 2014, 4, 13361–13366 RSC.
- F. Shojaei and H. S. Kang, J. Phys. Chem. C, 2014, 118, 25626–25632 CAS.
- H. M. Liu, X. L. Li, L. Chen, X. L. Wang, H. Z. Pan, X. M. Zhang and M. W. Zhao, J. Phys. Chem. C, 2016, 120, 3846–3852 CAS.
- A. J. Clough, J. M. Skelton, C. A. Downes, A. A. de la Rosa, J. W. Yoo, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2017, 139, 10863–10867 CrossRef CAS PubMed.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dinca, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dinca, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- J. S. Cui and Z. T. Xu, Chem. Commun., 2014, 50, 3986–3988 RSC.
- S. Biswas, C. S. Tiwary, S. Vinod, A. K. Kole, U. Chatterjee, P. Kumbhakar and P. M. Ajayan, J. Phys. Chem. C, 2017, 121, 8060–8069 Search PubMed.
- P. Kumbhakar, A. K. Kole, C. S. Tiwary, S. Biswas, S. Vinod, J. Taha-Tijerina, U. Chatterjee and P. M. Ajayan, Adv. Opt. Mater., 2015, 3, 828–835 CrossRef CAS.
- K. P. Wang, J. Wang, J. T. Fan, M. Lotya, A. O'Neill, D. Fox, Y. Y. Feng, X. Y. Zhang, B. X. Jiang, Q. Z. Zhao, H. Z. Zhang, J. N. Coleman, L. Zhang and W. J. Blau, ACS Nano, 2013, 7, 9260–9267 CrossRef CAS PubMed.
- N. Liaros, A. B. Bourlinos, R. Zboril and S. Couris, Opt. Express, 2013, 21, 21027–21038 CrossRef CAS PubMed.
- L. R. Dalton, A. W. Harper, B. Wu, R. Ghosn, J. Laquindanum, Z. Y. Liang, A. Hubbel and C. Z. Xu, Adv. Mater., 1995, 7, 519–540 CrossRef CAS.
- S. Ellinger, K. R. Graham, P. J. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. G. Mei, L. A. Padilha, T. R. Ensley, H. H. Hu, S. Webster, D. J. Hagan, E. W. Van Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805–3817 CrossRef CAS.
- P. K. Hegde, A. V. Adhikari, M. G. Manjunatha, C. S. S. Sandeep and R. Philip, Adv. Polym. Technol., 2011, 30, 312–321 CrossRef CAS.
- X. W. Zhan, Y. Q. Liu, D. B. Zhu, X. C. Liu, G. Xu and P. X. Ye, Chem. Phys. Lett., 2001, 343, 493–498 CrossRef CAS.
- M. Liu, H. S. Quah, S. C. Wen, J. Y. Wang, P. S. Kumar, G. Eda, J. J. Vittal and W. Ji, J. Mater. Chem. C, 2017, 5, 2936–2941 RSC.
- Y. Matsuzaki, M. Nakano, K. Yamaguchi, K. Tanaka and T. Yamabe, Chem. Phys. Lett., 1996, 263, 119–125 CrossRef CAS.
- X. W. Zhan, Y. Q. Liu, D. B. Zhu, X. C. Liu, G. Xu and P. X. Ye, Chem. Phys. Lett., 2002, 362, 165–169 CrossRef CAS.
- C. M. Amb, C. L. Heth, S. J. Evenson, K. I. Pokhodnya and S. C. Rasmussen, Inorg. Chem., 2016, 55, 10978–10989 CrossRef CAS PubMed.
- L. Pilia, M. Pizzotti, F. Tessore and N. Robertson, Inorg. Chem., 2014, 53, 4517–4526 CrossRef CAS PubMed.
- T. B. Faust, P. M. Usov, D. M. D'Alessandro and C. J. Kepert, Chem. Commun., 2014, 50, 12772–12774 RSC.
- X. B. Yang, L. Zhou, L. B. Huang, J. J. Xu, Y. Zhou, S. T. Han, Z. X. Xu, V. C. Y. Lau, M. H. W. Lam, W. Y. Wong and V. A. L. Roy, RSC Adv., 2013, 3, 12075–12079 RSC.
- L. Hu, J. Qin, N. Zhou, Y. F. Meng, Y. Xu, J. L. Zuo and X. Z. You, Dyes Pigm., 2012, 92, 1223–1230 CrossRef CAS.
- T. T. Bui, O. Thiebaut, E. Grelet, M. F. Achard, B. Garreau-deBonneval and K. I. M. C. Ching, Eur. J. Inorg. Chem., 2011, 2663–2676, DOI:10.1002/ejic.201001288.
