
Influence of molecular weight on PNIPAM brush modified colloidal silica particles†
Ben A.
Humphreys
 a,
Stuart W.
Prescott
b,
Timothy J.
Murdoch‡
a,
Andrew
Nelson
c,
Elliot P.
Gilbert
c,
Grant B.
Webber
a and
Erica J.
Wanless
*a
a,
Stuart W.
Prescott
b,
Timothy J.
Murdoch‡
a,
Andrew
Nelson
c,
Elliot P.
Gilbert
c,
Grant B.
Webber
a and
Erica J.
Wanless
*a
aPriority Research Centre for Advanced Particle Processing and Transport, University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: erica.wanless@newcastle.edu.au
bSchool of Chemical Engineering, UNSW Sydney, NSW 2052, Australia
cAustralian Centre for Neutron Scattering, ANSTO, Lucas Heights, NSW 2234, Australia
First published on 3rd December 2018
Abstract
The effect of molecular weight and temperature on the phase transition and internal structure of poly(N-isopropylacrylamide) brush modified colloidal silica particles was investigated using dynamic light scattering (DLS) and small angle neutron scattering (SANS) between 15 and 45 °C. Dry particle analysis utilising transmission electron microscopy (TEM), Fourier-transform infrared spectroscopy (FTIR) and thermogravimetric analysis (TGA) all confirmed the thickness of the polymer brush shell increased as a function of polymerisation time. Hydrodynamic diameter and electrophoretic mobility results revealed that the brush modified particles transitioned from swollen shells to a collapsed conformation between 15 and 35 °C. The dispersions were electrosterically stabilised over the entire temperature range investigated, with minimal thermal hysteresis recorded. Modelling of the hydrodynamic diameter enabled the calculation of a lower critical solution temperature (LCST) which increased as a function of brush thickness. The internal structure determined via SANS showed a swollen brush at low temperatures (18 and 25 °C) which decayed radially away from the substrate, while a collapsed block-like conformation with 60% polymer volume fraction was present at 40 °C. Radial phase separation was evident at intermediate temperatures (30 and 32.5 °C) with the lower molecular weight sample having a greater volume fraction of polymer in the dense inner region at these temperatures.
1. Introduction
Surface coatings possessing the ability to respond to external stimuli are becoming increasingly popular due to their wide range of possible applications.1 One method to achieve a ‘smart’ stimulus responsive surface is to end tether polymer chains to a substrate at a high enough grafting density to be in the polymer brush regime.2 With the appropriate synthetic method and choice of monomer, it is possible to create a polymer brush modified surface which possesses the ability to significantly alter its physicochemical properties such as hydrophobicity and lubricity along with the thickness of the polymer brush in response to small changes in environmental conditions such as temperature,3,4 light,5 pH,6 or solvent composition,7 as well as salt identity and concentration.8 Depending on the environmental trigger and the properties which enable the responsive behaviour of the brush, these physicochemical changes are often completely reversible leading to truly ‘smart’ surfaces. These behaviours are desired in applications such as targeted and controlled drug delivery,9 biotechnology,10,11 membranes,12 environmental remediation,13 and sensors.14Early work by Alexander15 and de Gennes16,17 proposed that polymers grafted to a planar surface are crowded by their neighbouring polymer chains and therefore adopt a non-Gaussian conformation where the brush thickness scales as a function of N, the degree of polymerisation. This model has since been extended to incorporate surface curvature where the crowding of neighbouring chains plays a slightly diminished role, resulting in a weaker scaling with respect to N.18–21 Polymer brush properties are also influenced by how closely packed the polymer chains are; defined as the grafting density.22 Therefore, other important factors to consider are the method of synthesis, i.e. ‘grafted to’ or ‘grafted from’, along with the curvature of the surface, where the grafting density can significantly change as a function of radial distance from the surface.23,24
Poly(N-isopropylacrylamide) (PNIPAM) (see Fig. S1 in ESI† for molecular structure) is a well-known thermoresponsive polymer that has gained significant attention due to its biologically relevant lower critical solution temperature (LCST) of ∼32 °C for free polymer in pure water.25,26 The exact temperature and breadth of this coil to globule transition is influenced by the molecular weight and the dispersity of the polymer chains, the identity of the endgroup present and solution additives such as salt.27–29 When PNIPAM is used in the preparation of a polymer brush, this abrupt LCST (of width 1–2 °C) broadens, due to the interactions between confined neighbouring polymer chains, into a temperature transition range spanning up to 25 °C.3,8,30 The influence of molecular weight, dispersity, endgroup identity and additives have similar effects on the temperature transition of a PNIPAM brush to that for free polymer, but now the grafting density of the tethered PNIPAM chains also plays an important role.31–35 PNIPAM brushes tethered to a curved substrate are complicated further still, where the grafting density is also influenced by the radius of curvature of the core particle and distance the polymer chains extend from the grafting surface.36 This allows the polymer layer to possess characteristics of free polymer and a polymer brush.8
On the basis of an analytical mean-field theory, this broad temperature transition is ascribed to a cooperative conformational transition as the solvent quality changes instead of a first-order phase transition.37 Furthermore, by incorporating an empirical concentration-dependent solvent quality term determined by Afroze and co-workers38 into numerical self-consistent field theory, it is predicted that a PNIPAM brush will not only have a broad temperature transition, but also undergo vertical phase separation above a critical grafting density.39,40 Therefore, at temperatures near the LCST, the brush layer may separate into an insoluble polymer-rich phase close to the substrate, and a solvated region at the periphery. However, this phenomenon is not predicted in theories that do not permit the solvent quality to vary throughout the brush layer.41 For planar substrates, indirect evidence of vertical phase separation for a PNIPAM brush has previously been provided by combining ellipsometry (which monitors overall brush thickness) with contact angle measurements (only sensitive to the brush periphery),3 while direct evidence of this phenomenon has been revealed through neutron reflectometry.30 Previous studies have utilised small angle neutron scattering (SANS) to probe the internal structure of dispersed colloidal particle systems. These include: the radial profile of a crosslinked PNIPAM shell surrounding a solid polystyrene core at 25 and 43 °C using solvent contrast variation,42 and examining the correlation length in crosslinked PNIPAM hydrogel shells surrounding silica particle cores of varying diameters and dispersities.43 However, the specific case of end grafted PNIPAM chains has received limited attention.
Here, we present a study on the internal structure of PNIPAM brush modified colloidal silica particles as a function of temperature and molecular weight where the curvature of the substrate influences both the structure and thermal response of the polymer brush layer. Curved substrates are of particular interest as close to the substrate the polymer chains are highly confined, similar to a brush on a planar substrate, but as the chain extends further from the particle the volume per chain rises allowing more conformational freedom as observed for free polymer in solution. The well-controlled, surface initiated activators continuously regenerated by electron transfer atom transfer radical polymerisation (SI-ARGET-ATRP) method44 was utilised with a sample of hybrid particles periodically removed from the reaction vessel up to 6 h after initiation of the polymerisation. The resulting hybrid particles were analysed by Fourier-transform infrared spectroscopy (FTIR), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). Each technique clearly showed an increase in PNIPAM brush thickness as a function of growth time. The temperature transition for the five hybrid particle samples was investigated using dynamic light scattering (DLS), with the resulting intensity average hydrodynamic diameter (Dhyd) used to define the polymer brush thickness as a function of temperature. The Dhyd results allowed a comparison of the LCST of the PNIPAM brush as a function of growth time. Further DLS experiments investigating the electrophoretic mobility (μ) of the hybrid particles between 15 and 45 °C highlighted a complex thermoresponsive behaviour. The temperature response of the 2 and 6 h hybrid particles was further probed using SANS in a H2O/D2O solvent mixture that highlights the scattering from the polymer brush shell (core contrast matched). This is the first time radial volume fraction profiles of the polymer brush layer as a function of brush thickness at 18–40 °C have been reported for PNIPAM brushes tethered to a curved substrate.
