
Impact of the synthesis method on the solid-state charge transport of radical polymers†
Yiren
Zhang‡
a,
Albert
Park‡
b,
Alicia
Cintora
a,
Stephen R.
McMillan
c,
Nicholas J.
Harmon
c,
Austin
Moehle
b,
Michael E.
Flatté
 c,
Gregory D.
Fuchs
*b and
Christopher K.
Ober
*a
c,
Gregory D.
Fuchs
*b and
Christopher K.
Ober
*a
aMaterials Science and Engineering, Cornell University, Ithaca, New York 14853, USA
bApplied and Engineering Physics, Cornell University, Ithaca, New York 14853, USA. E-mail: gdf9@cornell.edu
cOptical Science and Technology Center and Department of Physics and Astronomy, University of Iowa, Iowa City, Iowa 52242, USA
First published on 8th December 2017
Abstract
There are conflicting reports in the literature about the presence of room temperature conductivity in poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA), a redox active polymer with radical groups pendent to an insulating backbone. To understand the variability in the findings across the literature and synthetic methods, we prepared PTMA using three living methods – anionic, ATRP and RAFT polymerization. We find that all three synthetic methods produce PTMA with radical yields of 70–80%, controlled molecular weight, and low dispersity. Additionally, we used on-chip EPR to probe the robustness of radical content in solid films under ambient air and light, and found negligible change in the radical content over time. Electrically, we found that PTMA is highly insulating – conductivity in the range 10−11 S cm−1 – regardless of the synthetic method of preparation. These findings provide greater clarity for potential applications of PTMA in energy storage.
1 Introduction
Radical polymers, a class of polymers with robust radical pendent moieties along their backbone, have demonstrated suitable performance to meet the requirements for energy storage applications such as organic radical batteries (ORBs).1–6 Poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA) remains the most popular stable radical polymer for ORBs due to its facile preparation and fast redox kinetics of the 2,2,6,6-tetramethyl pipiridine-1-oxyl (TEMPO) stable radical moiety side group. The incorporation of TEMPO moieties into polymers also has a rich history in the study of such diverse applications as organic magnets7 and is currently being explored in antifouling coatings as a component to inhibit curing of biological adhesives.8 Such structures are also associated with certain types of stable radical polymerization.9Pristine PTMA has been largely considered to be an electrically insulating material in ORBs, which has been synthesized primarily via radical polymerization and subsequent oxidation.10–13 As a result, many attempts have been made to improve its charge transport capabilities in battery electrodes, such as by incorporating carbon black-based additives to the active cathode material.14–16 Recently, however, it has been reported that thin films of PTMA synthesized via reversible addition–fragmentation chain transfer (RAFT) can be considered as a novel class of highly-transparent polymers with a conductivity of ∼1 × 10−6 S cm−1, comparable to π-conjugated semiconducting polymers such as undoped poly(3-hexylthiophene) (P3HT).17–19 Motivated by the lack of agreement over the intrinsic conductivity of PTMA, we have set out to understand the impact of different synthetic preparation routes on charge transport.
Despite the versatility of synthetic techniques, the only prior reported preparation of conductive PTMA homopolymer follows a two-step process which involves the preparation of a precursor polymer, poly(2,2,6,6-tetramethyl-4-piperidyl methacrylate) (PTMPM) by RAFT and subsequent oxidation by peracid.17,20 Limited by the specific chemistry of polymerization or chemical treatment, the resulting PTMA is often accompanied by unwanted functionalities due to incomplete conversion or side reactions. These additional functionalities were found to have an effect on the measured solid state conductivity of PTMA thin films.20 Efforts to understand the charge transport in these systems found that the intrinsic conductivity of PTMA can be increased by a factor of two by incorporating the oxoammonium cation, which can be introduced either by chemical oxidation of PTMPM or as an external dopant. The conductivity enhancement was attributed to the oxoammonium cations serving as additional hopping sites for the nitroxide radical.20,21
In support of these results, charge transport in electrochemical systems has also been considered in the literature.22 TEMPO stable radicals pendent to an insulating, hydrocarbon backbone can effectively transport charge with the aid of an electrolyte solution. It has been suggested that in these systems, charge carriers travel from TEMPO group to TEMPO group via a variable range hopping mechanism enabled by chain motion.5,23,24 This model suggests that there can be a series of electron hops in the polymer from pendent site to pendent site with the aid of delocalized electrons. As such, elucidating the nature of the electron transport process in non-conjugated macromolecules with pendent stable radical groups could aid in opening new pathways to the designs of organic electronic materials.
Given the disagreement on the conductivity of non-conjugated stable radical polymers, direct comparison of PTMA prepared by different synthesis techniques is a crucial test of the robustness of charge transport phenomena that can reveal the role of structure and impurities in conductivity.
In this work, we have created a set of PTMA polymers synthesized via three different synthetic methods to understand how the variation of resulting pendent groups can affect the intrinsic conductivity of PTMA thin films. We used two living radical polymerization methods, RAFT and atom transfer radical polymerization (ATRP), for the preparation of two precursor polymers of PTMPM, which were then transformed to PTMA polymers (PTMA-RAFT, PTMA-H2O2, PTMA-mCPBA) by oxidation reactions. ATRP of 4-methacryloyloxy-1-((1′-phenylethyl)oxy-2,2,6,6-tetramethylpiperidine) (MPEOT) resulted in the precursor polymer PMPEOT, which was transformed to PTMA-O2 by oxidative C–O bond cleavage of the polyalkoxyamines25 (Scheme 1). For comparison, we also produced PTMA (PTMA-anionic) with both low molecular weight distribution and high TEMPO stable radical yield via anionic polymerization. Considering that this method can directly polymerize the TEMPO-bearing methacrylate monomer with few side reactions, PTMA-anionic can act as a suitable model polymer for physical characterization.