- X. Q. Wang, Q. Ren, J. W. Chen, W. T. Yu, H. L. Fang, T. B. Li, H. J. Cong, X. T. Liu, L. Y. Zhu, G. H. Zhang and D. Xu, Solid State Sci., 2011, 13, 896–903 CrossRef CAS.
- S. Dalgleish, J. G. Labram, Z. Li, J. P. Wang, C. R. McNeill, T. D. Anthopoulos, N. C. Greenham and N. Robertson, J. Mater. Chem., 2011, 21, 15422–15430 RSC.
- P. Basu, A. Nigam, B. Mogesa, S. Denti and V. N. Nemykin, Inorg. Chim. Acta, 2010, 363, 2857–2864 CrossRef CAS PubMed.
- X. Q. Wang, Q. Ren, H. L. Fan, J. W. Chen, Z. H. Sun, T. B. Li, X. T. Liu, G. H. Zhang, D. Xu and W. L. Liu, J. Cryst. Growth, 2010, 312, 2206–2214 CrossRef CAS.
- P. Deplano, L. Pilia, D. Espa, M. L. Mercuri and A. Serpe, Coord. Chem. Rev., 2010, 254, 1434–1447 CrossRef CAS.
- B. Garreau-deBonneval, K. I. M. C. Ching, F. Alary, T. T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- M. Iyoda and H. Shimizu, Chem. Soc. Rev., 2015, 44, 6411–6424 RSC.
- W. Q. Li, Q. Peng, H. L. Ma, J. Wen, J. Ma, L. A. Peteanu and Z. G. Shuai, Chem. Mater., 2017, 29, 2513–2520 CrossRef CAS.
- J. D. C. Gonzalez, M. Iyoda and J. P. Rabe, Nat. Commun., 2017, 8 Search PubMed.
- B. Holzer, J. Bintinger, D. Lumpi, C. Choi, Y. Kim, B. Stoger, C. Hametner, M. Marchetti-Deschmann, F. Plasser, E. Horkel, I. Kymissis and J. Frohlich, ChemPhysChem, 2017, 18, 549–563 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- B. Kirtman, V. Lacivita, R. Dovesi and H. Reis, J. Chem. Phys., 2011, 135, 154101 CrossRef PubMed.
- T. Miletic, A. Fermi, I. Orfanos, A. Avramopoulos, F. De Leo, N. Demitri, G. Bergamini, P. Ceroni, M. G. Papadopoulos, S. Couris and D. Bonifazi, Chem. – Eur. J., 2017, 23, 2363–2378 CrossRef CAS PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- S. Bonness, H. Fukui, K. Yoneda, R. Kishi, B. Champagne, E. Botek and M. Nakano, Chem. Phys. Lett., 2010, 493, 195–199 CrossRef CAS.
- R. Kishi, S. Bonness, K. Yoneda, H. Takahashi, M. Nakano, E. Botek, B. Champagne, T. Kubo, K. Kamada, K. Ohta and T. Tsuneda, J. Chem. Phys., 2010, 132, 094107 CrossRef PubMed.
- O. Loboda, R. Zalesny, A. Avramopoulos, J. M. Luis, B. Kirtman, N. Tagmatarchis, H. Reis and M. G. Papadopoulos, J. Phys. Chem. A, 2009, 113, 1159–1170 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128 Search PubMed.
- C. G. Liu, M. L. Gao and Z. J. Wu, RSC Adv., 2014, 4, 38300–38309 RSC.
- X. H. Xie, D. S. Hao and J. C. Yang, Chem. Phys., 2015, 461, 11–19 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. I. Lu, Theor. Chem. Acc., 2015, 134, 1589 CrossRef.
- D. Jacquemin, E. A. Perpete, M. Medved, G. Scalmani, M. J. Frisch, R. Kobayashi and C. Adamo, J. Chem. Phys., 2007, 126, 144105 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- L. Serrano-Andres, A. Avramopoulos, J. B. Li, P. Labeguerie, D. Begue, V. Kello and M. G. Papadopoulos, J. Chem. Phys., 2009, 131, 134312 CrossRef PubMed.
- H. Fukui, R. Kishi, T. Minami, H. Nagai, H. Takahashi, T. Kubo, K. Kamada, K. Ohta, B. Champagne, E. Botek and M. Nakano, J. Phys. Chem. A, 2008, 112, 8423–8429 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, Gaussian 09, Revision D.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H. D. Cohen and C. C. Roothaan, J. Chem. Phys., 1965, 43, S034–S039 Search PubMed.
- P. J. Davis and P. Rabinowitz, Numerical Intergration, Blaisdell, London, 1967 Search PubMed.
- W. Romberg, Kgl. Norske Vid. Selsk. Forsk, 1955, 28, 30–36 Search PubMed.
- H. Rutishauser, Num. Math., 1963, 5, 48–54 CrossRef.
- L. Salem and C. Rowland, Angew. Chem., Int. Ed., 1972, 11, 92 CrossRef CAS.
- J. Wirz, Pure Appl. Chem., 1984, 56, 1289–1300 CrossRef CAS.
- V. Bachler, G. Olbrich, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41, 4179–4193 CrossRef CAS PubMed.
- L. Serrano-Andres, D. J. Klein, P. Schleyer and J. M. Oliva, J. Chem. Theory Comput., 2008, 4, 1338–1347 CrossRef CAS PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- T. Kubo, A. Shimizu, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Org. Lett., 2007, 9, 81–84 CrossRef CAS PubMed.
- M. E. Casida and M. Huix-Rotllant, Annu. Rev. Phys. Chem., 2012, 63, 287–323 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpete, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS PubMed.
- D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpete and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed.
- C. A. Guido, S. Knecht, J. Kongsted and B. Mennucci, J. Chem. Theory Comput., 2013, 9, 2209–2220 CrossRef CAS PubMed.
- D. Geldof, A. Krishtal, F. Blockhuys and C. Van Alsenoy, J. Chem. Theory Comput., 2011, 7, 1328–1335 CrossRef CAS PubMed.
- P. Bultinck, C. Van Alsenoy, P. W. Ayers and R. Carbo-Dorca, J. Chem. Phys., 2014, 140, 144104 CrossRef PubMed.
- N. Otero, C. Van Alsenoy, C. Pouchan and P. Karamanis, J. Comput. Chem., 2015, 36, 1831–1843 CrossRef CAS PubMed.
- N. Otero, C. Van Alsenoy, P. Karamanis and C. Pouchan, Int. J. Comput. Theor. Chem., 2013, 1021, 114–123 CrossRef CAS.
- P. Karamanis, N. Otero, C. Pouchan, J. J. Torres, W. Tiznado, A. Avramopoulos and M. G. Papadopoulos, J. Comput. Chem., 2014, 35, 829–838 CrossRef CAS.
- P. Karamanis, N. Otero and C. Pouchan, J. Phys. Chem. C, 2015, 119, 11872–11885 CAS.
- C. van Alsenoy and A. Peeters, J. Mol. Struct. THEOCHEM, 1993, 286, 19–34 CrossRef.
- B. Rousseau, A. Peeters and C. Van Alsenoy, Chem. Phys. Lett., 2000, 324, 189–194 CrossRef CAS.
- D. M. Bishop and B. Kirtman, J. Chem. Phys., 1991, 95, 2646–2658 CrossRef CAS.
- D. M. Bishop and B. Kirtman, J. Chem. Phys., 1992, 97, 5255–5256 CrossRef CAS.
- D. M. Bishop, J. M. Luis and B. Kirtman, J. Chem. Phys., 1998, 108, 10013–10017 CrossRef CAS.
- D. M. Bishop, M. Hasan and B. Kirtman, J. Chem. Phys., 1995, 103, 4157–4159 CrossRef CAS.
- 1 a.u. of energy ≈ 27.21070 eV, ≈ 627.51 kcal mol−1, 1 a.u. of electric field ≈ 1.71514 × 107 esu, ≈ 5.14192 × 1011 V m−1, 1 a.u. dipole moment ≈ 2.54174 D, ≈ 8.47831 × 10−30 C m, 1 a.u. of polarizability ≈ 0.148176 × 10−24 esu, ≈ 0.164867 × 10−40 C2 m2 J−1, 1 a.u. of first hyperpolarizability ≈ 0.863993 × 10−32 esu, ≈ 0.320662 × 10−52 C3 m3 J−2, 1 a.u. of second hyperpolarizability ≈ 0.503717 × 10−39 esu, ≈ 0.623597 × 10−64 C4 m4 J−3.
- Y. W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS PubMed.
- E. Rudberg, P. Salek and Y. Luo, Nano Lett., 2007, 7, 2211–2213 CrossRef CAS PubMed.
- O. Hod and G. E. Scuseria, ACS Nano, 2008, 2, 2243–2249 CrossRef CAS PubMed.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS.
- Y. Su, X. Wang, L. Wang, Z. Zhang, X. Wang, Y. Song and P. P. Power, Chem. Sci., 2016, 7, 6514–6518 RSC.
- A. Avramopoulos, H. Reis, G. A. Mousdis and M. G. Papadopoulos, Eur. J. Inorg. Chem., 2013, 4839–4850 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc05047j |
| This journal is © The Royal Society of Chemistry 2018 |