2. Experimental
2.1. Materials and chemicals
The substrate for the brush synthesis was Ångströmsphere 120 nm nominal diameter silica particles (Fiber Optic Center, USA). Silane initiator functionalisation reagents (3-aminopropyl)triethoxysilane (APTES, >99%), 2-bromoisobutyryl bromide (BIBB, >99%) and triethylamine (Eth3N, 99%) were purchased from Sigma-Aldrich and used as received. Tetrahydrofuran (THF, Honeywell Burdick and Jackson, >99%) was dried for at least one day over 4 Å molecular sieves (ACROS Organics) before use. N-Isopropylacrylamide (NIPAM, Sigma-Aldrich, 98%) was stored below 4 °C and purified by recrystallisation in n-hexane (Merck, ≥98.0%) prior to use. Polymerisation reagents 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), copper(II) bromide (99.999%), and L-ascorbic acid (≥99.0%) were purchased from Sigma Aldrich and used as received. Methanol (Sigma Aldrich, anhydrous, 99.8%) was used as received, ethanol (Ajax Finechem, absolute) was distilled before use. The pH of the solutions was controlled at 8.5 ± 0.1 using potassium hydroxide (Chem-Supply Pty. Ltd AR grade). D2O was provided by ANSTO and filtered through 0.22 μm syringe filter prior to use and MilliQ™ water (Merck Millipore, 18.2 MΩ cm at 25 °C) was used throughout.2.2. PNIPAM brush synthesis on silica particles
PNIPAM brush modified core/shell particles were synthesised via the ‘grafted from’ ARGET ATRP method44 from surface bound bromine initiator sites. The grafting density of covalently bound ATRP initiators at the surface of the silica particle was estimated to be ∼2.4 sites nm−2 based on ∼2.36 wt% difference at 600 °C between the core silica particle and the initiator functionalised particles. The synthesis was performed with reagents, NIPAM/CuBr2/HMTETA/ascorbic acid in the molar ratios 900/1.5/15/10, dissolved in a solvent mixture of methanol/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) with the monomer to solvent mass ratio being 0.058/1. A 10 mL aliquot of the reaction dispersion was removed at 0.5, 1, 2, 4 and 6 h after the polymerisation was initiated. Full details of core silica particle preparation, covalent binding of surface initiator, and the PNIPAM brush synthesis procedure have previously been reported.8 The grafting density of the polymer chains was estimated to be ∼0.38 chains nm−2 based on the initiator efficiency of ∼16% previously reported for an ATRP synthesis of a PNIPAM brush from the same initiator covalently bound to a 70 nm silica core.45 This estimated grafting density is within the polymer brush regime.
:1 v/v) with the monomer to solvent mass ratio being 0.058/1. A 10 mL aliquot of the reaction dispersion was removed at 0.5, 1, 2, 4 and 6 h after the polymerisation was initiated. Full details of core silica particle preparation, covalent binding of surface initiator, and the PNIPAM brush synthesis procedure have previously been reported.8 The grafting density of the polymer chains was estimated to be ∼0.38 chains nm−2 based on the initiator efficiency of ∼16% previously reported for an ATRP synthesis of a PNIPAM brush from the same initiator covalently bound to a 70 nm silica core.45 This estimated grafting density is within the polymer brush regime.
2.3. Transmission electron microscopy (TEM)
Particle images were obtained using a JEOL 2100 HR TEM with an accelerating voltage of 200 kV. Samples were prepared from dilute particle dispersions with a small droplet placed on a carbon coated TEM mesh grid (ProSciTech., Australia) and dried in air at ambient temperature. The particle diameters from the TEM images were analysed using the Fiji variation of the ImageJ software46 with all reported values an average of at least 50 individual particles.2.4. Thermogravimetric analysis (TGA)
A PerkinElmer Diamond TG/DTA thermal analyser was used for the TGA of the samples. Aluminium pans were used for all experiments, which were performed from 30 to 600 °C at a ramp rate of 10 °C min−1 under constant flow of N2 at 30 mL min−1. The % mass loss of each sample between 180 and 500 °C (to ensure solvent was excluded), after subtracting the % mass loss that was recorded for the core silica particles over this temperature range, was then converted into a dry thickness of the polymer layer. The dry brush thickness calculations used a core particle radius of 613 Å (via TEM) and density of 1.89 g cm−3 (previously measured via pycnometry)24 with an assumed density of 1.07 g cm−3 for PNIPAM.2.5. Fourier-transform infrared spectroscopy (FTIR)
The infrared spectra were recorded on a PerkinElmer Spectrum Two FTIR spectrometer with a UATR Two attachment and were an average of four scans. The spectra were then normalised to the SiO2 peak at 1060 cm−1.2.6. Dynamic light scattering (DLS)
A Malvern Zetasizer Nano-ZSP was used to measure the hydrodynamic diameter (Dhyd) and electrophoretic mobility (μ) of the particles as a function of temperature in pure water. The DLS measurements collected the backscattered intensity at 173° with reported intensity average diameter values representing an average of at least 40 runs of 10 seconds each, with the standard deviation less than 5% of the reported values. The mobility results are an average of 10 measurements, each consisting of between 10 and 20 runs with the standard deviation less than 9% of the reported value of μ. The sample was held at each temperature for 10 min prior to each measurement to ensure thermal equilibrium was reached. The previously reported slight variations in the electrophoretic mobility of the particles as a function of temperature has been used to correct all results obtained in this study.82.7. Small angle neutron scattering (SANS)
SANS measurements at 18, 25, 30, 32.5, and 40 °C were performed on QUOKKA, the pinhole, small angle neutron scattering instrument at ANSTO.47,48 Three sample-detector distances of 1.3, 8, and 20 m were used to provide a Q range of 0.004 to 0.75 Å−1 where Q is the magnitude of the scattering vector, defined by Q = (4π/λ)sin(θ) for which the neutron wavelength, λ, was 5 Å (with resolution of 10%) and 2θ is the scattering angle. All data were corrected for blocked beam measurements, normalised, radially averaged and placed on an absolute scale, following attenuated direct beam measurements and using an empty cell measurement as background. For details of the sample preparation see ESI.† Contrast variation tests were performed in six different mixtures of H2O and D2O to determine the scattering length density (SLD) of the silica core.3. Methods
3.1. Particle dispersion preparation
The solution pH was first adjusted to 8.5 to enhance particle stability for all DLS and SANS experiments performed on the PNIPAM-brush-modified particle dispersions.8 At pH 8.5 the core silica particles had an electrostatically stabilising zeta potential greater than −45 mV in magnitude spanning the entire temperature range investigated (see Fig. S2, ESI†).3.2. Analysis of DLS Dhyd data
While free PNIPAM has an abrupt coil to globule transition at ∼32.5 °C,49 PNIPAM end tethered in the brush regime displays a broad swollen-to-collapsed transition spanning as much as 25 °C.8 As mentioned above, the curvature of the substrate of a polymer brush introduces an extra parameter into neighbouring polymer chain interactions where the volume that each chain can occupy increases radially from the core.24 Furthermore, the shape of this transition is significantly different to that for a polymer brush on a planar surface where a simple sigmoidal model is no longer able to adequately describe the transition.3 The LCST for each brush modified sample was defined as the midpoint of the temperature transition for Dhyd on the increasing temperature ramp. To achieve this, the maximum and minimum particle diameters were required and therefore five separate equations were used to smoothly model the results for each sample (see Fig. S3 and Table S1 in ESI† for full details).8 Fig. S3 (ESI†) details the modelling for the 1 h sample showing each section of the modelled transition, the maximum, minimum and half height particle diameters along with the LCST.3.3. SANS contrast variation study
