Understanding on the structural and electrochemical performance of orthorhombic sodium manganese oxides†
Received
10th September 2018
, Accepted 19th November 2018
First published on 22nd November 2018
Abstract
We investigate the orthorhombic Na0.67[NixMn1−x]O2 (x = 0 and 0.05) cathode materials that provide high capacity for prolonged cycles. X-ray absorption studies revealed that the redox activity of the Mn3+/4+ and Ni2+/3+ pairs is effective in suppressing the Jahn–Teller effect of Mn3+ ions because of the network with Ni2+ ions. This effect influenced the smooth voltage variations in the voltage profile for Na0.67[Ni0.05Mn0.95]O2, whereas several complicated voltage plateaus associated with the first-order phase transition were noticed in Na0.67MnO2. Operando synchrotron X-ray diffraction and transmission microscopy studies confirmed the simplicity of the phase transition for Na0.67[Ni0.05Mn0.95]O2 due to suppression of the Jahn–Teller effect of Mn3+ in the oxide lattice. These findings, along with the capacity retention during prolonged cycling and the acceptable thermal properties, make high-capacity sodium-ion batteries feasible, inexpensive, and safe for energy storage application.
Introduction
The price of lithium resources has dramatically increased because of the rapid spread of electric vehicles and mid-to-large scale energy storage devices using lithium ion batteries (LIBs) due to the limited lithium resources.1 Sodium is one of the most widespread elements and the intercalation chemistry of sodium is similar to that of lithium, therefore, sodium-based systems can effectively substitute conventional LIBs.2 Cathode materials for sodium ion batteries (SIBs), including layered NaxMO2 (x = 0.5–1 and M = transition metal),3–6 NASICON,7 and phosphates,8 have been investigated. Among them, sodiated manganese oxide (NaxMnO2) is of interest because its crystal structure varies with the sodium content. Earlier research by Parant et al.9 analyzed the various crystal structures of Na1−xMnO2 compounds and their ability for Na+ insertion/extraction from the crystal structure. Ma et al.10 first reported a high capacity cathode material for the monoclinic O′3-NaMnO2 (Na content close to 0.93), delivering approximately 197 mA h g−1 in the 2–3.8 V voltage range at a rate of C/30. Their subsequent work demonstrated a symmetric attraction of the two neighboring Jahn–Teller (JT) distorted 180° Na–O–Mn3+–O–Na strip formations in contrast with VNa–O–Mn3+–O–Na along the [100] axis.11 Structural imperfections rather than manganese dissolution are the reason for the gradual reduction in capacity observed during cycling in this system. Lowering the Na content to 0.7 in Na1−xMnO2 results in three different crystal structures depending on temperature: P3-type Na0.67MnO2 (R3m, <800 °C),12 P2-type Na0.67MnO2 (P63/mmc, 800–1000 °C),13,14 and orthorhombic P′2-type Na0.67MnO2 (Cmcm, 1000–1200 °C),13,15 where the prime (′) symbol denotes a distorted structure. Caballero et al.14 studied a reversible electrochemical reaction of P2–Na0.67MnO2 that delivers approximately 150 mA h g−1 but shows several voltage plateaus during the Na+-ion intercalation, although the resulting capacity decreased gradually upon cycling. Stoyanova et al.15 and Komaba et al.13 recently proved that the orthorhombic P′2-type Na2/3MnO2 (Na0.67[Mn3+2/3Mn4+1/3]O2), hereafter referred to as o-Na0.67MnO2, is active, has a high capacity of approximately 216 mA h g−1 (10 mA g−1) in the voltage range of 1.5–4.4 V, and shows a slow capacity decrease over 20 cycles, although the material needs improvement in its rate performance, having a capacity of approximately 55 mA h g−1 at the 2C (0.52 A g−1) rate. It should be noted that the JT distortion in six-coordinate Mn3+ prevails in the crystal structure of o-Na0.67MnO2, which causes large anisotropic volume changes and Na+/vacancy ordering during cycling, thus resulting in a decrease in capacity. Hence, the long-term cycling performances of Na–Mn–O compounds have not been reported in the literature. The JT distortion effect can be mitigated by doping or other approaches so that the resulting electrode performances can be greatly improved as we have experienced in earlier studies on spinel Li[Mn3+Mn4+]O4. For this reason, we, herein, focused on the partial replacement of Mn3+ in o-Na0.67[MxMn1−x]O2 (M: Ni, Co, Cu, Zn, Fe, and Al) to minimize the effect of JT distortion in the crystal structure. Ni-doped o-Na0.67[Mn0.95Ni0.05]O2 exhibited capacities of 222 mA h g−1 (13 mA g−1, 0.05C) and 100 mA h g−1 even at 5C rate. A full cell with o-Na0.67[Ni0.05Mn0.95]O2/hard carbon showed unprecedentedly good capacity retention with a high capacity of approximately 83% after 300 cycles (160 mA h g−1) at 0.2C (52 mA g−1). The synthesis, electrochemistry, and reaction process for o-Na0.67[M0.05Mn0.95]O2 are discussed in this paper.
Experimental
Material preparation
We prepared o-Na0.67[M0.05Mn0.95]O2 (M = Mn, Ni, Co, Cu, Zn, Fe, and Al) powders using spray pyrolysis. Stoichiometric amounts of metal nitrate hydrates (Sigma Aldrich) and sodium nitrate (Samchun) were used as the starting materials, and then they were dissolved in distilled water at a molar ratio of Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M of 0.67:1 at 25 °C. Citric acid (Junsei) as a chelating agent and sucrose (Samchun) as a particle growth inhibitor were added to the prepared aqueous solution with a starting material:citric acid:sucrose molar ratio of 1:0.2:0.05. The prepared aqueous solution was directly sprayed into a heated vertical reactor. The as-received precursor powders were calcined at various temperatures for 10 h in a furnace in dry air (300 mL min−1) and subsequently cooled to 25 °C. The obtained precursor and final products were stored in a glove box filled with Ar to prevent contamination by moisture.
:M of 0.67:1 at 25 °C. Citric acid (Junsei) as a chelating agent and sucrose (Samchun) as a particle growth inhibitor were added to the prepared aqueous solution with a starting material:citric acid:sucrose molar ratio of 1:0.2:0.05. The prepared aqueous solution was directly sprayed into a heated vertical reactor. The as-received precursor powders were calcined at various temperatures for 10 h in a furnace in dry air (300 mL min−1) and subsequently cooled to 25 °C. The obtained precursor and final products were stored in a glove box filled with Ar to prevent contamination by moisture.
Characterization
Synchrotron X-ray diffraction (XRD) measurements were carried out (2θ = 10–100°) to determine the crystal structure of the synthesized powders. Also, X-ray diffraction (XRD, PANalytical) with Cu Kα radiation was used to characterize the crystal structure of the synthesized powders and electrodes. The FullProf Rietveld software was used to analyze the observed XRD patterns.16 High-resolution transmission electron microscopy (HR-TEM, JEM-2010, JEOL) was used to observe the particle morphology. The direct volt-ampere method (CMT-SR1000, IT) was used to measure the direct current (DC) electrical conductivity, where disc samples were in contact with a four-point probe. Galvanostatic charge/discharge cycling tests were performed using R2032-type coin cells. Electrodes were fabricated by mixing the active powder (85 wt%), Super-P and KS-6 (3.75 wt% and 3.75 wt%) as the conductive material and polyvinylidene fluoride (PVDF, 7.5 wt%) in N-methyl-2-pyrrolidone (NMP). The prepared slurry was coated onto aluminum foil using a doctor blade and dried at 110 °C overnight in a vacuum oven prior to use. The loaded amount of active material was typically 5 mg cm−1. A thin sodium plate (Alfa Aesar Co.) and hard carbon were used as the negative electrode for the half and full cells, respectively. We used commercial hard carbon (Kureha). To eliminate adhered water and air-oxidized products on the surface of hard carbons, pre-treatment was done at 1000 °C for 2 h in Ar. After that, a direct contact was made between the hard carbon electrode and sodium metal in the presence of an electrolyte to minimize the unwanted capacity that appeared at the first discharge (sodiation process). The electrolyte solution used was 0.5 M NaPF6 in a mixture of propylene carbonate (PC) and fluorinated ethylene carbonate (FEC) with a volume ratio of 98:2. Glass filters (GB-100R) were used as separators. The assembled cells were charged/discharged by applying a constant current of 13 mA g−1 (0.05C) at 25 °C. In situ operando synchrotron XRD measurements using assembled 2032-type coin cells were carried out at beamline 9B of the Pohang Accelerator Laboratory (PAL, Pohang, South Korea). After charge and discharge processes, the cells were disassembled in an Ar-filled glove box, and the electrodes were washed with salt-free dimethyl carbonate for one day for ex situ X-ray absorption spectroscopy (XAS) analysis at the 8C Nano X-ray absorption fine structure (XAFS) beamline of the PAL. Density functional theory (DFT) calculations were performed by the projector-augmented plane-wave (PAW) method carried out using the Vienna Ab Initio Simulation Package (VASP).17 The Perdew–Burke–Ernzerhof (PBE) functional18 with on-site Coulomb interaction correction introduced by Dudarev et al.19 was utilized. We used U–J = 4.0 for Ni, which has been used in previous studies on bulk LiNiO2.20 To find the appropriate U–J value for Mn, we calculated the lattice parameters for bulk bare and Ni-doped NaMnO2 (unit cell of 6 × 4 × 1, 1 × 1 × 1 Monkhorst–Pack k-point mesh, 500 eV energy cutoff) using U–J = 3.0 and 4.0 eV (Table SI-3†).21–23 Because the lattice dimensions were found to be very similar for these two U–J values, we used U–J = 3.0 for Mn in this study. To calculate the lattice parameters for bare and Ni-doped Na0.67MnO2 and NaMnO2, we fully optimized the geometries and unit cell parameters. An orthorhombic P′2-type (Cmcm) structure was considered for the bare and Ni-doped Na0.67MnO2 and NaMnO2.