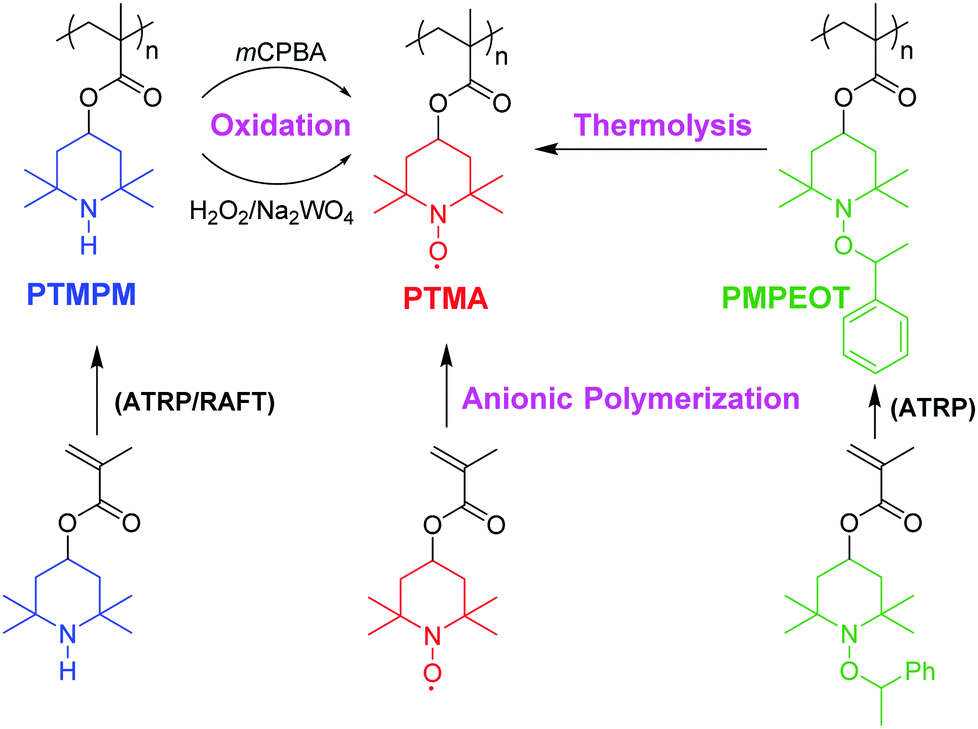 | ||
| Scheme 1 Synthesis of PTMA and precursor polymers via living polymerization and subsequent oxidation. | ||
To characterize conductivity, we used equipment designed for measuring high-resistance PTMA samples in a 4-wire geometry. This both avoids the effect of contact resistance and eliminates artifacts that can occur when the sample resistance is higher than the meter input impedance. In the following sections, we describe our studies on this comprehensive measurement of PTMA conductivity, which is a key step to the understanding of charge transport in a neat PTMA film. Finally, we analyse the results in the context of a variable range hopping theoretical analysis.
2 Experimental section
2.1 Materials
All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise noted. m-Chloroperbenzoic acid (mCPBA) was washed with DI water from diethyl ether and dried under vacuum. 2,2,6,6-Tetramethyl-4-piperidyl methacrylate (TMPM) and dibenzo-18-crown-6 were purchased from TCI America. Dibenzo-18-crown-6 was recrystallized from acetonitrile and dried under vacuum overnight. CuBr was stirred in acetic acid overnight, washed repeatedly with anhydrous ethanol and diethyl ether and dried under vacuum before use. The TEMPO methacrylate monomer6 and 2-phenyl-2-propylbenzodithioate26 were synthesized according to literature procedures.2.2 Characterization
Molecular weights (Mn) and polydispersities (Mw/Mn) of the PTMA samples and precursor polymers were dertermined by gel permeation chromatography (GPC). The GPC was performed on Waters Ambient Temperature GPC equipped with a Waters 410 differential refractive index detector. GPC columns were eluted at 1.0 mL min−1 with tetrahydrofuran (THF) at 40 °C. Polymer samples were dissolved in THF at 1 mg mL−1. Polystyrene standards with narrow molecular weight distribution were used for calibration. 1H-NMR spectra were recorded on a Varian INOVA 400 (1H, 400 MHz) spectrometer. Spectra were referenced to the residual chloroform (7.26 ppm) signal. The ATR-FTIR spectra ranging between wavenumbers 4000 and 400 cm−1 were recorded under vacuum with Bruker Vertex V80V Vacuum FTIR system. Spectra resolution was set at 4 cm−1.2.3 Polymer synthesis
2.4 Solution-state spin characterization
Solution-state measurements were performed in a Bruker X-band EPR spectrometer at room temperature. PTMA solutions for EPR measurements (1 mM of repeating unit concentration) were prepared by dilution of 384.6 μL of PTMA stock solutions to 4.00 mL of total volume with toluene. Stock solution was prepared by dissolving 10.0 mg of each polymer in 4.00 mL toluene overnight. The resulting EPR spectra were double-integrated and compared with the TEMPO standard (1 mM solution in toluene) to calculate radical yields. Error bars were generated by calculating the standard deviation of average radical yield from three parallel quantitative EPR tests in which the same PTMA stock solution was used to prepare independent test solutions.2.5 Solid-state spin characterization using a coplanar waveguide
Samples were prepared either by drop casting or spin coating a 10 mg mL−1 solution of PTMA directly onto the microfabricated coplanar waveguide meander.27 The waveguide was then wire-bonded into the microwave chip carrier, and the 6 GHz transmitted microwave power was monitored as a function of magnetic field with a microwave diode and a lock-in amplifier. Error bars were calculated by propagating errors including the uncertainties from the fitting and the noise in the lock-in voltage.2.6 Electrical characterization
Micro-fabricated 4-wire lateral templates consisting of 100 nm-thick, 2.5 mm-long gold electrodes were patterned on insulating, thermally oxidized Si substrates. The four electrodes were equally spaced at a distance of either 2, 5, 10, or 20 μm (Fig. 1). We used more than one electrode spacing for each PTMA type to validate that the results are not an artifact of the geometry. PTMA films were prepared by dropcasting PTMA solution (25 mg mL−1 in toluene) on a template, and heating at 70 °C for 15 min to remove the residual toluene. PTMA film thickness was measured using both an ellipsometer and Tencor AlphaStep 500 profilometer. After wire-bonding the template into a chip carrier, each sample was placed within a continuously pumped vacuum box to eliminate resistive shunting through a surface water layer. Current was applied to the outer electrodes and voltage measured between the inner electrodes. To eliminate a systematic error that can arise when measuring samples with very large resistance (a typical resistance is between 60 to 200 GΩ in our experiment) relative to the input impedance of a standard voltmeter, a Keithley 6514 electrometer that has an input impedance of 210 TΩ was used. Current was sourced using a Keithley 6610 precision current source, enabling current with sub-1 pA accuracy. The set-up was calibrated against standard Ohmite MOX resistors in the range 100 GΩ to 1 TΩ. (for the details of calibration please refer to the ESI†) As a control, the resistance of a 4-wire template in our set-up without a PTMA film under the same conditions was measured and the resistance was above 10 TΩ which exceeds the reasonable range for calibration. All measurements were done in a Faraday cage to prevent electrostatic artifacts. | ||