For the SANS, a solvent contrast variation study was performed on PNIPAM brush modified particle dispersions at 18 °C in six different mixtures of H2O and D2O to determine the SLD of the silica core. The SLD for the silica was predicted to be ∼2.99 × 10−6 Å−2 based on the measured density for the core particles of 1.89 g cm−3. The contrast match plot is presented in Fig. S4 in ESI† where the square root of the intensity (√IQ=0.004) is plotted as a function of % H2O in the H2O/D2O mixture. From this plot, the SLD of the silica core was approximated at 3.21 × 10−6 Å−2, equivalent to 45.5% H2O. The information gained from this contrast variation study was used to select the H2O/D2O solvent mixture for highlighting the scattering from the PNIPAM brush shell (core contrast matched) obviating the need for any polymer or core particle deuteration.3.4. Analysis of SANS data
SasView software version 4.1 was used to analyse the data utilising the spherical onion model with 3 shells.50,51 The scale factor (SF) used in this model incorporates the volume fraction of particles within the dispersion; however, with a thermoresponsive polymer brush, the SF varies significantly as a function of temperature. Therefore, the SF for the system where the polymer brush layer was swollen (low temperature), provided an upper constraint for the SF in the fitting for all other temperatures. After initial modelling, the radius and SLD of the silica core were fixed for all conditions at 650 Å and 3.2 × 10−6 Å−2 respectively and a 20 Å layer was included to account for small variations in core particle radii and the roughness between the substrate and the polymer brush. All other parameters were either manually set based on independently measured values or optimised iteratively using SasView to minimise the calculated χ2 values. A final analysis of the fits was conducted to ensure a constant amount of tethered polymer was present at all temperatures for each molecular weight.4. Results and discussion
4.1. Dry particle analysis
Preliminary dry experiments were performed on the core silica and PNIPAM brush modified particles to qualitatively and quantitatively determine the presence of the PNIPAM brush layer. The infrared spectra for the core silica particles and the series of five PNIPAM brush modified samples are presented in Fig. S5 in ESI.† This highlights the presence of a PNIPAM brush layer surrounding the core silica particles through the peaks assigned to the amide carbonyl stretch (∼1630 cm−1), N–H bending (∼1550 cm−1), C–H bending (∼1460 cm−1) and the C–N stretch (twin peaks at ∼1390 cm−1 and ∼1370 cm−1). The intensity of these peaks progressively increased for the PNIPAM brush modified samples from 0.5 h up to 6 h as the thickness of the polymer shell increased.TEM images for the core silica particles, 0.5, 2, 4, and 6 h samples are displayed in Fig. 1. The core silica particles (Fig. 1(a) and (b)) provide evidence of a monodisperse spherical sample with a number average diameter of 1226 ± 67 Å prior to the addition of the polymer shell. Images in Fig. 1 (c) through to (j) are of the polymer brush modified particles which show clear evidence of a polymer shell present on all samples with the thickness of this film increasing as a function of polymerisation time. Polymer bridging between neighbouring particles is also observed for all PNIPAM brush modified samples with the degree of bridging and bridge thickness increasing as a function of polymer shell thickness. These bridges are the result of drying as DLS measurements, detailed later, and do not indicate particle aggregation when in good solvent conditions. The average TEM diameters for all samples imaged are presented in Table 1.
 | ||
| Fig. 1 TEM images of (a and b) silica core particles, (c and d) 0.5 h sample, (e and f) 2 h sample, (g and h) 4 h sample and (i and j) 6 h sample. | ||
| sample | TEM diameter (Å) | TGA thickness (Å) | TGAa diameter (Å) |
|---|---|---|---|
| a The TGA derived diameter was calculated as twice the dry thickness added to the TEM silica core diameter (1226 Å) for comparison. | |||
| Silica core | 1226 ± 67 | N/A | N/A |
| 0.5 h | 1260 ± 71 | 23 | 1272 |
| 1 h | 1265 ± 64 | 25 | 1276 |
| 2 h | 1275 ± 61 | 31 | 1288 |
| 4 h | 1307 ± 84 | 41 | 1308 |
| 6 h | 1312 ± 66 | 53 | 1332 |
TGA analysis of the five PNIPAM brush modified samples was also performed to accurately determine the weight % of the polymer coating present (Fig. S6 in ESI†). The weight % of polymer was then used to estimate PNIPAM brush thickness, h, for each sample using eqn (1), where r is the silica particle radius, mi is the polymer mass fraction and ρp and ρb are the densities of the particle and polymer brush, respectively.24
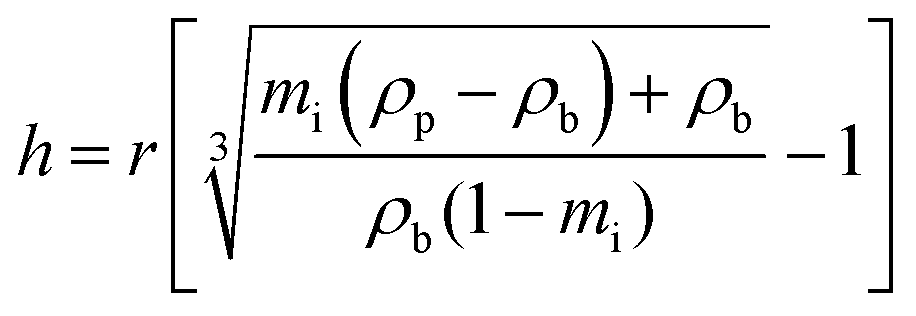 | (1) |
This equation assumes an idealised core–shell hybrid particle with a core particle radius of 613 Å (measured by TEM) and density of 1.89 g cm−3 (measured by pycnometry) while the polymer brush density was set at 1.07 g cm−3 (density of untethered polymer25). The TGA-derived thickness of the polymer brush layer displayed in Table 1 was used in conjunction with the core silica particle diameter to define the diameter of the dry PNIPAM brush modified samples (also presented in Table 1). The particle diameters determined from TEM and TGA are in excellent agreement.
4.2. Thermoresponsive particle dispersions
For DLS measurements, the intensity average hydrodynamic diameter (Dhyd) was measured as a function of both increasing and decreasing temperature between 15 and 45 °C for dispersions of the five different samples (see Fig. 2). The values of Dhyd reported account for the temperature dependent viscosity of water. | ||
| Fig. 2 Intensity average hydrodynamic diameter (Dhyd) as a function of increasing (closed symbols) and decreasing (open symbols) temperature for the five PNIPAM brush modified particle samples dispersed in water adjusted to pH 8.5. Inset displays the calculated LCST for each sample. For reference, the Dhyd for the bare silica particles was measured to be 1310 ± 20 Å, concordant with the core particle diameter used in the SANS analysis. | ||
All five samples in Fig. 2 displayed temperature responsive behaviour between 15 and 45 °C with minimal hysteresis between increasing and decreasing temperature ramps. The shape of the temperature transition is similar for all samples where two regions are present, a phenomenon previously reported for PNIPAM brush modified colloidal particles with a similar grafting density.4,8,45 Looking at the increasing temperature results (closed symbols), a swollen polymer brush layer is present at 15 °C, well below the LCST of free PNIPAM, followed by a gradual decrease in particle diameter as the polymer brush starts to collapse. A significant change in brush thickness occurs between 30 and 35 °C at which point, now above the LCST for free polymer, the brush layer is fully collapsed. Also, consistent with the dry sample analysis, the thickness of the hydrated polymer brush shell increased as a function of polymerisation time, with the greatest variations between samples being at low temperatures when the PNIPAM brush layer is highly swollen. At 15 °C the diameter of the 0.5 h sample was 500 Å less than the 6 h sample, while at 45 °C when the brush layer is collapsed the difference in diameter between the two samples was only 21 Å.