Thermal properties
Electrochemical desodiation was performed in coin cells using sodium metal as an anode. After charging to 4.3 V, the coin cells were disassembled in a glove box, and the electrodes were washed by immersion in salt-free dimethyl carbonate for one day. The electrodes were centrifuged in NMP to remove the PVDF and the conducting carbon, leaving only the desodiated powders after vacuum drying at 80 °C for one day. The electrochemically desodiated powders were used for thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and in situ high-temperature X-ray diffraction (HT-XRD). The loaded samples (10 mg) were heated from 25 to 600 °C for TGA (DGT-60, Shimadzu, Japan) measurements under a heating and cooling rate of 1 °C min−1 and were held at selected temperatures for 10 min before data collection. The in situ HT-XRD (XRD, PANalytical) data were obtained using a PANalytical X'pert diffractometer (Bragg–Brentano geometry) with Cu Kα radiation (λ1 = 1.5406 Å and λ2 = 1.5444 Å) in the 2θ range of 10–60° (0.026°/10 s per step) at each temperature setting. For the DSC experiments, the electrochemically desodiated bare and coated powders (3–5 mg) were loaded into stainless-steel-sealed pans with gold-plated copper seals (which can resist 150 atm of pressure before rupturing and have a capacity of 30 μL). These measurements were carried out in a Pyris 1 calorimeter (Perkin-Elmer) at a temperature ramp rate of 1 °C min−1. During the experiments, there were no unwanted leakage and weight change.
Results and discussion
Characterization
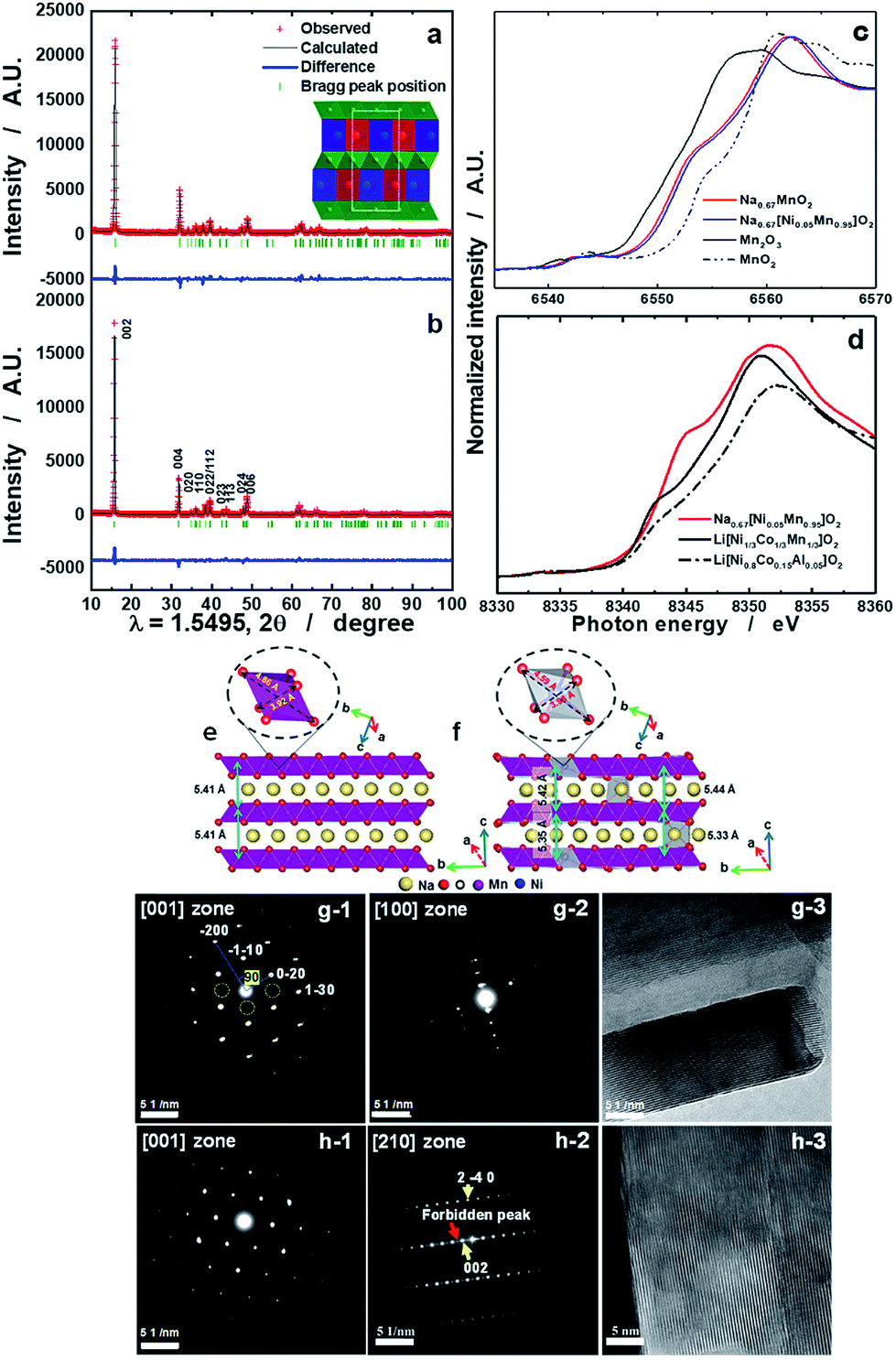
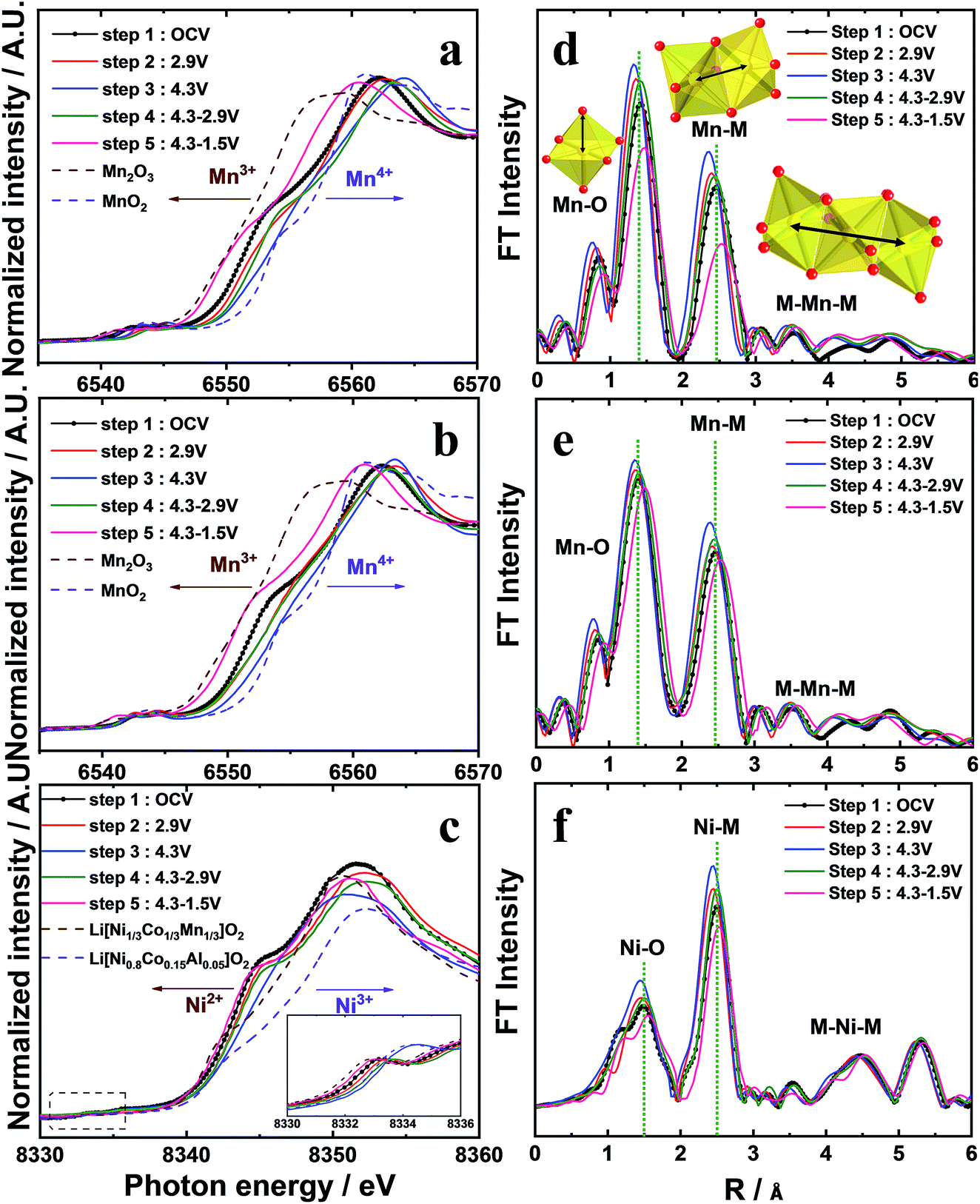
Details of the phase evolution to form orthorhombic sodium manganese oxide are shown in Fig. SI-1.† Rietveld refinement data of the synchrotron XRD (S-XRD) patterns are summarized in Fig. 1a and b, and Tables S1 and S2,† respectively. The crystal structures of both materials were refined using the Cmcm space group, and the results agree with earlier reports by Laure Bordet-Le Guenne et al.24 and Stoyanova et al.15 Interestingly, better refinement results were obtained when we assumed that Ni could partially occupy the prismatic Naf(1) and Nae(2) sites and the Mn sites (Fig. 1b and Table SI-1†). The final structure can be expressed as [Na0.636Ni0.026][Mn0.974Ni0.026]O2 (hereafter, referred to as o-Na0.67[Ni0.05Mn0.95]O2). As listed in Table SI-2,† Ni doping resulted in variations in the lattice parameters. These changes are substantially dependent on the ionic radius of the dopant. The oxidation states of Mn for both compounds are close to 3.4+, and there is a slight shift of photon energy toward higher energy for the Ni-doped compound (Fig. 1c). Interestingly, the oxidation state of Ni is +2 (Fig. 1d). Assuming the oxidation state of Ni to be +2, we can explain the reason for the increase in the lattice parameters for o-Na0.67[Ni0.05Mn0.95]O2. o-Na0.67MnO2 can be ideally written as Na0.67[Mn3+0.6Mn4+0.4]O2. Because of the similarities in ionic radius between Ni2+ (0.69 Å) and Mn3+ (0.65 Å), Ni2+ preferentially occupies the Mn3+ sites rather than Mn4+ (0.53 Å) in Na0.67[Ni2+0.05Mn3+0.55Mn4+0.4]O2, which is responsible for the increase in the lattice parameters, except for the b-axis. Kumakura et al.13 explained the lattice distortion using the ratio bortho/aortho = √3 in the hexagonal lattice. According to this equation, the present o-Na0.67MnO2 has a bortho/aortho ratio that is greater than √3, approximately 7.0%, whereas the distortion ratio was lowered to approximately 4.5% for the Ni-doped one as a result of the reduction in the b-axis length. This suggests that the anisotropy of the Mn3+–O bond is mitigated by the partial doping of o-Na0.67MnO2. We carried out DFT calculations to validate our assumptions on the presence of Ni in the prismatic Na sites (Fig. SI-2, SI-3 and Tables SI-3, SI-4†). Na occupancies similar to our experimental XRD results (Na(1): 0.25 and Na(2): 0.4375) were used for Na0.67MnO2. Two different Na arrangements (labeled (i) and (ii)) were considered (Fig. SI-2-i and ii†). After optimization, the energy of structure i was 0.14 eV (per formula unit) more