| Fig. 1 (a) Schematics of circuit for conductivity measurement. (b) Optical image of fabricated template with 2 μm spacing between electrodes. | ||
3 Results and discussion
3.1 Polymerization
Scheme 1 summarizes our synthetic strategy of PTMA synthesis in this study. In previous reports, PTMA synthetic routes mainly fall into two categories: (i) direct polymerization of TEMPO-containing methacrylate monomer using reactions that are compatible with stable radicals, such as anionic polymerization28 and group-transfer polymerization;29 and (ii) synthesis of precursor polymers via radical polymerization, which can be terminated by nitroxide radical, and subsequent radical regeneration via chemical treatment. To overcome the intrinsic drawback of traditional radical polymerizations, including lack of control over the molar mass and the dispersity, reversible-deactivation polymerizations allow the creation of well controlled PTMA precursor polymer due to their fast initiation rates and consistent concentration of active sites. To minimize the influence of dispersity, here we used only living polymerization, including anionic polymerization, ATRP and RAFT.Table 1 summarizes the corresponding polymerization methods, chemical oxidation, molecular weights and dispersity index (PDI) of synthesized PTMA samples. All of the PTMA products show low PDI of 1.15–1.28, which is in agreement with previous reports. Among them, PTMA-anionic show a narrow dispersity, indicating that the TEMPO bearing methacrylate monomers can be polymerized with high suppression of side reactions on nitroxide radical via sodium-mediated anionic polymerization. Such controlled polymer structures are desired in exploration of the effects of different existing pendent groups due to minimization of molecular weight and distribution effects.
| Sample | Polymerization | Oxidation | M n (kg mol−1) | Đ |
|---|---|---|---|---|
| PTMA-anionic | Anionic | — | 19.9 | 1.22 |
| PTMA-mCPBA | ATRP | mCPBA | 24.7 | 1.15 |
| PTMA-H2O2 | ATRP | H2O2 | 24.4 | 1.16 |
| PTMA-O2 | ATRP | O2 | 14.2 | 1.17 |
| PTMA-RAFT | RAFT | mCPBA | 17.8 | 1.28 |
3.2 Oxidation
Due to the fact that modern synthetic chemistry of stable nitroxides developed historically from piperidine derivatives, the oxidation of sterically hindered secondary amines by peroxides and peracids has been the most popular preparative method for the synthesis of PTMA. Though various reaction conditions have been used, including H2O2/EDTA/Na2WO4 combination and mCPBA, the oxidation of amine actually occurs by a similar mechanism.30 In the first stage, the hydroxylamino derivative (N–OH) is generated as a major product, which is converted into an oxoammonium moiety (+N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) via subsequent oxidation. The consumption of +NO, the intermediate product, is realized by reaction with reducing species, which is hydroxylamino derivative (N–OH) for mCPBA system and H2O2 derivative for H2O2/EDTA/Na2WO4 system.
O) via subsequent oxidation. The consumption of +NO, the intermediate product, is realized by reaction with reducing species, which is hydroxylamino derivative (N–OH) for mCPBA system and H2O2 derivative for H2O2/EDTA/Na2WO4 system.
Meanwhile, thermolysis of alkoxyamine under aerated conditions can also be a reliable method for efficient TEMPO recovery, considering its simpler mechanism and fewer intermediate species compared with the oxidation of amines. Here, optimized oxidation conditions using either H2O2/Na2EDTA/Na2WO4 combination and mCPBA were adopted for our PTMPM samples, while PMPEOT polymer were deprotected in tert-butylbenzene at 135 °C under oxygen atmosphere. After oxidation, all the PTMA samples were pale orange solid, while corresponding polymer solutions exhibited the characteristic absorption of TEMPO moiety at 463 nm, suggesting the successful generation of TEMPO radical in all the PTMA samples.
Prevention of unintentional oxoammonium (+NO) production is important to our study, since we want to establish the base conductivity of pristine PTMA, and oxoammonium has been reported previously as a conductivity enhancing dopant.21,22,31,32 We used attenuated total internal reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy to study the existence of +NO species in our PTMA samples as shown in Fig. 2. Compared with their precursor polymers, all the PTMA samples exhibit the characteristic peak of N–O˙ at 1365 cm−1, while only PTMA oxidized by mCPBA show an observable single peak at 1540 cm−1, which corresponds to the +NO. Beside the disproportionation and over-oxidation by mCPBA, this can also be explained by the inefficient reduction of intermediate +NO formed in amine oxidation by polymer-bounded N–OH, while reduction by excess H2O2 in PTMA-H2O2 is enough for complete +NO reduction. The unavoidable presence of +NO is consistent with lower radical conversion in PTMA oxidized by mCPBA as compared with other methods. In the samples not oxidized with mCPBA, we conclude that the pendent groups consist only of TEMPO stable radicals and a small fraction of corresponding precursor groups. These materials with their high purity and range of synthetic strategies appear to be suitable systems for the subsequent conductivity tests.