The LCST for the five samples, as shown in the inset to Fig. 2, are a function of brush thickness; ranging from 30.7 °C for the 0.5 h sample up to 32.2 °C for the 6 h sample. It is proposed that the ability of the hydrophobic bromine end group of the polymer chains to reduce the LCST becomes less significant as the growth time, and therefore molecular weight of the polymer, increases. Similar results have previously been reported by Schweizerhof and co-workers for molecular weight effects on free PNIPAM and PNIPAM brushes on gold nanorods with a hydrophobic benzyl endgroup.52
The electrophoretic mobility (μ) was also investigated in the temperature range 15 to 45 °C for the five PNIPAM brush modified particle dispersions (Fig. 3). Owing to the complex behaviour of brush modified particles, where the motion of the particles is also influenced by the polymer chain conformation, the raw mobility results have been reported instead of the zeta potential. At low temperatures when the polymer brush layer is swollen, the viscous drag of the particle is at its highest. Thus, the influence of the applied electric field is minimal with μ = −1.10 × 10−8 m2 V−1 s−1 at 15 °C for the 0.5 h sample with increasingly more viscous drag as the PNIPAM brush thickness increased down to μ = −0.26 × 10−8 m2 V−1 s−1 for the 6 h sample. As the temperature is increased, the polymer brush layer collapses and the viscous drag is reduced. This is evident by an increase in mobility at higher temperatures. The variation in μ between samples is also reduced as the polymer brush layer collapses because the difference in diameter between samples, as highlighted in Fig. 2, is minimal.
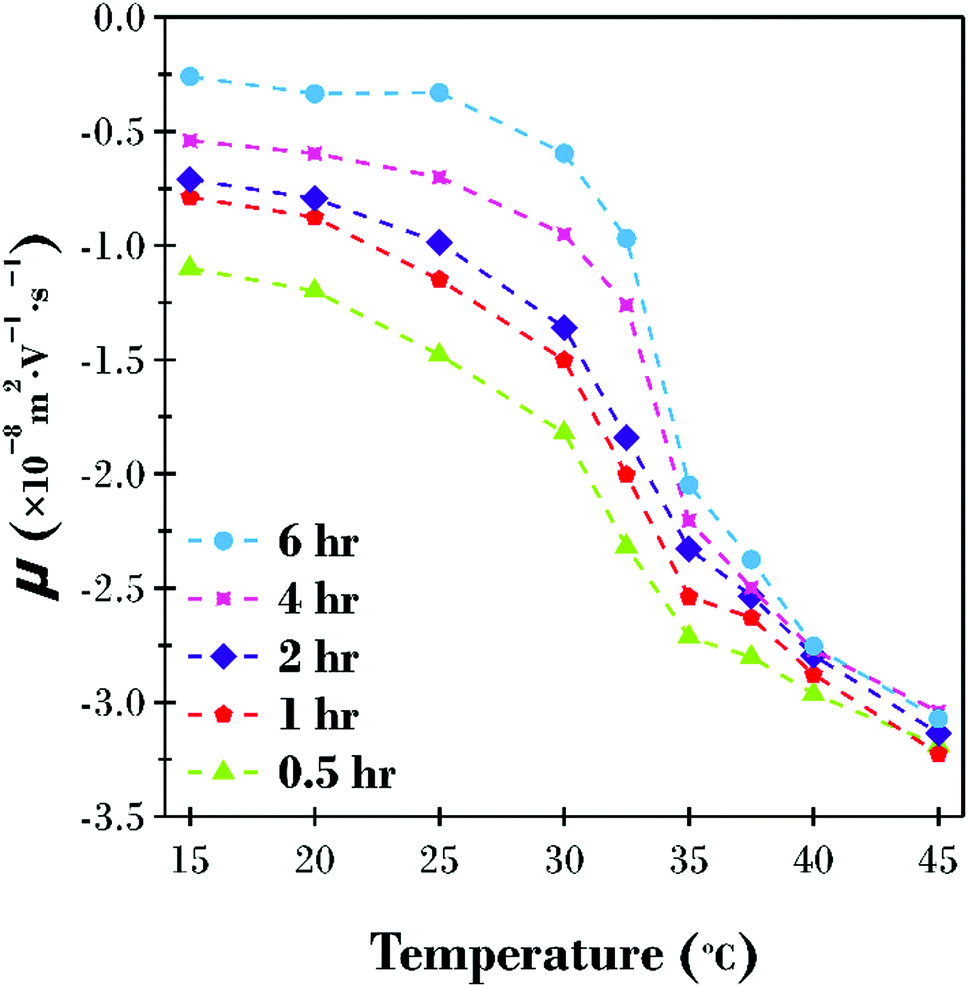 | ||
| Fig. 3 Electrophoretic mobility (μ) as a function of increasing temperature for the five PNIPAM brush modified particle samples dispersed in water adjusted to pH 8.5. | ||
It is of particular interest that all five samples formed stable dispersions over the entire temperature range investigated, even at temperatures above the LCST when polymer–polymer interactions are expected to be favoured.25 It has previously been reported that PNIPAM brush modified particle aggregation occurs in the presence of as little as 50 mM monovalent salt.8 Therefore, it is proposed that a combination of electrostatic and steric stabilisation exist for this system. The particles are sterically stabilised at low temperatures when the polymer brush is swollen, and polymer–solvent interactions are favoured. Conversely, the particles are electrostatically stabilised at high temperatures when the PNIPAM brush shell is collapsed and polymer–polymer interactions dominate, yet the underlying particle surface charge (see Fig. S2, ESI†) is high enough to prevent particles getting sufficiently close to aggregate. As previously reported, the addition of relatively low concentrations of salt reduces the Debye length below the thickness of the collapsed brush shell, and particle–particle interactions are possible leading to aggregation.8
A SANS investigation was performed to probe the underlying structure of the PNIPAM brush corona as a function of temperature and molecular weight. The 2 and 6 h PNIPAM brush modified samples were chosen for the SANS investigation, with measurements performed from low to high temperature at 18, 25, 30, 32.5, and 40 °C. The increasing temperature Dhyd results (from Fig. 2) and the modelled brush transition for the two samples (from Table S1 in ESI†) are displayed in Fig. 4. Vertical dashed lines at the temperatures investigated using SANS are included to highlight the differences between the 2 and 6 h samples at these points of interest. Although the two samples exhibit a similarly shaped transition there are subtle differences, particularly in the range 30–35 °C in the vicinity of the LCST. In this range, the 6 h sample has a sharper decrease in thickness with increasing temperature.