favorable than that of (ii). The sodium occupancy of structure (i) (Fig. 1e) after relaxation was Na(1): 0.188 and Na(2): 0.500, which is comparable to the experimental results. To model Ni-doped Na0.67MnO2 with 5% Ni doping at the Na and Mn sites, we considered three different configurations for the Na occupancies according to our experimental results. The most favorable optimized structure is illustrated in Fig. 1f, and its Na occupancy numbers are listed in Fig. SI-2-iii to v† and are, again, in good agreement with the experimental occupancy data. To model NaMnO2, we considered a similar occupancy as indicated by the experimental results. However, after optimization, all site (1) Na+ cations moved to site (2), which is located at the center of the O-prisms, as shown in Fig. SI-3.† To model [Na0.96Ni0.02][Mn0.96Ni0.02]O2, [Na0.90Ni0.02][Mn0.96Ni0.02]O2, and [Na0.85Ni0.02][Mn0.96Ni0.02]O2, we studied two different configurations. The most favorable structure of [Na0.96Ni0.02][Mn0.96Ni0.02]O2 is illustrated in Fig. 4f. The calculated lattice parameters (a, b, and c) for the bare o-Na0.67MnO2 are in agreement with the experimental values with deviations of 1.76%, 2.85%, and −0.71%, respectively (Fig. 1e and Table SI-4†). Ni doping increased the lengths of the a- (2.08%) and c-axes (0.45%) but decreased that of the b-axis (−3.69%) (Fig. 1f and Table SI-4†). These results are in good agreement with the experimental changes of 0.35, 0.62, and −2.08%, respectively. The increase in the a-axis length is due to the expansion of the equatorial bonds (from an average value of 3.92 to 4.14 Å) in the octahedral complexes where Mn3+ is replaced by Ni2+. The reduction in the b-axis length is, however, due to the shortening of axial bonds (from 4.56 to 4.25 Å) in the octahedral complexes. Our calculated value for the equatorial to axial length ratio for NiO6 is 4.25/4.14 = 1.02, and 4.56/3.92 = 1.16 for MnO6. The reason for the increase in the c-axis length is also the expansion of the equatorial bonds. Not only doping with Ni but also doping with Co, Cu, Zn, Fe, or Al was successful for Na0.67[M0.05Mn0.95]O2, as shown in Fig. SI-4 and Table SI-2.† All these cases exhibited similar trends, that is, a decrease in the b-axis parameters with decreasing bortho/aortho ratio. Therefore, the partial substitution of Mn can reduce the lattice distortion (Table SI-2†). Fig. 1g-1 and g-2 show the selected area electron diffraction (SAED) patterns of the as-prepared o-Na0.67MnO2 observed normal and parallel to the direction of the layer planes. The reflections in Fig. 1g-1 were indexed to the [001] zone of the P′2 structure but also contained faint superlattice peaks (circled in yellow), suggesting some vacancy ordering in the Na plane. Note that the electron diffraction patterns taken parallel to the layer planes in Fig. 1g-2 showed no extra peaks in the (00l) direction. Hence, there are no stacking defects as seen in Fig. 1g-3. For o-Na0.67[Ni0.05Mn0.95]O2, the [001] zone pattern, shown in Fig. 1h-1, did not contain any superlattice peaks; however, extra peaks corresponding to (±001), (±003), (±005), and (±00l)…, which are forbidden in the Cmcm space group, were clearly observed from the [210] zone diffraction pattern in Fig. 1h-2. Ni doping apparently destroyed the screw axis along the c-axis, which requires the absence of (00l) reflections where l = 2n + 1. Because Mn ions occupy the 4a sites in the lattice, which are the high-symmetry Wyckoff positions, Ni ions replacing Mn would not affect the screw axis symmetry along the c-axis. Therefore, the appearance of the forbidden peaks suggests that Ni ions also occupied the vacant sites in the Na layer. The structural difference in the stacking of the layer planes between o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2 can be clearly seen in the high-resolution image in the [210] direction in Fig. 1h-3 when compared with Fig. 1g-3. Another advantage of Ni doping is the improvement in the electrical conductivity to 2.2 × 10−5 S cm−1 compared to the conductivity of Na0.67MnO2, which was found to be approximately 6.8 × 10−6 S cm−1 using the four-point DC method (Table SI-1†).