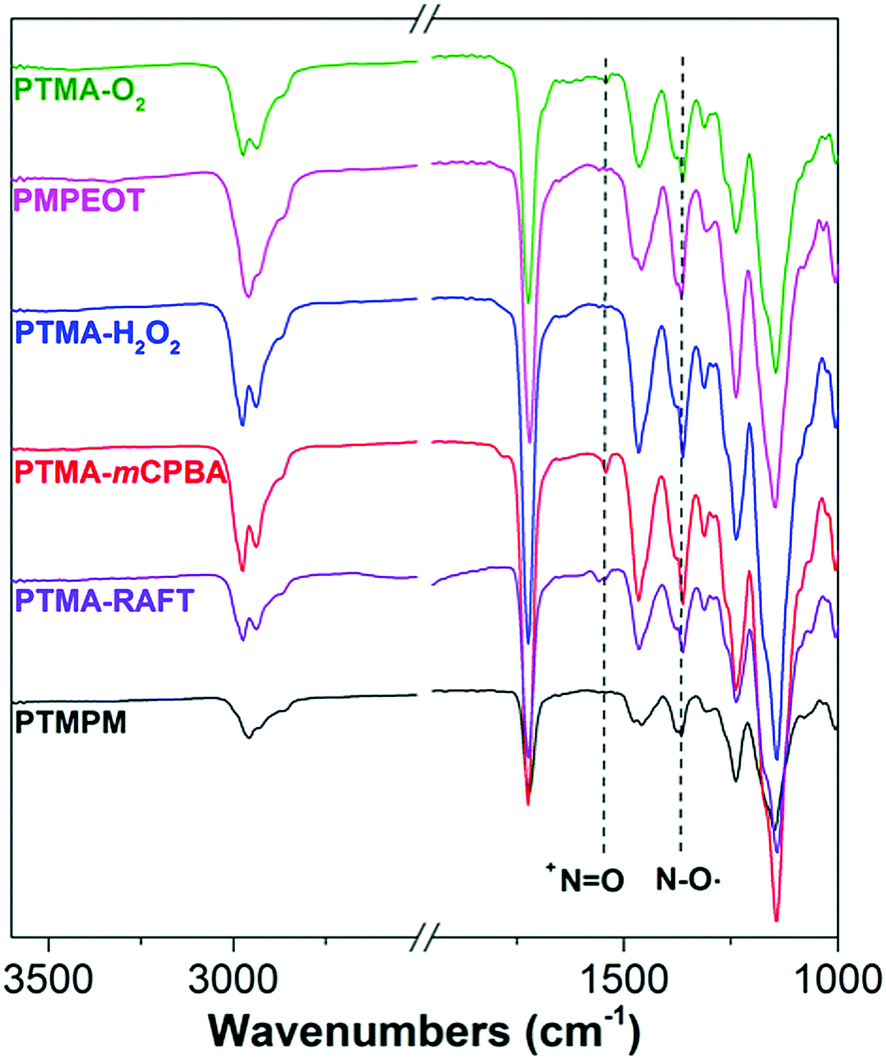 | ||
| Fig. 2 FTIR of PTMA samples and the precursor polymers. The peak at 1365 cm−1 is associated with N–O˙, while the peak at 1540 cm−1 is associated with +NO. The +NO peak is only observable in mCPBA-oxidized PTMA (PTMA-RAFT, PTMA-mCPBA). | ||
3.3 Spin characterization
We measured radical yields using quantitative solution-state electron paramagnetic resonance (EPR). Fig. 3(b) shows that the radical yield of PTMA, regardless of synthesis approach, varied by only 10 percent within the range 70 to 80%. PTMA-anionic had the highest average yield of 77%, while PTMA-ATRP showed lowest yield of 70%. Although we prepared PTMA using different polymerization and oxidation methods, we found no significant variation in radical content among all PTMA samples (Fig. 3(b)). The absence of a triplet signal, which originates either from free-floating TEMPO molecules or isolated radical groups along the polymer chain, further confirms high radical contents and good purity in our PTMA samples. Consistent with FTIR results and with our understanding of the synthetic methods, we expect that most of the side-groups that are not converted to radicals are not electrically active oxoammonium.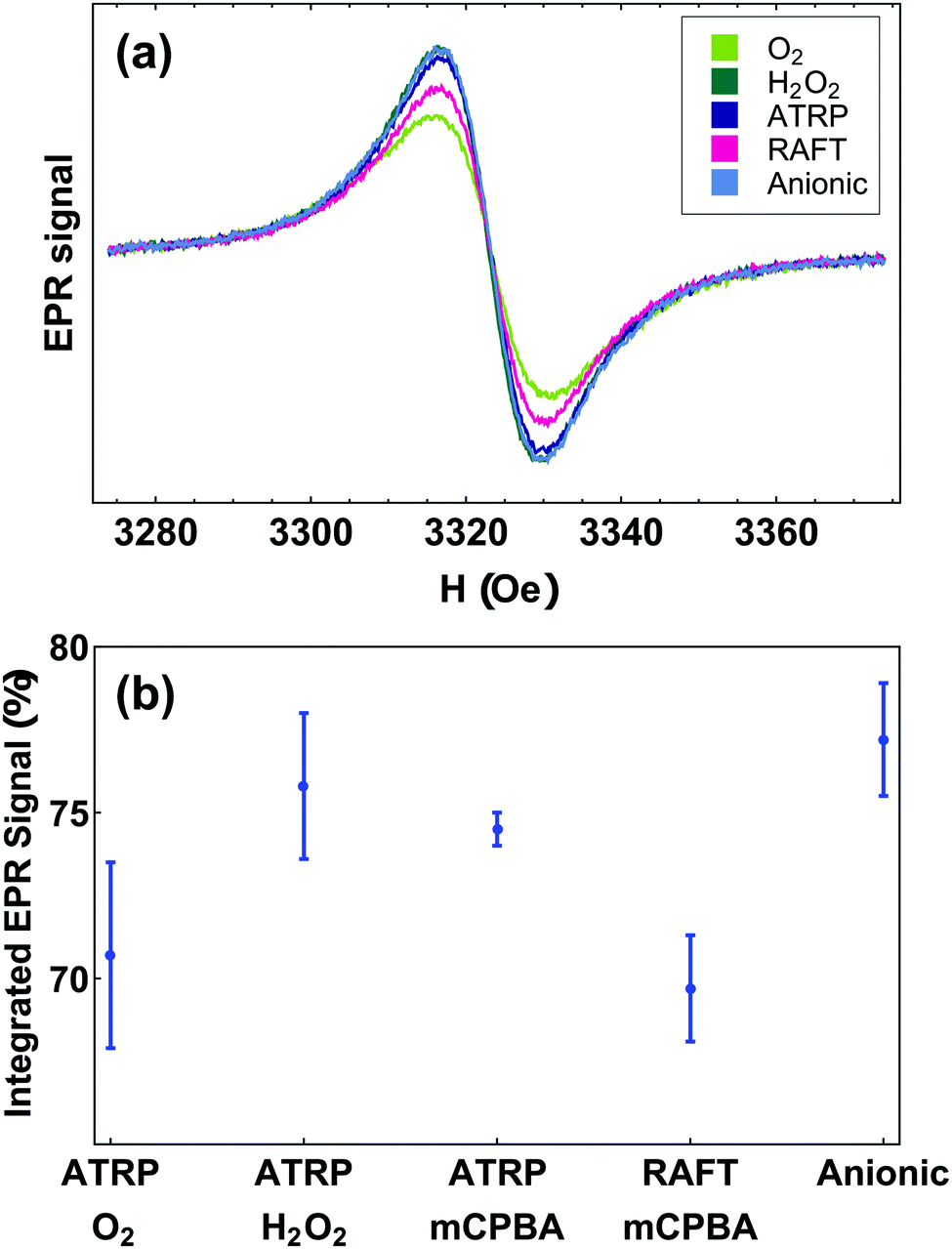 | ||
| Fig. 3 (a) EPR lineshapes of PTMA samples. (b) Radical yield of PTMA samples determined by liquid-state EPR. | ||
Motivated to understand the stability of the radical spins in solid films, which is relevant for applications, we also performed room temperature EPR of solid PTMA films using a broadband, on-chip coplanar waveguide set-up shown in Fig. 4a and b. The transmitted microwave power, P, that passes through a meandering coplanar waveguide is demodulated with respect to a 6 G, 587 Hz magnetic field, H, modulation. This is a measurement of dP/dH which rejects H independent microwave power transmission. As the static component of H is swept through the spin resonance, we expect a single, derivative-of-Lorentzian line-shape because of the high radical yield in our materials and because of the decreased radical inter-chain spacing within a solid film. We verified that the signal scales with film thickness as expected for thin films, and that spin-coating and drop-casting give similar amplitudes once normalized for film thickness.