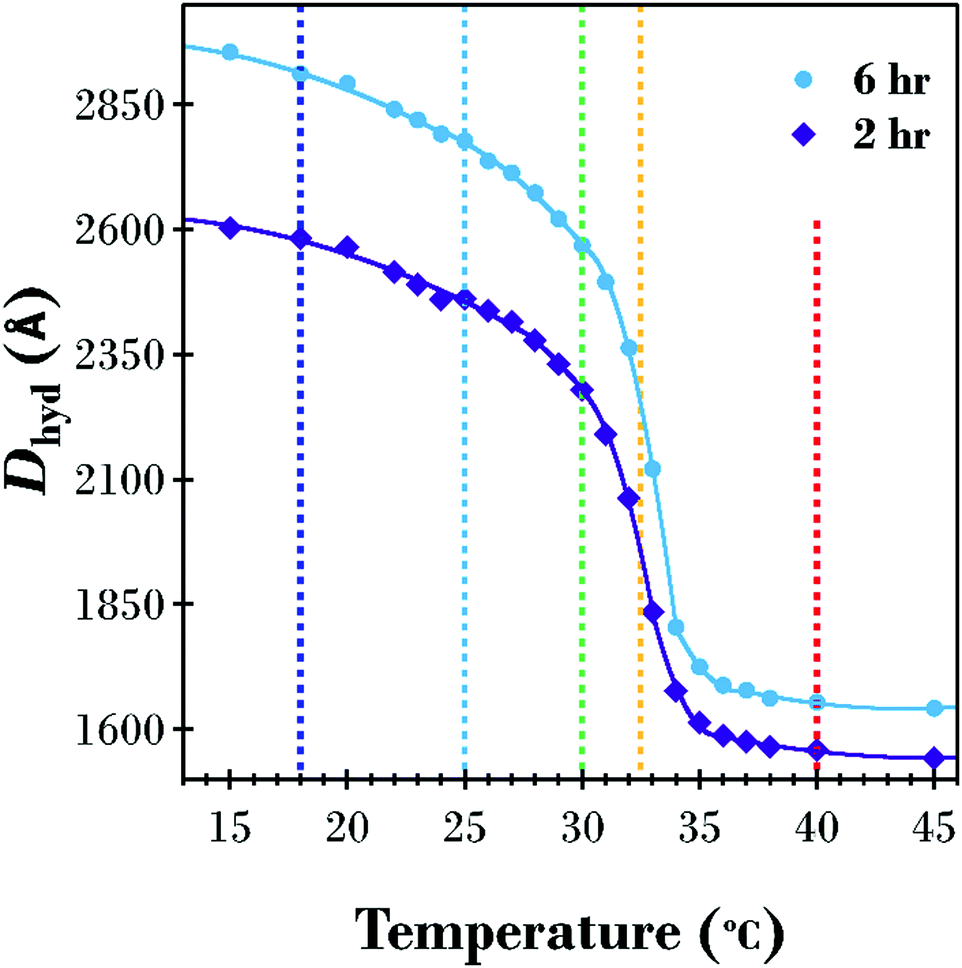 | ||
| Fig. 4 DLS Dhyd increasing temperature results and modelled temperature transitions for the 2 and 6 h PNIPAM brush modified samples with vertical dashed lines indicating SANS measurement temperatures. | ||
The 2 and 6 h brush modified samples were prepared by dispersing a low volume fraction of particles in a H2O/D2O mixture. At the particle volume fractions investigated (maximum 0.5 volume % and decreasing with temperature), structure factors effects were not considered in the modelling. For full details of the SasView spherical onion model description see ESI.† All of the parameters used in modelling the 2 and 6 h samples, including χ2 quality of fit, are presented in Tables S3 and S4 in ESI.†
The SANS data and modelled fits for the two samples are presented in Fig. 5(a and c). Data were only modelled in the Q range between 0.004 and 0.04 Å−1 as higher Q data were relatively featureless due to the overall size of the particles (see Fig. S7 in ESI† for full data for 2 and 6 h samples at 18 °C). Oscillations in the data are evident under all conditions, with the maximum intensity for the two samples decreasing as the temperature was increased. The maximum intensity for the 6 h sample reduced from 2.03 cm−1 at 18 and 25 °C down to 2.01, 1.79 and finally 1.05 cm−1 at 30, 32.5 and 40 °C respectively. The reduction in measured intensity is due to the polymer brush layer collapsing at higher temperatures, resulting in the overall volume fraction of the particles being significantly reduced.
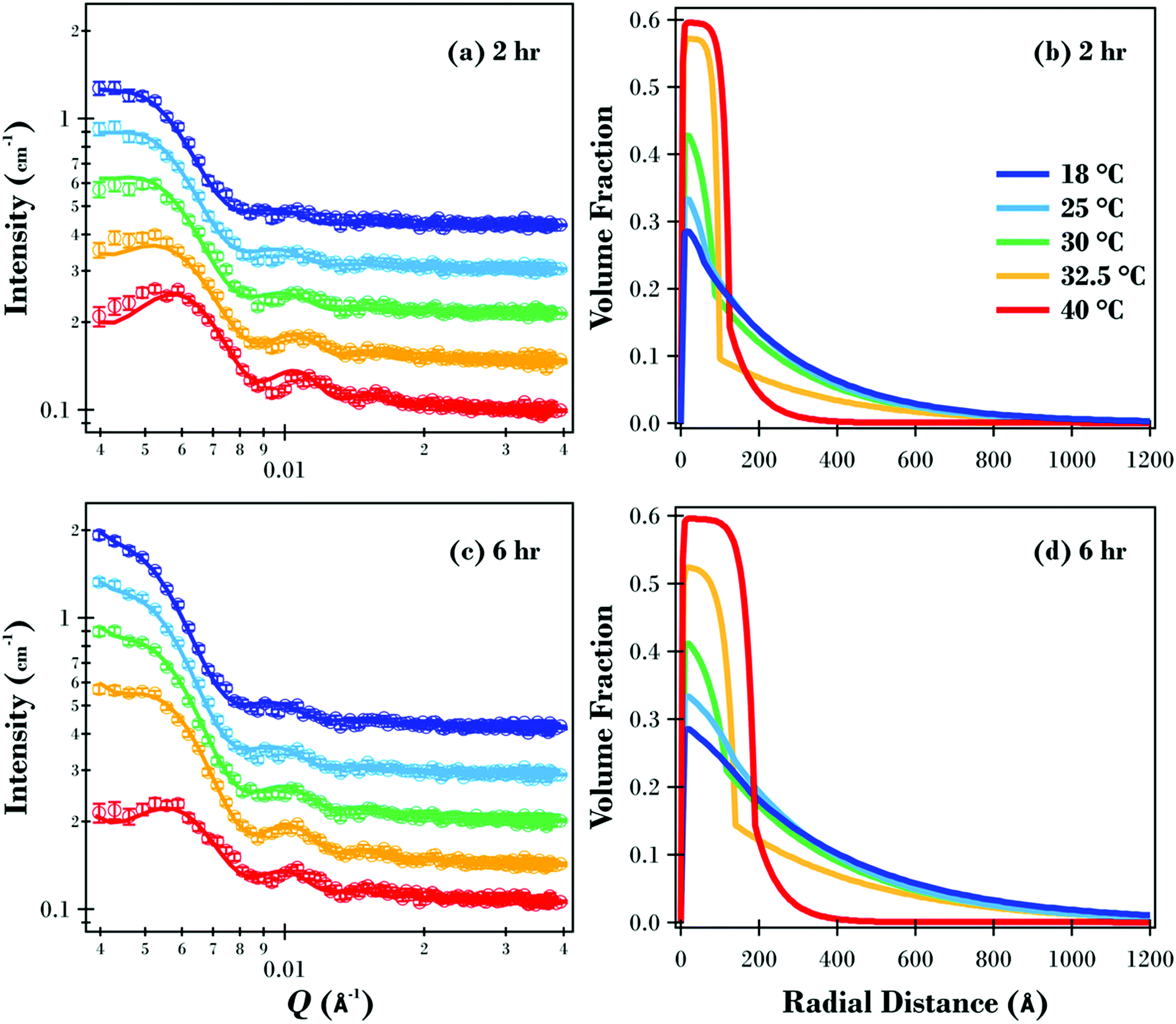 | ||
| Fig. 5 SANS results as a function of temperature (open circles) for (a) 2 h and (c) 6 h sample including modelled fits (solid lines) with the curves offset on the intensity axis for clarity. The corresponding volume fraction profiles (2 h and 6 h samples – (b) and (d) respectively) for the polymer brush layer as a function of radial distance from the substrate. | ||
This SANS study has elucidated the structure of the underlying PNIPAM brush layer and emphasised the effects of molecular weight on the radial volume fraction profile of the polymer brush shell. The radial volume fraction profiles for the two polymer brush samples are displayed in Fig. 5. For the 2 h sample (Fig. 5b), the brush is swollen at 18 and 25 °C with the volume fraction of polymer exponentially decaying radially from the core silica particle surface. As the temperature was increased, the initial volume fraction of polymer at this interface increased from ∼0.3 (18 °C) up to ∼0.6 at 40 °C where the polymer brush forms a collapsed dense region. The change in the level of hydration as a function of temperature for the PNIPAM brush modified particles was similar to previous PNIPAM brush experiments on planar substrates.30 At 30 and 32.5 °C where the temperature was close to the LCST, radial phase separation is evident with a dense inner region transitioning to a dilute, solvated tail region. As discussed previously, this phase separation is predicted by theory for a neutral temperature responsive polymer brush, and has been reported for PNIPAM brushes on planar substrates.30,33,53Fig. 5(d) shows the volume fraction profiles for the 6 h sample. Like the 2 h sample, the brush layer for the 6 h sample was swollen well below the LCST up to 25 °C and collapsed above the LCST at 40 °C with radial phase separation present at 30 and 32.5 °C.