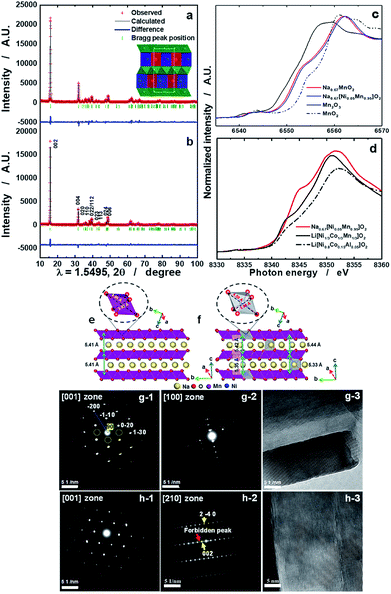 |
| | Fig. 1 Synchrotron XRD patterns of (a) o-Na0.67MnO2 and (b) o-Na0.67[Ni0.05Mn0.95]O2 calcined at 1200 °C for 10 h in a dry air atmosphere; (c) Mn K edge XANES spectra of o-Na0.67MnO2, o-Na0.67[Ni0.05Mn0.95]O2, Mn2O3, and MnO2, and (d) Ni K edge XANES spectra of o-Na0.67[Ni0.05Mn0.95]O2, Li[Ni1/3Co1/3Mn1/3]O2, and Li[Ni0.8Co0.15Al0.05]O2. Side views of bare o-Na0.67MnO2 (e) and o-[Ni0.02Na0.625][Ni0.02Mn0.96]O2 (f). Octahedral MnO6 and NiO6 complexes, as well as equatorial and axial bonds, are shown. SAED patterns from the as-prepared o-Na0.67MnO2 cathode observed from directions (g-1) normal and (g-2) parallel to the layer planes and (g-3) high-resolution image. SAED patterns from the as-prepared o-Na0.67[Ni0.05Mn0.95]O2 cathode observed from directions (h-1) normal and (h-2) parallel to the layer planes and (h-3) high-resolution image. | |
Electrochemical performance in half cells
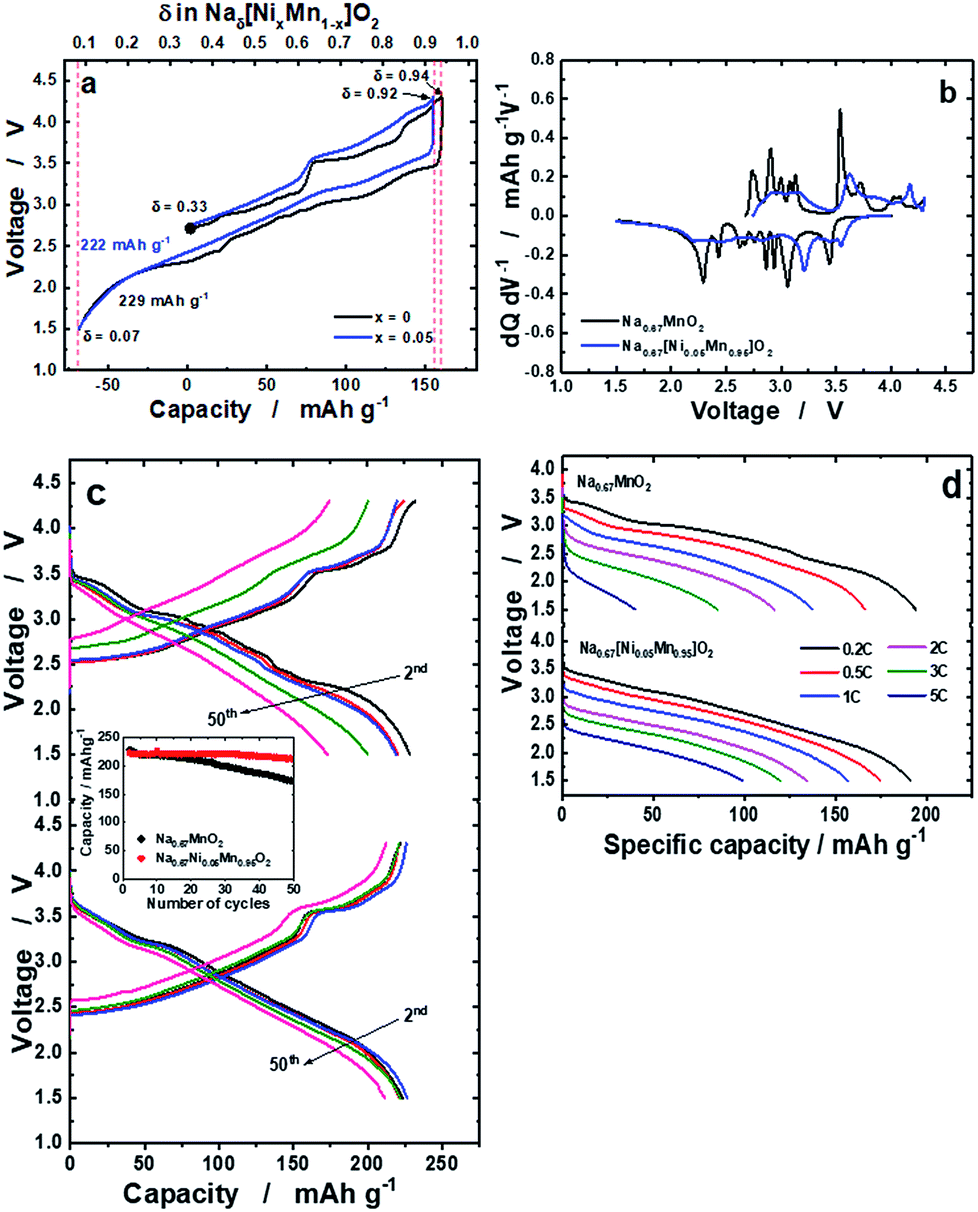
The delivered charge and discharge capacities for o-Na0.67MnO2 were 160 and 228 mA h g−1, respectively (Fig. 2a). Slightly lower capacities were observed for o-Na0.67[Ni0.05Mn0.95]O2: 155 mA h g−1 on charge and 222 mA h g−1 on discharge. It should be noted that o-Na0.67[Ni0.05Mn0.95]O2 exhibits reduced polarization during the first cycle. Many plateaus are related to several phase transitions for o-Na0.67MnO2, whereas such complicated reactions were strikingly suppressed by the Ni doping (Fig. 2b). The resulting capacity retention was approximately 75% for o-Na0.67MnO2 over 50 cycles (Fig. 2c). By contrast, Ni doping results in an improvement in the capacity retention of more than 95%, which delivered a capacity of over 210 mA h g−1 even after 50 cycles. As shown in Fig. SI-5,† M (M = Mn, Ni, Co, Cu, Zn, Fe, or Al) doped Na0.67[M0.05Mn0.95]O2 showed different electrochemical behaviors during 50 cycles; 228, 223, 202, 178, 128, 166, and 177 mA h g−1 for the first discharge capacities and the resulting capacity retentions approximately 75, 95, 92, 87, 87, 69, and 85%, respectively. It is evident that several voltage plateaus observed in o-Na0.67MnO2 were diminished by different metal dopings as shown in the charge–discharge curves (Fig. SI-5 and Table SI-2†). Specifically, introduction of Ni, Co, and Cu into o-Na0.67[M0.05Mn0.95]O2 contributed to not only lowering the degree of structural distortion from 7.0% to ∼5.0% but also increasing the electrical conductivity. As a result, improved electrode performances were seen for Ni, Co, and Cu-doped o-Na0.67[M0.05Mn0.95]O2. Besides, the added Fe, Zn, and Al dopants in o-Na0.67[M0.05Mn0.95]O2 do not likely assist in increasing the electrical conductivity or structural distortion particularly for the Al element. These physical properties resulted in inferior electrode performances for the Fe, Zn, and Al-doped o-Na0.67[M0.05Mn0.95]O2 in terms of failure in suppressing the phase transition, low capacity, and capacity retention. From these results, it is thought that both structural perfectness and electrical conductivity are the main parameters for the improvement in the electrochemical performances of o-Na0.67[M0.05Mn0.95]O2. As anticipated, o-Na0.67MnO2 delivered small capacities of approximately 116 mA h g−1 at 2C (520 mA g−1) and 40 mA h g−1 at 5C (1.3 A g−1), whereas the Ni-doped o-Na0.67[Ni0.05Mn0.95]O2 had high capacities of approximately 134 mA h g−1 at 2C and 100 mA h g−1 at 5C on discharge (Fig. 2d). Compared with earlier studies25 that obtained a capacity of approximately 100 mA h g−1 at a rate of 2C (520 mA g−1), such an improvement in the case of o-Na0.67[Ni0.05Mn0.95]O2 could be related to the higher electric conductivity. It is also likely that the suppression of the multi-step phase transition facilitates Na+-ion diffusion, even at high rates for o-Na0.67[Ni0.05Mn0.95]O2.