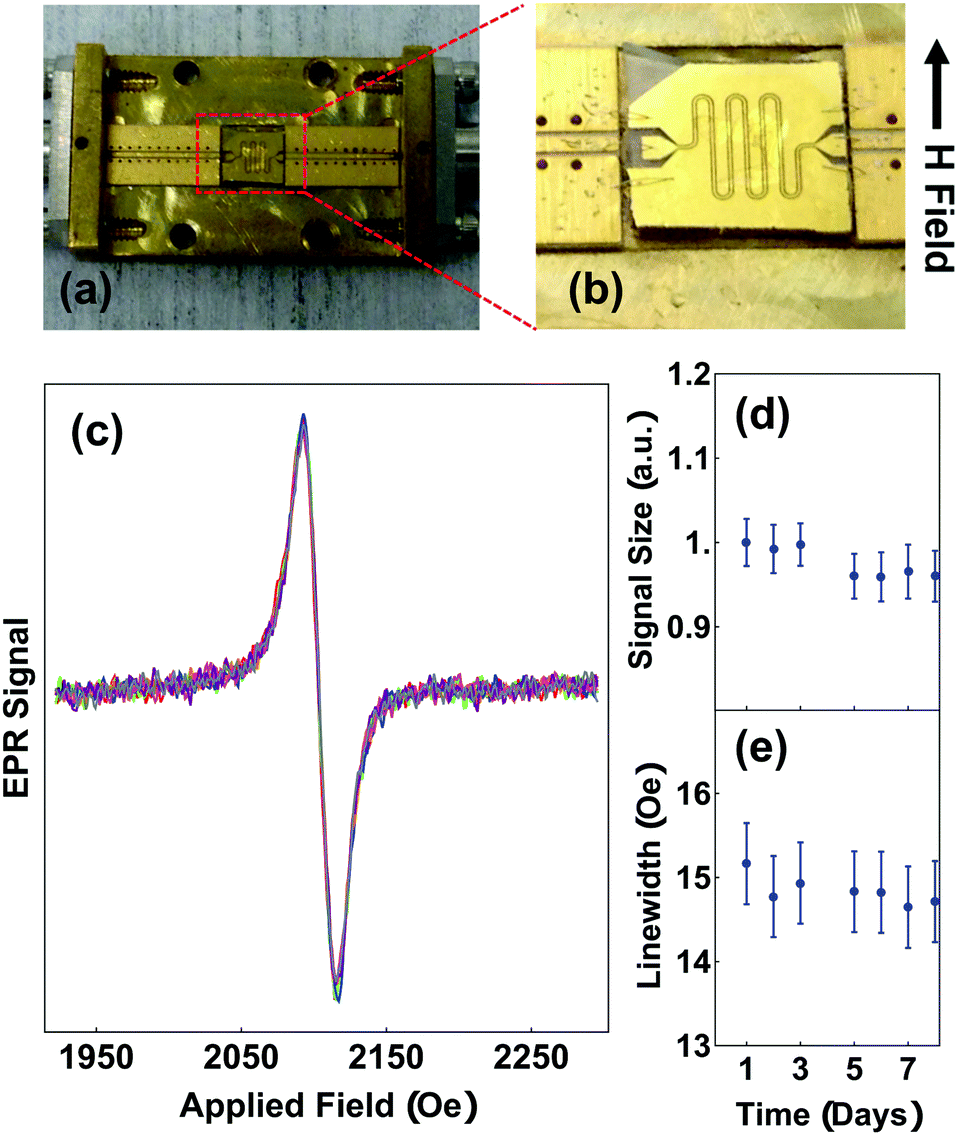 | ||
| Fig. 4 EPR (a) Brass box with coplanar waveguide for microwave experiments (b) RF waveguide wire bonded to a box with sample drop casted on top. (c) Overlay of on-chip EPR curves taken for 8 days (d and e) change in size of the signal and linewidth over the course of 8 days. | ||
The robustness of radical spins in PTMA exposed to ambient light and air is a particular concern for many applications, including for conductive polymer materials.33–36 Although the radical spins in PTMA films are generally believed to be stable, there has been no prior quantitative test in solid films, which is the relevant form of the material for applications. Fig. 4(c) shows our measurement of waveguide EPR, which is representative of all of our measurements of PTMA materials in this study. Because we measure the solid-state form of PTMA, the singlet linewidth is larger than that measured in solution state due to a closer spacing between radical groups, which enhances exchange narrowing.37,38 Without breaking any microwave contacts, we repeated our measurement of the solid-state EPR over a period of eight days (Fig. 4d and e), over which the sample was exposed to ambient fluorescent lighting and air. We found a negligible decrease in the EPR signal amplitude over that period. We also re-measured PTMA samples after 5 months without a significant change to the EPR amplitude. However, we note that there are small variations in signal amplitudes that originate from breaking and re-making microwave contacts that adds uncertainty to quantitative comparisons. From these measurements, we conclude that radical groups in PTMA solid films are robust to ambient conditions.
3.4 Conductivity measurement
We performed electrical measurements of solid PTMA films to understand how different synthetic methods influence the steady-state electrical conductivity of this material. We chose a lateral, 4-wire geometry in this measurement for two reasons: first, it eliminates contact resistance, which may vary significantly depending on the contact electrode materials