The volume fraction brush profiles presented in this study, when the solvent quality is good (18 and 25 °C) and when the solvent quality is poor (40 °C), are in good agreement with the SCF calculations of Wijmans and Zhulina for polymer brushes on curved substrates.54 This provides further evidence that the brush modified particles prepared in this study have end tethered PNIPAM chains with a grafting density in the polymer brush regime. However, the profiles around the LCST at 30 and 32.5 °C, where vertical phase separation is evident, deviate from this model's predictions indicating that the behaviour of a PNIPAM brush tethered to a curved substrate requires a more complex model. Future research focusing on a quantitative comparison between the findings in this study and a self-consistent field model based on a modified Flory–Huggins free energy could provide a more accurate free energy expression, compared with the one previously proposed by Afroze et al.38
The volume fraction profiles for the two samples are directly compared at each temperature in Fig. 6 together with a schematic representation of the cross section of the brush modified particles at each temperature. Fig. 6 highlights how the response of the brush and the underlying structure are influenced by the molecular weight of the polymer. Although the profiles are quite similar for the swollen and collapsed brush conformations, apart from the distance the brush extends from the surface, there are significant variations for the 30 and 32.5 °C profiles. Due to the method utilised in the synthesis of the PNIPAM brush modified samples, where equal aliquots were systematically removed from a single reaction vessel as a function of polymerisation time, the grafting density is consistent for all samples, evident when looking at the swollen and collapsed profiles for both samples in Fig. 6(a and e) where the volume fraction of polymer at the interface is the same independent of the polymer molecular weight. The radial volume fraction profile decreases faster for the 2 h sample at all temperatures investigated, due to the lower molecular weight of the tethered polymer chains.
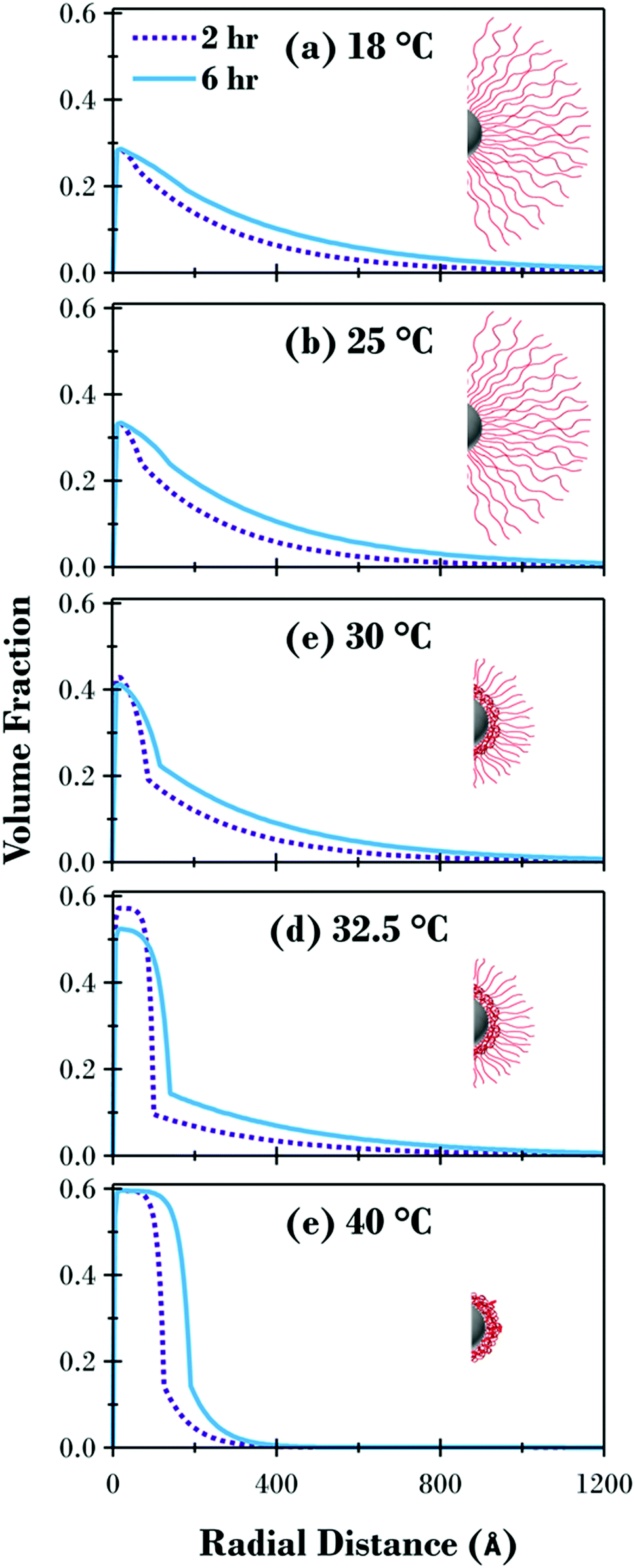 | ||
| Fig. 6 Volume fraction profiles for the 2 h (dashed line) and 6 h (solid line) PNIPAM brush modified samples presented as the radial distance from the substrate at (a) 18, (b) 25, (c) 30, (d) 32.5 and (e) 40 °C. Insets schematically illustrate the state of the brush at each temperature. | ||
While the detailed curve analysis of the DLS data showing the temperature-dependence of Dhyd in Fig. 2 provided evidence that the 2 h sample had an LCST 0.4 °C lower that the 6 h sample, the SANS comparison in Fig. 6 further clarifies the effect of molecular weight variation on the polymer brush volume fraction profile. When comparing the two samples in Fig. 4, the 2 h sample has a transition which starts at a slightly lower temperature, accentuated by the greater degree of polymer brush collapse between the highlighted 30 and 32.5 °C points. Fig. 6 provides further evidence of the LCST decrease for the thinner sample where the volume fraction of polymer at the interface is slightly higher at 30 °C and therefore less hydrated; a small amount of radial phase separation is evident for both samples but there is slightly more of the dehydrated collapsed region at the substrate surface for the 2 h sample. The difference between the two samples is greatest in Fig. 6(d) at 32.5 °C (the temperature closest to the LCST for both samples) where there is significantly more of the polymer brush as a percentage of the entire area under the curve in the phase separated collapsed region, for the 2 h sample (66%) compared to the 6 h sample (57%).
The average brush thickness, L1st, was determined for both samples at each temperature by twice the normalised first moment of the volume fraction profiles in Fig. 5:
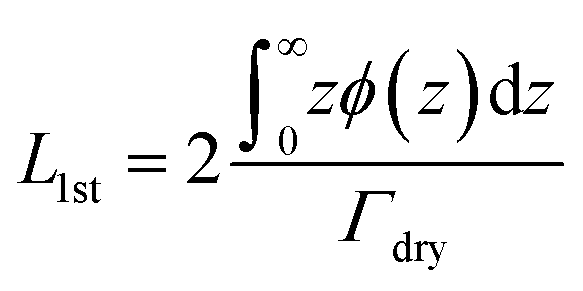 | (2) |
A factor of 2 is used in eqn (2) as this corresponds to the thickness of a step-density profile with the same normalised first moment.55 This method has previously been utilised to provide comparable results between neutron reflectometry and ellipsometry results for thermoresponsive brushes on planar substrates.30,56 A DLS and SANS comparison of the effect of molecular weight on the thermoresponse of the PNIPAM brush modified particles reported as a radial brush thickness is presented in Fig. 7. The results are remarkably similar between the two techniques with the DLS results showing a slightly thicker, swollen brush due to the increased sensitivity of this technique to the periphery of the solvated brush. This concordance of the results confirms the validity of the SasView core–shell onion model used in this study.
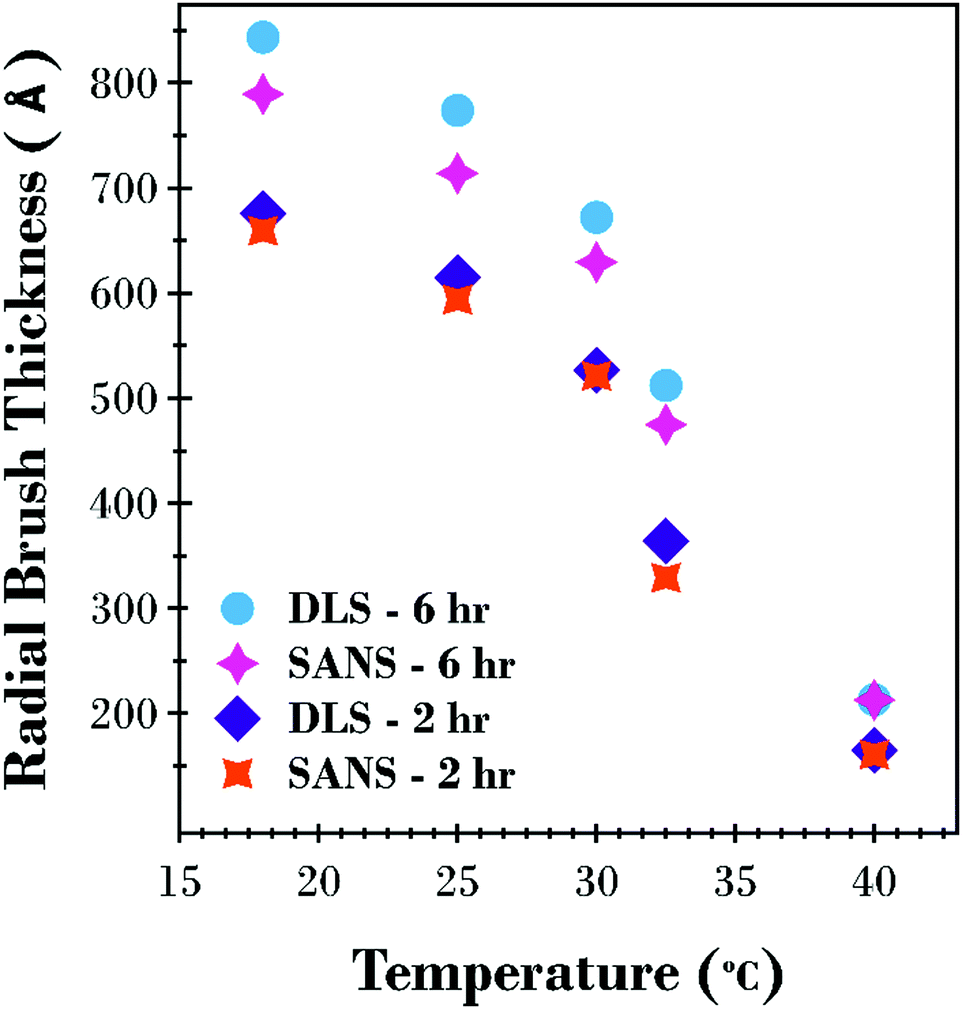 | ||
| Fig. 7 Comparison of calculated thickness as a function of temperature from SANS (L1st) and DLS ((Dhyd – core diameter)/2) results for the 2 and 6 h PNIPAM brush modified silica particles. | ||
5. Conclusions
The temperature transition and internal structure of the PNIPAM brush modified particles as a function of molecular weight has been investigated utilising DLS and SANS. Dry particle analysis employing TEM, FTIR and TGA confirmed the presence of the PNIPAM brush shell surrounding the core silica particle which increased in thickness as a function of polymerisation time.DLS measurements confirmed that the PNIPAM brush modified particles formed stable dispersions over the temperature range investigated (15 to 45 °C) with very little swelling hysteresis evident. The brush layer was swollen at 15 °C with the overall particle diameter increasing as a function of growth time between 2450 Å for the 0.5 h sample up to 2960 Å for the 6 h sample. All brush modified samples displayed a gradual decrease in diameter between 15 and ∼30 °C followed by a relatively abrupt decrease in temperature up to 35 °C where all samples were collapsed to ∼1600 Å (only 21 Å difference between the 0.5 and 6 h). The LCST increased as a function of molecular weight, ranging from 30.7 °C (0.5 h) up to 32.2 °C (6 h). The electrophoretic mobility of the particles increased as the polymer brush collapsed with the thickness of the polymer brush layer responsible for the degree of viscous drag associated with the particle.
The underlying structures of the 2 and 6 h samples were interrogated via SANS and modelled utilising a sphere core/shell onion model. The results showed a swollen brush profile at low temperature (18 and 25 °C) which decays out from the surface, and a block like collapsed conformation containing 60% volume fraction of polymer at 40 °C. The intermediate temperatures of 30 and 32.5 °C displayed evidence of vertical phase separation with a collapsed, polymer rich phase adjacent to the substrate transitioning to a dilute solvated tail region. This is the first PNIPAM brush modified particle study utilising SANS to detail the volume fraction profile to characterise the thermoresponse of the polymer shell.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. A. H. and T. J. M. thank Australian Government Research Training Program (RTP scholarship) and AINSE Ltd (PGRA Award) for providing financial assistance. The Australian Research Council is thanked for LE140100090. This work was supported by the Australian Centre for Neutron Scattering proposal grants P5945 and P5950 and Edwin Johnson (University of Newcastle), Isaac Gresham and Zengyi Wei (UNSW) are thanked for their assistance throughout the SANS experiment. The Electron Microscope X-ray Unit (EMX) and Dr Huiming Zhang at the University of Newcastle are thanked for assistance with TEM imaging. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No. 654000.References
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS.
- S. Minko, J. Macromol. Sci., Polym. Rev., 2006, 46, 397–420 CrossRef CAS.
- B. A. Humphreys, J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Phys. Chem. Chem. Phys., 2016, 18, 6037–6046 RSC.
- T. J. Murdoch, B. A. Humphreys, E. C. Johnson, G. B. Webber and E. J. Wanless, J. Colloid Interface Sci., 2018, 526, 429–450 CrossRef CAS.
- J. Xiang, X. Tong, F. Shi, Q. Yan, B. Yu and Y. Zhao, J. Mater. Chem. B, 2018, 6, 3531–3540 RSC.
- J. D. Willott, B. A. Humphreys, T. J. Murdoch, S. Edmondson, G. B. Webber and E. J. Wanless, Phys. Chem. Chem. Phys., 2015, 17, 3880–3890 RSC.
- S. Li, L. Feng, H. Lu and S. Feng, New J. Chem., 2017, 41, 1997–2003 RSC.
- B. A. Humphreys, E. J. Wanless and G. B. Webber, J. Colloid Interface Sci., 2018, 516, 153–161 CrossRef CAS.
- M. Das, S. Mardyani, W. C. W. Chan and E. Kumacheva, Adv. Mater., 2006, 18, 80–83 CrossRef CAS.
- M. M. Ali, S. Su, C. D. Filipe, R. Pelton and Y. Li, Chem. Commun., 2007, 4459–4461 RSC.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- M. Ulbricht, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 113–125 CrossRef CAS.
- D. Parasuraman and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 2732–2737 CrossRef CAS.
- G. R. Hendrickson and L. A. Lyon, Soft Matter, 2009, 5, 29–35 RSC.
- S. Alexander, J. Phys., 1977, 38, 983–987 CrossRef CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- P. G. de Gennes, J. Phys., 1976, 37, 1445–1452 CrossRef CAS.
- E. B. Zhulina, T. M. Birshtein and O. V. Borisov, Eur. Phys. J. E: Soft Matter Biol. Phys., 2006, 20, 243–256 CrossRef CAS.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143–9150 CrossRef CAS.
- D. Dukes, Y. Li, S. Lewis, B. Benicewicz, L. Schadler and S. K. Kumar, Macromolecules, 2010, 43, 1564–1570 CrossRef CAS.
- M. Daoud and J. P. Cotton, J. Phys., 1982, 43, 531–538 CrossRef CAS.
- H. Huang, S. E. Rankin, L. S. Penn, R. P. Quirk and T. H. Cheong, Langmuir, 2004, 20, 5770–5775 CrossRef CAS.
- S. Minko, in Polymer Surfaces and Interfaces: Characterization, Modification and Applications, ed. M. Stamm, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 215–234 DOI:10.1007/978-3-540-73865-7_11.
- B. T. Cheesman, A. J. G. Neilson, J. D. Willott, G. B. Webber, S. Edmondson and E. J. Wanless, Langmuir, 2013, 29, 6131–6140 CrossRef CAS PubMed.
- H. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- Y. Xia, N. A. Burke and H. D. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- Y. Zhang and P. S. Cremer, Annu. Rev. Phys. Chem., 2010, 61, 63–83 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- T. J. Murdoch, B. A. Humphreys, J. D. Willott, K. P. Gregory, S. W. Prescott, A. Nelson, E. J. Wanless and G. B. Webber, Macromolecules, 2016, 49, 6050–6060 CrossRef CAS.
- N. Ishida and S. Biggs, Macromolecules, 2010, 43, 7269–7276 CrossRef CAS.
- B.-C. Choi, S. Choi and D. Leckband, Langmuir, 2013, 29, 5841–5850 CrossRef CAS.
- H. Yim, M. Kent, S. Mendez, G. Lopez, S. Satija and Y. Seo, Macromolecules, 2006, 39, 3420–3426 CrossRef CAS.
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS.
- E. S. Kooij, X. Sui, M. A. Hempenius, H. J. Zandvliet and G. J. Vancso, J. Phys. Chem. B, 2012, 116, 9261–9268 CrossRef CAS PubMed.
- P. W. Zhu and D. H. Napper, Colloids Surf., A, 1996, 113, 145–153 CrossRef CAS.
- E. Zhulina, O. Borisov, V. Pryamitsyn and T. Birshtein, Macromolecules, 1991, 24, 140–149 CrossRef CAS.
- F. Afroze, E. Nies and H. Berghmans, J. Mol. Struct., 2000, 554, 55–68 CrossRef CAS.
- A. Halperin and M. Kröger, Macromolecules, 2011, 44, 6986–7005 CrossRef CAS.
- V. A. Baulin and A. Halperin, Macromol. Theory Simul., 2003, 12, 549–559 CrossRef CAS.
- S. Mendez, J. G. Curro, J. D. McCoy and G. P. Lopez, Macromolecules, 2005, 38, 174–181 CrossRef CAS.
- N. Dingenouts, S. Seelenmeyer, I. Deike, S. Rosenfeldt, M. Ballauff, P. Lindner and T. Narayanan, Phys. Chem. Chem. Phys., 2001, 3, 1169–1174 RSC.
- M. Karg, S. Wellert, S. Prevost, R. Schweins, C. Dewhurst, L. M. Liz-Marzán and T. Hellweg, Colloid Polym. Sci., 2011, 289, 699–709 CrossRef CAS.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS.
- T. Wu, Y. Zhang, X. Wang and S. Liu, Chem. Mater., 2008, 20, 101–109 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
- E. P. Gilbert, J. C. Schulz and T. J. Noakes, Physica B, 2006, 385–386, 1180–1182 CrossRef CAS.
- K. Wood, J. P. Mata, C. J. Garvey, C.-M. Wu, W. A. Hamilton, P. Abbeywick, D. Bartlett, F. Bartsch, P. Baxter, N. Booth, W. Brown, J. Christoforidis, D. Clowes, T. d'Adam, F. Darmann, M. Deura, S. Harrison, N. Hauser, G. Horton, D. Federici, F. Franceschini, P. Hanson, E. Imamovic, P. Imperia, M. Jones, S. Kennedy, S. Kim, T. Lam, W. T. Lee, M. Lesha, D. Mannicke, T. Noakes, S. R. Olsen, J. C. Osborn, D. Penny, M. Perry, S. A. Pullen, R. A. Robinson, J. C. Schulz, N. Xiong and E. P. Gilbert, J. Appl. Crystallogr., 2018, 51, 294–314 CrossRef CAS.
- X. Zheng, Z. Tong, X. Xie and F. Zeng, Polym. J., 1998, 30, 284–288 CrossRef CAS.
- M. Doucet, J. H. Cho, G. Alina, J. Bakker, W. Bouwman, P. Butler, K. Campbell, M. Gonzales, R. Heenan, A. Jackson, P. Juhas, S. King, P. Kienzle, J. Krzywon, A. Markvardsen, T. Nielsen, L. O'Driscoll, W. Potrzebowski, R. Ferraz Leal, T. Richter, P. Rozycko and A. Washington, SasView version 4.1. Zenodo, 2017, March 25, http://10.5281/zenodo.438138 Search PubMed.
- L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Plenum Press, New York, 1987 Search PubMed.
- S. Schweizerhof, D. E. Demco, A. Mourran, H. Keul, R. Fechete and M. Möller, Macromol. Rapid Commun., 2017, 38, 1700362 CrossRef.
- L. C. Elliott, B. Jing, B. Akgun, Y. Zhu, P. W. Bohn and S. K. Fullerton-Shirey, Langmuir, 2013, 29, 3259–3268 CrossRef CAS.
- C. Wijmans and E. B. Zhulina, Macromolecules, 1993, 26, 7214–7224 CrossRef CAS.
- J. Habicht, M. Schmidt, J. Rühe and D. Johannsmann, Langmuir, 1999, 15, 2460–2465 CrossRef CAS.
- T. J. Murdoch, B. A. Humphreys, E. C. Johnson, S. W. Prescott, A. Nelson, E. J. Wanless and G. B. Webber, Polymer, 2018, 138, 229–241 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular structure for PNIPAM; zeta potential measurements for core silica particles; details about the modelling for the Dhyd increasing temperature results including an example for the 1 h sample and a table listing all parameters; SANS contrast match plots for square root of I (Q = 0.004) vs. % H2O in D2O/H2O mixture; SANS PNIPAM brush modified particle dispersion preparation; dry particle analysis including the FTIR spectra and % weight loss as a function of synthesis time from TGA; SasView model description; and full list of SasView sphere core/shell onion model parameters for the 2 and 6 h samples along with the full Q range data for the 18 °C SANS experiments. See DOI: 10.1039/c8sm01824c |
| ‡ Current address: Department of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. |
| This journal is © The Royal Society of Chemistry 2019 |