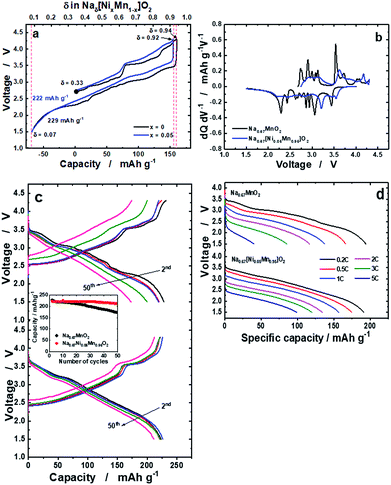 |
| | Fig. 2 (a) First charge and discharge curves of o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2 measured at a current of 13 mA g−1 in the voltage range of 1.5–4.3 V at 25 °C, (b) the resulting differentiated curves for o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2, (c) continuous cycling curves of o-Na0.67MnO2 (top), o-Na0.67[Ni0.05Mn0.95]O2 (bottom), and cyclability (middle), and (d) rate performances measured up to a 5C rate (1300 mA g−1). | |
In situ operando synchrotron XRD analysis
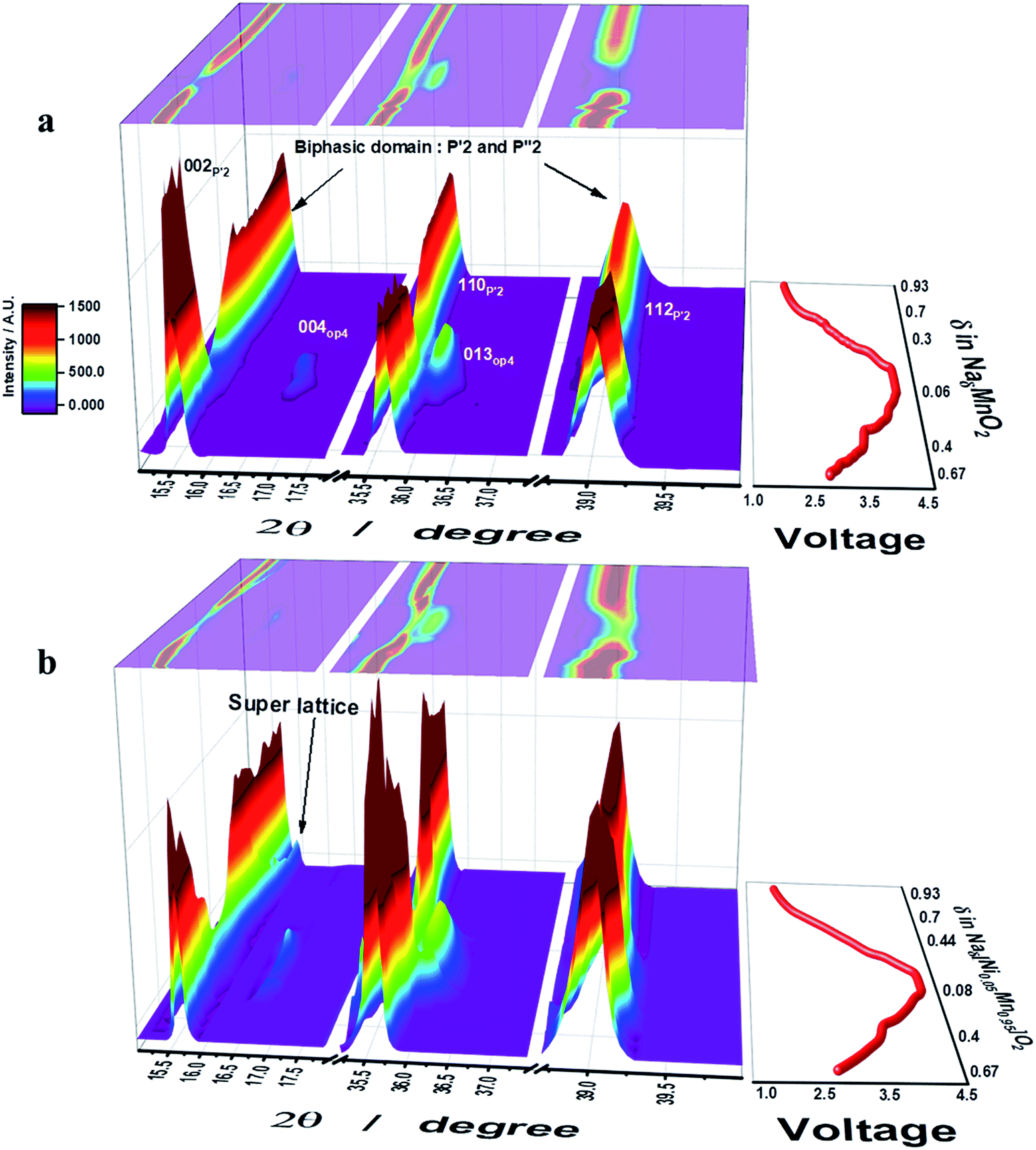
Operando S-XRD measurements were employed to study the structural evolution at the first cycle for both o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2. As desodiation was initiated, the (002) peak began to split into two peaks on the lower plateau region, 0.4 ≤ δ ≤ 0.67 in NaδMnO2, which is indicative of a two-phase domain (Fig. 3a and S6a†). Indeed, five small voltage plateaus were observed on the lower 2.6–3.5 V plateau region of the dQ/dV curve (Fig. 2b), indicating that a biphasic reaction was dominantly progressing. For 0.3 ≤ δ ≤ 0.4 in NaδMnO2 (3–3.5 V), the upper plateau still showed the presence of the biphasic domain. However, the relative diffraction intensity of the (002) peak somehow decreased. It should be noted that the appearance of the OP4 (004) peak was noticed at 2θ = 16.5 and 36°, which is in agreement with the recent research by Kumakura et al.13 Further desodiation caused the emergence of the OP4 phase but the relative intensity of the P′2 phase greatly lowered on the upper voltage plateau on charge. The (004) OP4 peak shifted gradually toward higher angle on charge, implying a single-phase solid solution reaction in this range. This decrease in the interslab distance is usually observed in highly delithiated O3-type compounds and is attributed to the reduction in the metal–oxygen distance (Fig. SI-7†), which simultaneously increases the covalent character in the crystal structure. On discharge, the (002) P′2 peak was evidently restored followed by the backshift of the (004) OP4 peak, which was extinguished at δ = 0.3 in NaδMnO2 (3.3 V). Meanwhile, the (002) P′2 peak appeared again at δ = 0.25 in NaδMnO2 and merged with the peak at δ = 0.48 in NaδMnO2. Afterward, the (002) peak moved monotonously toward higher angles for δ = 0.7 in NaδMnO2 (2.4 V). For the 0.3 ≤ δ ≤ 0.7 range in NaδMnO2 (3.3–2.4 V), slight changes were continuously observed in the S-XRD patterns because of the consecutive phase transitions when 2θ = 36–40°, although the resulting (002) peak became abnormally broad. From that point on, there were no more changes in the position of the (002) peak. This broadening hinders the clear separation of the (002) peak that has been determined to be another P′2 phase (2θ = 16.3°) resulting from a biphasic reaction and became dominant when further sodiated at 1.5 V, as proposed by Kumakura et al.13 Unlike o-Na0.67MnO2, o-Na0.67[Ni0.05Mn0.95]O2 exhibits different structural variations during desodiation (Fig. 3b). As desodiation was initiated, the (002) peak gradually shifted toward lower angles via a one-phase reaction, forming a solid solution without the peak splitting in the 0.4 ≤ δ ≤ 0.67 range for o-Naδ[Ni0.05Mn0.95]O2, and the resulting voltage varied monotonously in this region. It should be noted that the biphasic process resulted in several voltage plateaus and was dominant in this range for o-Na0.67MnO2 (Fig. 3a). Similar to o-Na0.67MnO2, the (002) peak splits when 0.33 ≤ δ ≤ 0.4 in o-Naδ[Ni0.05Mn0.95]O2, that is, during the short voltage plateau (3.6 V) located in the upper plateau. Afterward, the two peaks merged and the relative intensity reduced; this was accompanied by the formation of the OP4 phase, whose peaks moved toward higher angles because of its covalent character in the highly desodiated state. On discharge, the peaks corresponding to the formed OP4 phase shifted toward lower angles, whereas the relative intensity of the (002) peak of the P′2 phase increased as sodiation progressed to δ = 0.67 in o-Naδ[Ni0.05Mn0.95]O2 in the upper voltage plateau, below which the OP4 phase was no longer detected. The (002) peak appeared and was maintained as a single peak from δ = 0.44 to 0.75 in o-Naδ[Ni0.05Mn0.95]O2. Afterward, strikingly, the (002) peak separated into two peaks, and the right-side shoulder peak grew and shifted toward higher angles in the δ = 0.44–0.8 range for o-Naδ[Ni0.05Mn0.95]O2. At the same time, the parent (002) peak and that corresponding to the resulting P′2 phase also shifted together in this range. As seen from the discharge curves (Fig. 2a), the voltage decay progressed smoothly in this range, without new voltage plateaus. However, the peak splitting was not clear for 0.7 ≤ δ in o-NaδMnO2 (2.4–1.5 V) but abnormally broadened (see the 002 P′2 peak in Fig. 3a). It is evident that the Ni doping of o-Na0.67MnO2 likely relieves the stress caused by the biphasic reaction in the oxide lattice.