and surface preparation. Second, it eliminates the potential for unintentional parallel conductance pathways that could occur in a vertically stacked sandwich structure18–21,32 (e.g. bottom electrode, PTMA film, top electrode deposited on top via evaporation). For example, these structures are susceptible to pin-holes and uncontrolled diffusion of the top electrode into the polymer film, which provide a shunt resistive path. Furthermore, because the polymer film is not over-coated in metal, residual solvent could persist in the polymer if it is not fully baked out after PTMA film deposition. Finally, we note that unintentional conductivity is difficult to check for in vertically-stacked structures. Thus, we choose to eliminate these uncertainties by working in a lateral measurement geometry, consistent with some prior studies.17Table 2 summarizes our findings for PTMA-anionic, PTMA-H2O2 and PTMA-RAFT. Note that of the three ATRP-synthesized PTMA materials, we studied only PTMA-H2O2 electrically because it had the largest radical yield of that group. We found that while the conductivity of PTMA prepared using different synthetic methods varies by a factor of ∼5, all samples were highly insulating, independent of synthetic approach. The conductivity values that we report, between 1.09 ± 0.76 × 10−11 and 5.59 ± 0.78 × 10−11 S cm−1, are orders of magnitude smaller than some prior reports18–21 but in agreement with others.32 This could indicate that while ionic transport between TEMPO moieties can lead to much higher conductivity as compared to PMMA, which has conductivity in range of 10−14–10−15 S cm−1, it is far below the conductivity of organic semiconductors with a π-conjugated backbone. For confirmation of this statement, we also measured the resistance of an undoped regioregular P3HT film, which is a common conjugated polymer. We found a conductivity of 4.5 × 10−8 S cm−1, which is 2 orders of magnitude smaller than typically reported39 but which is consistent with the lack of crystallinity from drop casting and with normal sample aging.40 Nonetheless, we found that the P3HT film is 3 orders of magnitude more conductive than PTMA films, regardless of synthetic preparation.
| Sample | Polymerization | Oxidation | Conductivity (S cm−1) |
|---|---|---|---|
| PTMA-anionic | Anionic | — | 1.09 ± 0.76 × 10−11 |
| PTMA-H2O2 | ATRP | H2O2 | 5.59 ± 0.78 × 10−11 |
| PTMA-RAFT | RAFT | mCPBA | 2.54 ± 0.31 × 10−11 |
| P3HT | — | — | 4.46 ± 2.03 × 10−8 |
To understand these results in the context of radical-mediated electronic transport mechanisms, here we theoretically estimate the number of radical sites involved in electronic transport. We consider charge transport due to phonon-assisted tunnelling between sites with Mott variable range hopping,41 which assigns hopping rates depending on both the wave function overlap and also any thermal activation required to hop. An increase in site separation, or a difference in site energy, both lead to lower hopping rates42 by reducing the density of states g(μ), near the Fermi level μ. The concentration of conducting sites corresponds to Nc = 2g(μ)εopt0, where the optimal bandwidth
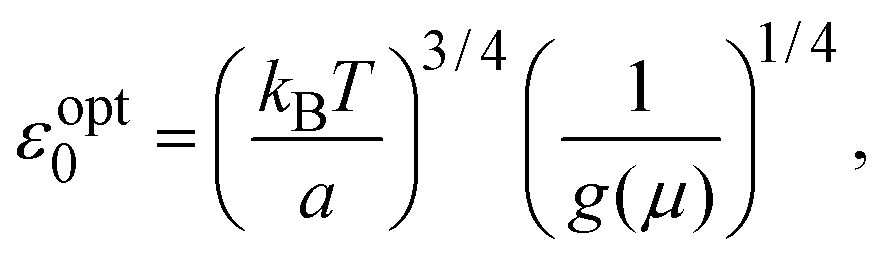 | (1) |
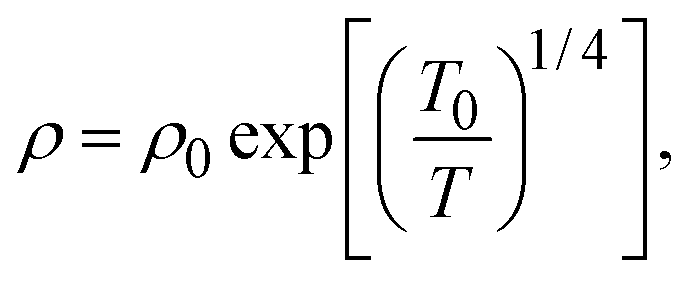 | (2) |
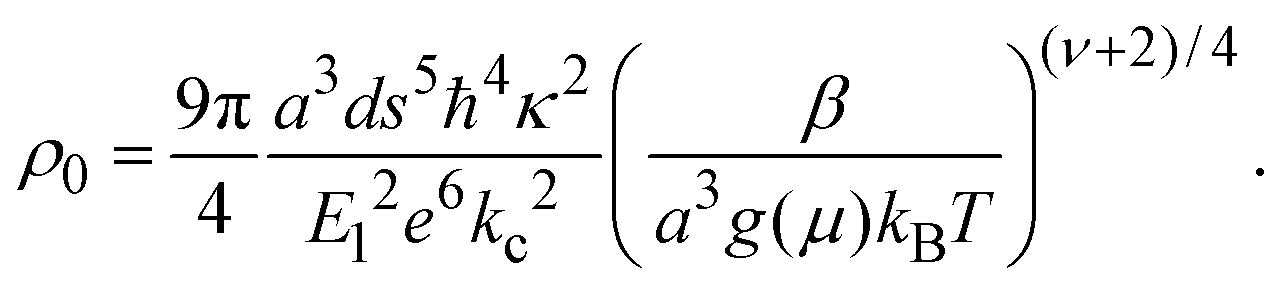 | (3) |
In the above expression d is the mass density of the dielectric, s is the speed of sound in the dielectric,43κ is the dielectric constant,44E1 is the deformation potential,45e is the electronic charge, kc is the Coulomb constant, and ν is a numerical constant determined through percolation simulations.41 An estimate of the density of states is made by equating eqn (2) with measured values of the resistivity (see ESI†). For the anionic PTMA sample with ρ = 9.19 × 1010 Ω cm, g(μ) = 2.5 × 1020 eV−1 cm−3, and from eqn (1), εopt0 = 0.52 eV. It is assumed that each monomer of PTMA contains one side chain and given a mass density of μs = 1.018 g cm−3 and the molecular weight of a monomer w = 240.32 Da, the average spacing between sites is r = 4.5 Å. The concentration of conducting sites is Nc = 2.5 × 1020 cm−3 and so the ratio of conducting sites to insulating sites, f, is
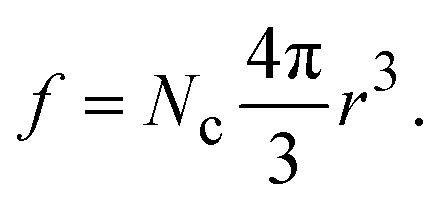 | (4) |
4 Conclusions
In this study, we have prepared a series of PTMA materials of controlled molecular weight and dispersity using a range of living polymerization methods. We have used both controlled radical and anionic polymerizations to make the target polymers. Anionic polymerization enables formation of narrow dispersity polymer directly from the stable radical monomer. Controlled radical methods require that we polymerize from a precursor amine to make a polymer and then oxidize the polymer to form the stable radical. Using solution EPR methods to evaluate the stable radical content in each polymer, we find that all synthetic methods result in a TEMPO content per repeat of between 70 and 80 percent. Only in the case of peracid oxidation can we observe by IR spectroscopy an overoxidation to form an oxoammonium species. We also used an on-chip EPR technique to probe the stability of solid PTMA films, finding no appreciable change in the radical content for more than a week. Having established the high-quality synthesis of stable radical polymers, we then investigate the electrical conductivity in these materials using lateral template devices that enable a four-point measurement. We find that independent of synthetic method, these polymers are highly insulating, with conductivities ranging from 1.1 × 10−11 to 5.6 × 10−11 S cm−1. We have compared these results with variable range hopping theory and find that very few radical sites are electrically active. These results have implications for the production of energy storage systems and motivate future research seeking to increase conductivity in stable radical containing materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the Department of Energy Office of Basic Energy Science (Grant DE-SC0014336). We acknowledge use of the facilities of the Cornell NanoScale Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by the National Science Foundation (NSF) (Grant ECCS-1542081). One of us (AC) is grateful to the National Science Foundation for a Graduate Research Fellowship. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650441. We also acknowledge the use of shared facilities of the Cornell Center for Materials Research which is supported through the NSF MRSEC program (Grant DMR-1719875) and the shared facilities of the National Biomedical Center for Advanced Electron Spin Resonance Technology, which is supported by the National Institute of General Medical Sciences, a part of the National Institutes of Health (Grant P41GM103521)Notes and references