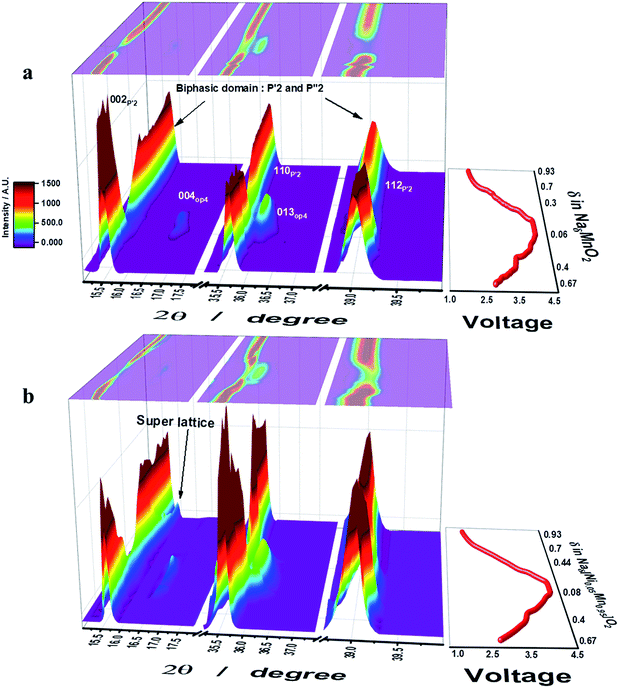 |
| | Fig. 3 The initial charge–discharge curves (left) and contour maps of operando XRD patterns (right) for (a) o-Na0.67MnO2 and (b) o-Na0.67[Ni0.05Mn0.95]O2 electrodes recorded during one cycle in 0.5 M NaPF6 in PC:FEC (98:2 v/v mixture). | |
Microstructural study with DFT calculations of cycled electrodes
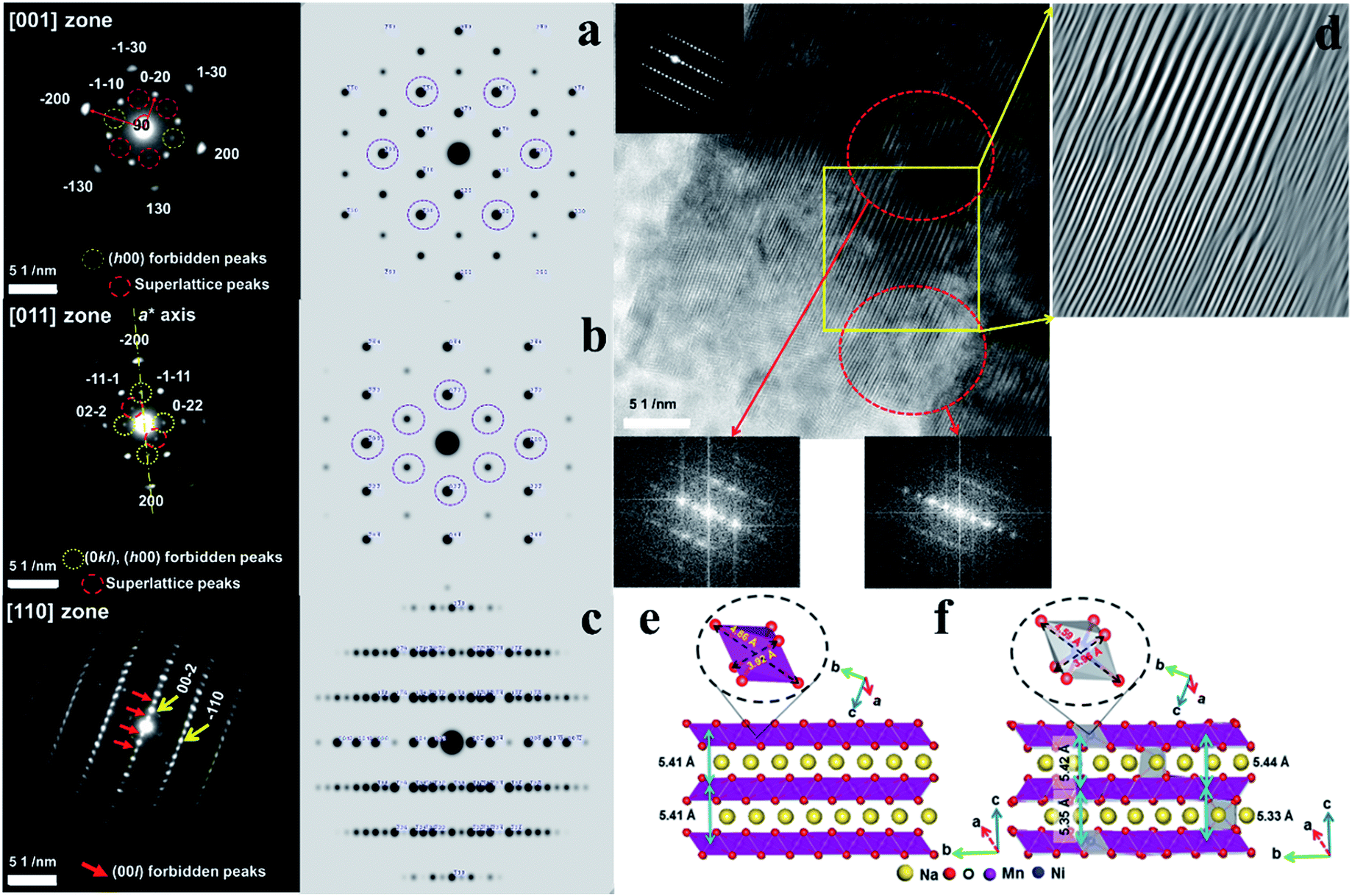
Fig. 4a presents the [001] diffraction pattern of o-Na0.67[Ni0.05Mn0.95]O2 after the first discharge with almost fully occupied Na layers. In comparison with the ideal diffraction pattern, there are forbidden (h00) peaks, where h = 2n + 1, and a set of superlattice peaks (±1/2, ±3/2, 0). The appearance of these peaks implies symmetry breaking and cation ordering within the layer planes, which are likely to be the Na planes. The diffraction pattern for o-Na0.67[Ni0.05Mn0.95]O2 in the [011] zone in Fig. 4b also contained forbidden (h00) peaks, where h = 2n + 1, and (0kl) reflections where k = 2n + 1, as well as (−1/2, 1/2, −1/2) and (1/2, −1/2, 1/2) superlattice reflections. In addition, there was asymmetry about the a-axis (or h00 direction) in the [011] zone pattern arising from the absence of the (−1/2, −1/2, 1/2) and (1/2, 1/2, −1/2) reflections. Because the Mn layers have mirror symmetry in the a-axis, whereas the Na layers do not possess such mirror symmetry, the [011] zone diffraction unequivocally indicates that the Ni ions occupy the Na sites in a highly ordered fashion. Fig. 4c shows the [110] zone pattern with the calculated pattern and shows the forbidden (00l) reflections, where the l = 2n + 1 reflections became stronger. The high-resolution image in the [110] direction in Fig. 4d suggests two co-existent phases. As indicated, the Fourier transform (FT) of the upper region showed no extra peaks, whereas the lower region contained the (00l) peaks that should have been extinct. These data reveal the layer planes that appear to split into two because of the ordered Ni occupancy in the Na layer, which is more distinct when fully sodiated compared to that with the fresh electrode (Fig. 1h-3). DFT calculations support the TEM results (Fig. 4e and f). For Na concentrations higher than 0.67 (Fig. SI-7†), the a- and b-axes expanded because of the increase in Na+–Na+ repulsion (more Na+ ions with shorter Na+–Na+ separation) along the Na layers. However, the c-axis shrunk due to the stronger Na+–O2− attraction (higher number of Na+ cations) for higher Na concentrations. The calculated lattice parameters for Na0.92MnO2 are closer to the experimental values (after the first discharge) compared to Na1.00MnO2, which showed that the concentration of Na after the first discharge is less than 1. Ni doping did not significantly change the lattice dimension along the a-axis in [Ni0.02Na0.96][Ni0.02Mn0.96]O2 (Fig. 4e and f), which is in agreement with the experimental data (Fig. SI-7, SI-9 and Tables SI-4, SI-5†). This is because of the small change in the equatorial bonds in NiO6. However, the large decrease in axial bond length (by 0.27 Å) led to a large decrease in the b-axis length. The shortening of the b-axis was also observed experimentally (Table SI-4†). Both our experiments and calculations show that the c-axis length decreases because of Ni doping. Our DFT calculations overestimated this decrease for NaMnO2 and Na0.92MnO2. However, the calculated c-axis value for the latter was closer to the experimental values. The reason for the small decrease in the c-axis length is the smaller size of Ni2+ ions (0.69 Å) compared to Na+ ions (1.02 Å), as well as the stronger attraction between Ni2+ and O2−. The slight increase in equatorial bond length cannot compete with the former effect. In comparison, the discharged Na0.67MnO2 electrode did not exhibit any of the complex structures observed in the Ni-doped sample (Fig. SI-8a†). The [210] zone pattern did not contain any forbidden peaks and matched well with the structure observed in the as-prepared state (Fig. 1g-2), as expected from the full occupancy of the Na layer. The corresponding high-resolution image revealed well-formed lattice fringes, attesting to the high crystallinity and lack of structural defects in the cathode when no doping material was introduced. The [110] zone diffraction pattern of the o-Na0.67[Ni0.05Mn0.95]O2 cathode after 50 cycles showed streaks instead of discrete diffraction spots as a result of the significant site disordering within the layer planes from repeated Na insertion and removal (Fig. SI-8b†). However, the strong (±002) reflections suggest that the layer structure was maintained after 50 cycles.
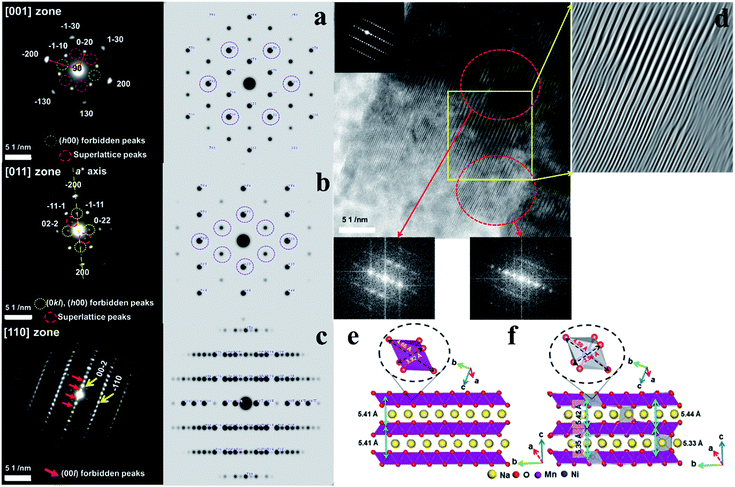 |
| | Fig. 4 SAED patterns of the o-Na0.67[Ni0.05Mn0.95]O2 cathode observed in the (a) [001] zone axis, (b) [011] zone axis, and (c) [110] zone axis of o-Na0.67[Ni0.05Mn0.95]O2 after the 1st cycle and calculated patterns and (d) high-resolution image for the two co-existent phases. Side views of bare o-Na1.00MnO2 (e) structures, as well as Ni-doped o-Na1.00MnO2 (f) structures. The octahedral MnO6 and NiO6 complexes, as well as equatorial and axial bonds, are shown. | |
XANES and EXAFS analyses of o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2 electrodes
The Mn K-edge for o-Na0.67MnO2 (Fig. 5a) gradually shifted toward higher energies on charging (oxidation), indicating an increase in the average Mn oxidation state close to +4. On discharge (reduction), the Mn K-edge spectrum shifted back to the Mn2O3 reference Mn K-edge spectrum at 1.5 V, demonstrating that the oxidation state of Mn was reduced to Mn3+. The progressive increase in the oxidation state demonstrates that desodiation did not induce oxidative decomposition of the parent compound but resulted in the extraction of Na+ ions from the host structure. Similarly, the reduction led to a monotonous decrease in the oxidation state of Mn to +3, which was associated with the appearance of two phases: the P′2 phase and another P′2 phase (P′′2). This is indicated by the broadened P′2 peaks, which might be ascribed to the overlapping of the reflections corresponding to the two phases. As a result, a large discharge capacity of approximately 228 mA h g−1 was obtained. The broadening of the S-XRD peaks is understandable because of the almost complete filling of both face- and edge-sharing sites, Naf and Nae, respectively, on discharge, as shown in Fig. SI-3, SI-9 and Table SI-6.† Despite the different structural behavior of o-Na0.67[Ni0.05Mn0.95]O2 compared to o-Na0.67MnO2, the oxidation states of Mn varied similarly, that is, Mn4+ in the charged state and Mn3+ in the fully discharged state (Fig. 5b). In addition, Ni2+ was oxidized to Ni3+. However, the variation appears dominant when 0.5 ≤ δ in o-Naδ[Ni0.05Mn0.95]O2, whereas, on discharge, the opposite happens (Fig. 5c). This variation in oxidation demonstrates that both o-Na0.67MnO2 and o-Na0.67[Ni0.05Mn0.95]O2 are activated by the Mn3+/4+ redox reaction, whereas the reversible Ni2+/3+ redox couple additionally contributes to the capacity of o-Na0.67[Ni0.05Mn0.95]O2, particularly when 0.5 ≤ δ in o-Naδ[Ni0.05Mn0.95]O2. Local structures of o-Na0.67[NixMn1−x]O2 (x = 0 and 0.05) were investigated by extended X-ray absorption fine structure (EXAFS) measurements during desodiation (Fig. 5d–f). The two intense peaks at the Mn and Ni K-edges in the 1–3 Å range are assigned to the metal–oxygen bonds in the MO6 octahedral units in the first coordination sphere and to the metal–metal bonds in the second coordination sphere.26 The amplitudes of the bonds associated with Mn for o-Na0.67MnO2 developed during desodiation, reflecting the change in the Mn oxidation state from +3 to +4, but the opposite happened during sodiation (Fig. 5d). In contrast, the variation in amplitude is less notable for o-Na0.67[Ni0.05Mn0.95]O2 (Fig. 5f). From these results, it is thought that the smaller change in the FT amplitude for o-Na0.67[Ni0.05Mn0.95]O2 mitigates the cooperative JT effect caused by Mn3+ in the following sequence, O–Mn3+–O–Ni2+–O–Mn4+–O, in the a–b plane of the transition metal layers. Hence, the anisotropic elongation of the Mn3+–O bond can be suppressed by the presence of the oxygen atoms shared with Ni2+. In addition, the changes in the interatomic distance and the lowered FT amplitude of the Ni K-edge indicate that the electrochemical activity of Ni is driven by the Ni2+/3+ redox couple in o-Na0.67[Ni0.05Mn0.95]O2. Therefore, smooth voltage variation appeared throughout the operating range, that is, the suppression of the cooperative JT distortion along a specific direction in the MnO6 octahedra. For these reasons, it is thought that o-Na0.67[Ni0.05Mn0.95]O2 exhibited excellent cycling performance, even at high rates (Fig. 2c and d). Furthermore, the cycled o-Na0.67[Ni0.05Mn0.95]O2 electrode did not show structural degradation in comparison with the pristine o-Na0.67[Ni0.05Mn0.95]O2 but showed a continuous superlattice peak, while peak broadening and altering of the XRD pattern were observed for the extensively cycled o-Na0.67MnO2 (Fig. SI-9 and Table SI-6†). This behavior supports our assumption regarding the improvement of the structural stability, particularly the mitigation of the cooperative JT distortion arising from the presence of Ni2+O6 octahedra in the crystal structure.
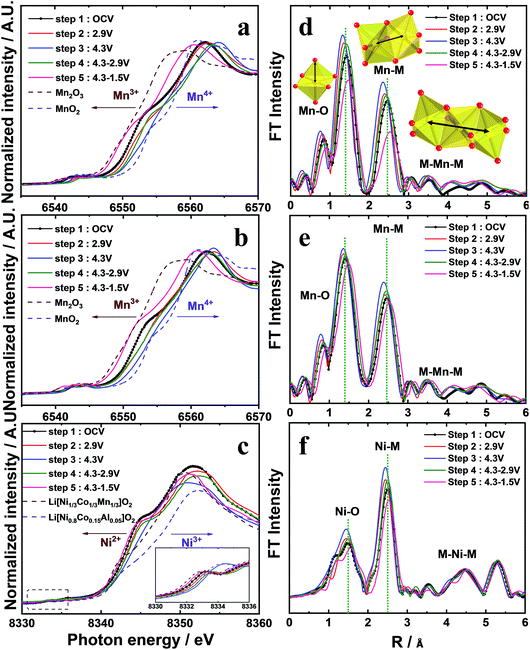 |
| | Fig. 5 XANES spectra at the (a and b) Mn and (c) Ni K-edge and the K3-weighted Fourier transform magnitudes of the (d and e) Mn and (f) Ni K-edge EXAFS spectra during the sodium intercalation process. | |
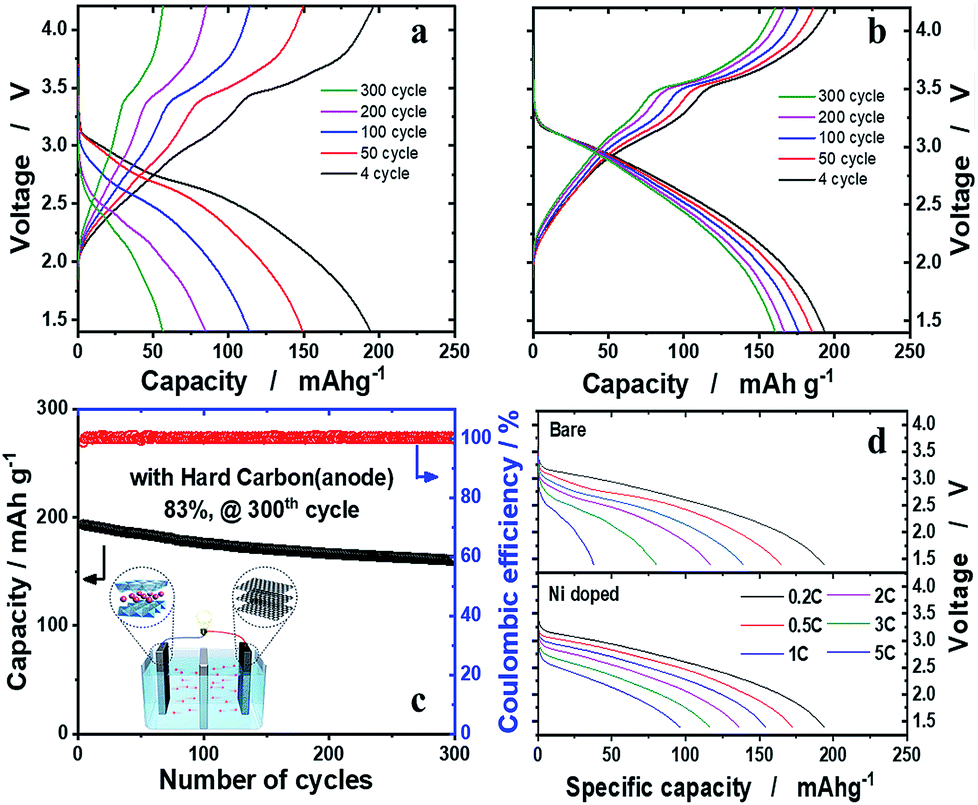
Electrochemical performance in full cells
The hard carbon anode was paired with the o-Na0.67[Ni0.05Mn0.95]O2 cathode to verify our assumption that, provided that Ni is effective in the structural stabilization of o-Na0.67[Ni0.05Mn0.95]O2 in the presence of the JT distortion, the material is able to retain high capacity upon prolonged cycling tests. Both o-Na0.67[NixMn1−x]O2 (x = 0 and 0.05) were presodiated by full sodiation via two electrochemical cycles and, particularly, direct contact with sodium metal in the presence of the electrolyte for hard carbon anodes, to balance the N/P (negative versus positive electrode capacity) ratio to 1.15. The electrochemical properties of hard carbon are shown in Fig. SI-10.† The o-Na0.67MnO2/hard carbon cell exhibited disappointing cell performance, as expected from the half-cell tests (Fig. 6a). In contrast, the o-Na0.67[Ni0.05Mn0.95]O2/hard carbon cell demonstrated unprecedentedly stable cycling performance, showing approximately 83% retention of the initial capacity, that is, 160 mA h (g-oxide)−1 after 300 cycles at 0.2C (52 mA g−1) (Fig. 6b and c). Capacity fading was evident mainly in the 2.5–1.5 V voltage range on discharge, where the Mn3+ ions associated with the JT distortion were dominant. These phenomena are common in Na–Mn–O compounds.13 However, capacity fading is inevitable in this range due to the Jahn–Teller distortion of Mn3+O6 octahedra in the crystal structure. The aim of this work was how to minimize the Mn3+ Jahn–Teller effect, and we suggested that mitigation of the Jahn–Teller distortion is possible by introduction of Ni2+ into the crystal structure of o-Na0.67MnO2 in this work. The delivered capacities at 3C and 5C rates were approximately 116 and 96 mA h g−1, respectively (Fig. 6d). It is worth mentioning that such superior capacity retention in the layered cathode materials for a Na cell has been achieved for the first time in the present o-Na0.67[Ni0.05Mn0.95]O2/hard carbon system. Further work is necessary to raise the operation voltage above 2.5 V by altering the redox pairs from Mn4+/3+ to M4+/3+ (M: transition metal) in the P′2 structure. For practical application, an approach using sodium containing sacrificial salts is known to be the most effective process to have extra sodium to compensate for the deficient charge capacity. We added a sacrificial salt, NaNO2, in o-Na0.67[Co0.05Mn0.95]O2 (Fig. SI-11†). As anticipated, the charge capacity was able to increase almost to a similar level to the discharge capacity, followed by an oxidative decomposition reaction: NaNO2 → NO2 + Na+ + e−, so that additional Na+ is provided to compensate the insufficient Na+ from the NaNO2. This action improves the first abnormal coulombic efficiency (CE) close to 1 in Fig. SI-11.† Indeed, the first CEs were approximately 1.423 and 1.432 for o-Na0.67MnO2 and o-Na0.67[M0.05Mn0.95]O2, respectively. Therefore, it is thought that sacrificing salts are applicable to these kinds of sodium deficient cathode materials to overcome the limitation of insufficient charge capacity. Based on our experience with Li systems, Ni-rich cathode materials showing a high discharge capacity of approximately 200 mA h g−1 always suffer violent exothermic reactions, which threaten the safety of batteries. Mn-based cathode materials are known to have impressive thermal properties when they are highly delithiated, as explained in Fig. SI-12 and SI-13.†
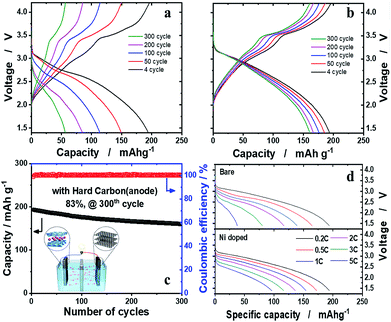 |
| | Fig. 6 Full cell performance of the o-Na0.67[NixMn1−x]O2 (x = 0 and 0.05) cathode with the hard carbon anode. The cathode and anode were first pretreated to calculate the N/P ratio, which was balanced to 1.15. Voltage profiles of (a) o-Na0.67MnO2 and (b) o-Na0.67[Ni0.05Mn0.95]O2 at 0.2C (52 mA g−1). (c) Cyclability and coulombic efficiency plots of the o-Na0.67[Ni0.05Mn0.95]O2 full cell over 300 cycles. (d) Rate capability using a constant current density between 0.2C (52 mA g−1) and 5C (1300 mA g−1) from 1.4 to 4.2 V. | |
Conclusions
In summary, divalent dopants were effective in lowering the bortho/aortho = √3 relationship in the hexagonal lattice of o-Na0.67[M0.05Mn0.95]O2 (M = Mn, Ni, Co, Cu, Zn, Fe, and Al). Among them, the most promising electrode performances were observed for Ni-doped o-Na0.67[Ni0.05Mn0.95]O2. XANES analysis illustrated the electrochemical activities of both Mn3+/4+ and Ni2+/3+, and EXAFS measurements demonstrated the mitigation of the cooperative JT effect of Mn3+ in the oxide lattice arising from the presence of Ni in o-Na0.67[Ni0.05Mn0.95]O2, which showed not only a high rate of Na+-ion insertion ability but a stable cyclability for 300 cycles, retaining 83% (ca. 160 mA h g−1) of the initial capacity in the o-Na0.67[Ni0.05Mn0.95]O2/hard carbon full cell. The presence of Ni further increases the thermal stability in highly desodiated states, accompanied by a decreased oxygen evolution from the crystal structure. This work represents a significant step in creating high capacity cathode materials with excellent cyclability for SIBs. The present chemistry satisfies the safety and high capacity goals while demonstrating the feasibility of high-energy SIBs for energy storage applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2015M3D1A1069713, NRF-2017R1A2A2A05069634).
References
- M. D. Slater, D. H. Kim, E. J. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- A.-S. Nagelberg and W.-L. Worrell, J. Solid State Chem., 1979, 29, 345–354 CrossRef CAS.
- J. Deng, W.-B. Luo, X. Lu, Q. Yao, Z. Wang, H.-K. Liu, H. Zhou and S.-X. Dou, Adv. Energy Mater., 2017, 1701610 Search PubMed.
- P.-F. Wang, Y. You, Y.-X. Yin, Y.-S. Wang, L.-J. Wan, L. Gu and Y.-G. Guo, Angew. Chem., Int. Ed., 2016, 55, 7445–7449 CrossRef CAS.
- C.-Y. Yu, J.-S. Park, H.-G. Jung, K.-Y. Chung, D. Aurbach, Y.-K. Sun and S.-T. Myung, Energy Environ. Sci., 2015, 8, 2019–2026 RSC.
- A. Konarov, J. U. Choi, Z. Bakenov and S.-T. Myung, J. Mater. Chem. A, 2018, 6, 8558–8567 RSC.
- Z. Jian, Y.-S. Hu, X. Ji and W. Chen, Adv. Mater., 2017, 1601925 CrossRef.
- Z. Jian, W. Han, W. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- J. P. Parant, R. Olazcuaga, M. Devalette, C. Fouassier and P. J. Hagenmuller, J. Solid State Chem., 1971, 3, 1–11 CrossRef CAS.
- X. Ma, H. Chen and G. J. Ceder, J. Electrochem. Soc., 2011, 158, A1307–A1312 CrossRef CAS.
- A. Mendiboure, C. Delmas and P. Hagenmuller, J. Solid State Chem., 1985, 57, 323–331 CrossRef CAS.
- M. Yoncheva, R. Stoyanova, E. Zhecheva, E. Kuzmanova, M. Sendova-Vassileva, D. Nihtianova, D. Carlier, M. Guignard and C. Delmas, J. Mater. Chem., 2012, 22, 23418–23427 RSC.
- S. Kumakura, Y. Tahara, K. Kubota, K. Chihara and S. Komaba, Angew. Chem., Int. Ed., 2016, 55, 12760–12763 CrossRef CAS.
- A. Caballero, L. Hernan, J. Morales, L. Sanchez, J. Santos Pena and M. A. G. Aranda, J. Mater. Chem., 2002, 12, 1142–1147 RSC.
- R. Stoyanova, D. Carlier, M. Sendova-Vassileva, M. Yoncheva, E. Zhecheva, D. Nihtianova and C. Delmas, J. Solid State Chem., 2010, 183, 1372–1379 CrossRef CAS.
-
T. Roisnel and J. Rodriguez-Carjaval, Fullprof Manual, Institut Laue-Langevin, Grenoble, France, 2002 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- S. Laubach, S. Laubach, P. C. Schmidt, D. Ensling, S. Schmid, W. Jaegermann, A. Thissen, K. Nikolowski and H. Ehrenberg, Phys. Chem. Chem. Phys., 2009, 11, 3278–3289 RSC.
- C. Zheng, B. Radhakrishnan, I.-H. Chu, Z. Wang and S. P. Ong, Phys. Rev. Appl., 2017, 7, 064003 CrossRef.
- H. Kim, D. J. Kim, D.-H. Seo, M. S. Yeom, K. Kang, D. K. Kim and Y. Jung, Chem. Mater., 2012, 24, 1205–1211 CrossRef CAS.
- I. D. Seymour, S. Chakraborty, D. S. Middlemiss, D. J. Wales and C. P. Grey, Chem. Mater., 2015, 27, 5550–5561 CrossRef CAS.
- B.-L. Guenne, P. Deniard, P. Biensan, C. Siret and R. Brec, J. Mater. Chem., 2000, 10, 2201–2206 RSC.
- N. Yabuuchi, M. Kajiyama, J. Iwatata, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS.
- K.-W. Nam, M. G. Kim and K.-B. Kim, J. Phys. Chem. C, 2007, 111, 749–758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, DFT, cell performance, HT-XRD, DSC and TGA results of o-sodium manganese oxide compounds, lattice parameters from DFT and Rietveld refinement results (fresh and after 50 cycles). See DOI: 10.1039/c8ta08796b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Chong Seung
Yoon
b,
Qian
Zhang
c,
Payam
Kaghazchi
a,
Chong Seung
Yoon
b,
Qian
Zhang
c,
Payam
Kaghazchi