- T. Suga, H. Ohshiro, S. Ugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS
.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed
- H. Nishide and T. Suga, Electrochem. Soc. Interface, 2005, 14, 32–36 CAS
- K. Nakahara, K. Oyaizu and H. Nishide, Chem. Lett., 2011, 40, 222–227 CrossRef CAS
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed
- H. Nishide, Adv. Mater., 1995, 7, 937–941 CrossRef CAS
-
B. Wenning, PhD thesis, Cornell Univerisity, 2016
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed
- J. K. Kim, G. Cheruvally, J. W. Choi, J. H. Ahn, S. H. Lee, D. S. Choi and C. E. Song, Solid State Ionics, 2007, 178, 1546–1551 CrossRef CAS
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS
- J.-K. Kim, Y. Kim, S. Park, H. Ko and Y. Kim, Energy Environ. Sci., 2016, 9, 1264–1269 CAS
- J.-K. Kim, J. Scheers, J.-H. Ahn, P. Johansson, A. Matic and P. Jacobsson, J. Mater. Chem. A, 2013, 1, 2426–2430 CAS
- C. M. Liu, J. Chen, F. Q. Wang and B. L. Yi, Russ. J. Electrochem., 2012, 48, 1052–1057 CrossRef CAS
- Y. Kim, C. Jo, J. Lee, C. W. Lee and S. Yoon, J. Mater. Chem., 2012, 22, 1453–1458 RSC
- W. Guo, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2012, 5, 5221–5225 CAS
- L. Rostro, A. G. Baradwaj and B. W. Boudouris, ACS Appl. Mater. Interfaces, 2013, 5, 9896–9901 CAS
- A. G. Baradwaj, L. Rostro and B. W. Boudouris, Macromol. Chem. Phys., 2016, 217, 477–484 CrossRef CAS
- A. G. Baradwaj, L. Rostro, M. A. Alam and B. W. Boudouris, Appl. Phys. Lett., 2014, 104, 213306 CrossRef
- L. Rostro, S. H. Wong and B. W. Boudouris, Macromolecules, 2014, 47, 3713–3719 CrossRef CAS
- A. G. Baradwaj, S. H. Wong, J. S. Laster, A. J. Wingate, M. E. Hay and B. W. Boudouris, Macromolecules, 2016, 49, 4784–4791 CrossRef CAS
- C. Karlsson, T. Suga and H. Nishide, ACS Appl. Mater. Interfaces, 2017, 9, 10692–10698 CAS
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, J. Am. Chem. Soc., 2008, 130, 14459–14461 CrossRef CAS PubMed
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS
- F. Behrends, H. Wagner, A. Studer, O. Niehaus, R. Pöttgen and H. Eckert, Macromolecules, 2013, 46, 2553–2561 CrossRef CAS
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300–8306 CrossRef CAS
- Y. Wiemann, J. Simmendinger, C. Clauss, L. Bogani, D. Bothner, D. Koelle, R. Kleiner, M. Dressel and M. Scheffler, Appl. Phys. Lett., 2015, 106, 193505 CrossRef
- T. Sukegawa, H. Omata, I. Masuko, K. Oyaizu and H. Nishide, ACS Macro Lett., 2014, 3, 240–243 CrossRef CAS
- L. Bugnon, C. J. H. Morton, P. Novak, J. Vetter and P. Nesvadba, Chem. Mater., 2007, 19, 2910–2914 CrossRef CAS
-
L. B. Volodarsky, V. A. Reznikov and V. I. Ovcharenko, Synthetic Chemistry of Stable Nitroxides, CRC Press, Boca Raton, 1993 Search PubMed
- T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. C, 2014, 118, 17213–17220 CAS
- T. Suga, S. Takeuchi, T. Ozaki, M. Sakata, K. Oyaizu and H. Nishide, Chem. Lett., 2009, 38, 1160–1161 CrossRef CAS
- K. Lee, J. Y. Kim, S. H. Park, S. H. Kim, S. Cho and A. J. Heeger, Adv. Mater., 2007, 19, 2445–2449 CrossRef CAS
- A. P. Monkman, H. D. Burrows, M. da G. Miguel, I. Hamblett and S. Navaratnam, Chem. Phys. Lett., 1999, 307, 303–309 CrossRef CAS
- K. Z. Xing, N. Johansson, G. Beamson, D. T. Clark, J. Bredas and W. R. Salaneck, Adv. Mater., 1997, 13, 1027–1031 CrossRef
- R. D. Scurlock, B. Wang, P. R. Ogilby, J. R. Sheats and R. L. Clough, J. Am. Chem. Soc., 1995, 117, 10194–10202 CrossRef CAS
- M. S. Davis, C. Mao and R. W. Kreilick, Mol. Phys., 1975, 29, 665–672 CrossRef CAS
- C. R. Mao and R. W. Kreilick, Chem. Phys. Lett., 1975, 34, 447–450 CrossRef CAS
- I. R. Gearba, C. Y. Nam, R. Pindak and C. T. Black, Appl. Phys. Lett., 2009, 95, 173307 CrossRef
- Y. Wang and M. F. Rubner, Synth. Met., 1990, 39, 153–175 CrossRef CAS
-
B. I. Shklovskii and A. L. Efros, Electronic Properties of Doped Semiconductors, Springer, Berlin, 1984 Search PubMed
- A. Miller and E. Abrahams, Phys. Rev., 1960, 120, 745 CrossRef CAS
-
J. E. Mark, Physical Properties of Polymers Handbook, Springer, New York, 2007 Search PubMed
- T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. C, 2015, 119, 21369–21375 CAS
- J. E. Northrup, Appl. Phys. Lett., 2011, 99, 062111 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc04645f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |