
Propofol adsorption at the air/water interface: a combined vibrational sum frequency spectroscopy, nuclear magnetic resonance and neutron reflectometry study†
Petru
Niga
 *a,
Petra M.
Hansson-Mille
a,
Agne
Swerin
ab,
Per M.
Claesson
ab,
Joachim
Schoelkopf
c,
Patrick A. C.
Gane
cd,
Jing
Dai
e,
István
Furó
e,
Richard A.
Campbell
fg and
C. Magnus
Johnson
*b
*a,
Petra M.
Hansson-Mille
a,
Agne
Swerin
ab,
Per M.
Claesson
ab,
Joachim
Schoelkopf
c,
Patrick A. C.
Gane
cd,
Jing
Dai
e,
István
Furó
e,
Richard A.
Campbell
fg and
C. Magnus
Johnson
*b
aRISE Research Institutes of Sweden – Chemistry, Materials and Surfaces, Box 5607, SE-114 86 Stockholm, Sweden. E-mail: petru.niga@ri.se
bKTH Royal Institute of Technology, Department of Chemistry, Division of Surface and Corrosion Science, SE-100 44 Stockholm, Sweden. E-mail: magnusj@kth.se
cOmya International AG, Baslerstrasse 42, CH-4665 Oftringen, Switzerland
dAalto University, School of Chemical Technology, Department of Bioproducts and Biosystems, FI-00076 Aalto, Helsinki, Finland
eKTH Royal Institute of Technology, Department of Chemistry, Division of Applied Physical Chemistry, SE-100 44 Stockholm, Sweden
fInstitut Laue-Langevin, 71 Avenue des Martyrs, CS20156, 38042 Grenoble Cedex 9, France
gDivision of Pharmacy and Optometry, University of Manchester, Manchester M13 9PT, UK
First published on 8th November 2018
Abstract
Propofol is an amphiphilic small molecule that strongly influences the function of cell membranes, yet data regarding interfacial properties of propofol remain scarce. Here we consider propofol adsorption at the air/water interface as elucidated by means of vibrational sum frequency spectroscopy (VSFS), neutron reflectometry (NR), and surface tensiometry. VSFS data show that propofol adsorbed at the air/water interface interacts with water strongly in terms of hydrogen bonding and weakly in the proximity of the hydrocarbon parts of the molecule. In the concentration range studied there is almost no change in the orientation adopted at the interface. Data from NR show that propofol forms a dense monolayer with a thickness of 8.4 Å and a limiting area per molecule of 40 Å2, close to the value extracted from surface tensiometry. The possibility that islands or multilayers of propofol form at the air/water interface is therefore excluded as long as the solubility limit is not exceeded. Additionally, measurements of the 1H NMR chemical shifts demonstrate that propofol does not form dimers or multimers in bulk water up to the solubility limit.
Introduction
Alcohol/water mixtures are widely used as industrial solvents and chemical reagents, and the need of a better understanding of the interfacial behaviour of aqueous alcohol solutions at different interfaces has led to increasing interest in fundamental research.1,2 The behaviour of alcohols at solid interfaces plays a key role in applications like cleaning, etching, and electrochemical reactions.3,4 Let alone the industrial applications, the properties of alcohols at buried interfaces have important implications in cell membrane function. The functioning of intrinsic membrane proteins can be altered by introduction of short chain alcohols (e.g. propofol) at the membrane surface.5,6 As a result, propofol has, for example, been shown to induce a significant change in membrane permeability.7 This is potentially important since low molecular weight anaesthetics are used daily in hospitals, but yet the molecular mechanism of general anaesthesia remains to some extent ambiguous.8In spite of the importance of interfacial properties of alcohol/water mixtures in a wide variety of applications9,10 the study of such interfaces remains complex, mainly due to their dynamic nature and the lack of surface-specific experimental techniques. The development of nonlinear optical methods has contributed to overcoming some of these problems and facilitated better molecular understanding of such interfaces. Specifically, vibrational sum frequency spectroscopy (VSFS) has proven to be a powerful technique for studying a wide variety of aqueous interfaces11 due to its capability of yielding surface-specific information at a molecular level.
Different types of alcohols have been studied using VSFS with respect to the stretching vibrations of the hydrocarbon chains,11–14 the fingerprint region15 and the water of hydration.16–20 It was found that for a series of increasing chain length alcohols (C1–C8), at the neat air/alcohol interface, the tails point out into the gas phase due to the amphiphilic character of alcohols, and that the presence of gauche defects is chain length dependent. At the same time a very well ordered interfacial hydrogen bonding network was detected.21 Alcohols with different chain lengths have been shown to affect the surface water structure in different ways regarding hydrogen bonding strength and the orientation of the water molecules.20 Indeed in mixtures with water, glycerol was found to partition to the interface with air, whereby disturbing the topmost water layers.18 At a hydrophobic solid surface, methanol adsorbed from its mixture with water to the interface with the C–O bond aligned to the surface normal while the water band at 3200 cm−1 weakened in strength with increasing methanol concentration.16
The knowledge of small hydrophobic but weakly amphiphilic drugs at the interface is rather limited compared with the more amphiphilic molecules. Amphiphilic molecules such as surfactants,22 proteins,23 block co-polymers24 and peptides25 have been the subject of extensive investigations. By contrast there is a lack of body of work on the surface properties of small hydrophobic drugs. For example, for ibuprofen there has been a study on its interactions with lipid bilayers26 and cholesterol27 but to our knowledge not on its adsorption properties at the air/water interface. A recent study looked at the interactions of testosterone enanthate with surfactants at the air/water interface,28 but again to our knowledge the surface properties of the drug alone have not been examined explicitly. Further, for propofol the present authors have conducted a study on its interactions with phospholipid monolayers29 but an analogous study of its adsorption properties at the air/water interface is missing. To address this shortcoming, we have decided to study its adsorption at the air/water interface.
Propofol is a small alcohol that contains both hydrophobic and hydrophilic parts and has interesting biochemical properties, e.g., it is used commonly in general anaesthesia to induce a state of reduced consciousness in patients during medical procedures.30–32 Similar in structure to propofol, phenol has been briefly characterised by VSFS at the air/water interface,15 by focusing on only the carbon–oxygen stretching region of the spectrum. It was found that phenol is present at the interface at both low and high pH, and at high pH there are both phenol and phenolate ions. Surprisingly however, according to our knowledge, there are no detailed studies of propofol at the air/water or water/hydrophobic interface despite its fundamental interest as a small amphiphilic drug with important biomedical applications.
In the present work we evaluate the adsorption isotherm from surface tension data, and the adsorbed amount is also determined using neutron reflectometry (NR). NR and VSFS data are then combined to gain structural information on the adsorbed layer and its interaction with water. We also report a bulk study of the chemical shifts of hydrogen nuclei over the entire solubility range of propofol in water in order to detect possible multimeric structures. The results represent a necessary first step in resolving the driving forces for the interactions of propofol with interfaces, and they provide complementary information to that which can be obtained from other studies involving its interactions in more complex systems (including supported lipid bilayers33,34 and membrane proteins35,36) that are closer in nature to those in practical applications of the drug.
Materials and methods
Materials
Propofol (European Pharmacopoeia) with purity higher than 99% was bought from Sigma Aldrich. Its chemical structure is presented in Fig. 1, where also the NMR chemical shifts are listed. A Millipore Milli-Q Plus system was used as water supply, providing purified water with a resistivity of 18.2 MΩ cm.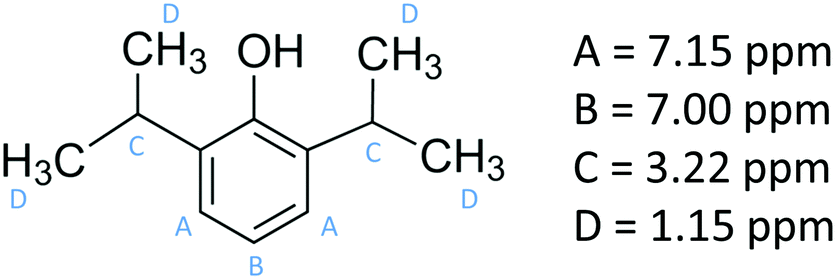 | ||
| Fig. 1 The chemical structure of propofol together with the 1H NMR peak assignments. | ||
Nuclear magnetic resonance
1H NMR spectra of propofol dissolved in D2O were obtained for a concentration series on a Bruker Advance III 500 MHz spectrometer using a 5 mm Bruker DIFF 30 probe. All experiments were performed at 293 K and the chemical shifts of the different propofol peaks were recorded; as spectral reference, we used the 1HDO signal (set to 4.75 ppm).Surface tensiometry
A Du Noüy ring instrument (Krüss) was used to determine the surface tension of aqueous propofol solutions. An independent solution was prepared for each concentration. To estimate the surface excess from the surface tension measurements, we used the Gibbs equation assuming only a single adsorbing neutral species,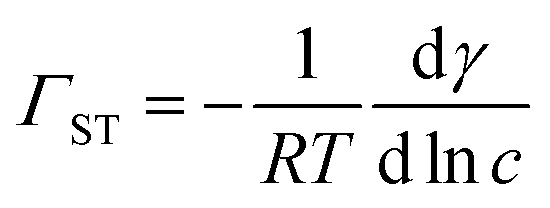 | (1) |
The solutions were prepared by dropwise adding liquid propofol to water, then gently shaking and hand warming for a few minutes until the propofol was dissolved. At least 10 min was allowed before the measurements were recorded. For higher concentrations it took longer time for propofol to dissolve completely.
Neutron reflectometry (NR)
NR measurements were performed using the time-of-flight reflectometer FIGARO at the Institut Laue-Langevin (Grenoble, France).37 The neutron reflectivity, R, determines the ratio of the number of neutrons in the specular reflection to those in the incident beam with respect to the momentum transfer, qz,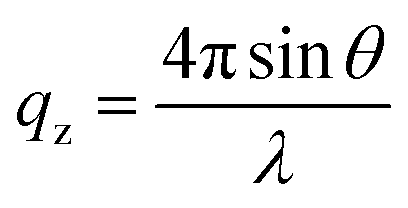 | (2) |
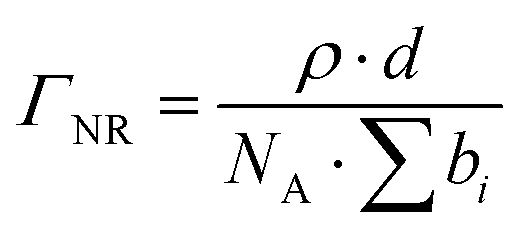 | (3) |
Two different data acquisition approaches were used to determine: (1) the adsorption isotherm and (2) the interfacial structure of the adsorbed molecules at the air/water interface, following the methodology adopted in ref. 39. In the first case, measurements were made only in ACMW, and the data were reduced only over 4.5–12 Å to restrict the qz-range to 0.01–0.03 Å−1. This approach reduces the sensitivity of the analysis to details of the structure at the interface.40 Note that the surface excess of propofol could be resolved accurately even though it was used in its normal, non-deuterated form thanks to the high flux at low-qz of FIGARO. The interfacial roughness values were fixed to 3.1 Å, consistent with the outcome of the structural analysis (note that for this low-qz analysis even neglect of the roughness values resulted in a change in surface excess of <0.5%). The background was not subtracted from these data in this analysis, but its accurate determination is essential to the precise quantification of the scattering excess: its value was iterated from the lowest value possible where a layer of zero surface excess could be fitted to a measurement of pure ACMW.41
In the second case, for the structural analysis, data were recorded at a propofol concentration of 0.89 mM over a broader qz-range with measurements at θ = 0.623° and θ = 3.78° both in ACMW and D2O. The background was subtracted from these data with the use of the 2D detector on the instrument. A single-layer structural model adequately described the data. In this case the fitting parameters were the thickness of the layer and its volume fraction. The roughness values of the air/propofol and propofol/water interfaces were constrained to be equal to each other and consistent with capillary wave theory, following an approach described recently.42 The data analysis was performed exclusively using Motofit.43
Vibrational sum frequency spectroscopy
The experimental VSF spectrometer has been described previously.44 A 1064 nm laser beam generated from a Nd:YAG laser system (PL-2251A-20, Ekspla) is used to pump the optical parameter generator/optical parameter amplifier OPG/OPA (LaserVision) system, which produces a fixed visible beam (532 nm) and a tuneable IR beam (1000–4000 cm−1). These two laser beams overlap at the sample surface with incident angles of 55° and 63° from the surface normal for the visible and IR beams, respectively. The VSFS beam is collected and filtered both optically and spatially before being sent to a photomultiplier tube (Jobin Yvon). The signal is integrated in a boxcar and finally processed by a computer program.The theory behind VSFS is well described in the literature.45–49 It is a nonlinear optical technique, which is able to provide surface specific information such as the nature and orientation of the species present at an interface. The intensity of the collected beam is proportional to the intensities of the incoming visible and infrared beams and the square of the effective second order nonlinear susceptibility χ(2)eff. χ(2)eff is in turn proportional to the number density multiplied by the orientationally averaged molecular hyperpolarisability 〈β(2)〉 of the probed species. We note that in order for a molecule or part of a molecule to be VSFS active it has to be both IR and Raman active, according to eqn (4):
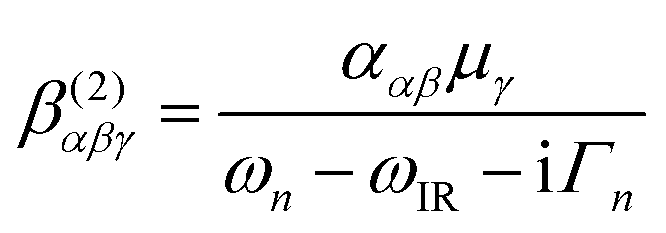 | (4) |
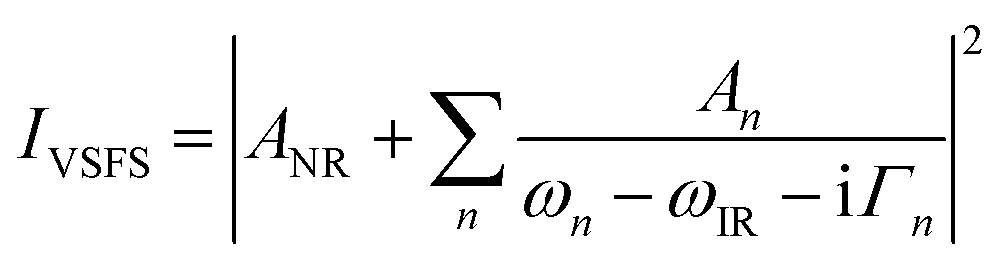 | (5) |
Orientational information about the probed molecular species is given by elements of the second order nonlinear susceptibility tensor, specifically by using different polarisation combinations. In this study we have used SSP, PPP, and SPS, where P refers to light polarised parallel to the plane of incidence and S refers to light polarised perpendicular to the plane of incidence. The first letter corresponds to the polarisation of the sum frequency beam, the second to that of the visible beam, and the third to the infrared beam.
Results and discussion
Propofol in aqueous bulk solution
The 1H NMR spectrum of propofol has been published before,51,52 and therefore it is not shown here. As is clear from Fig. 2, the chemical shifts of the hydrogen atoms in propofol remain constant in the concentration range 0.02–0.89 mM. Since propofol contains an aromatic moiety with large resulting “ring current” effects, any aggregation should have a large effect on the observed shifts.53,54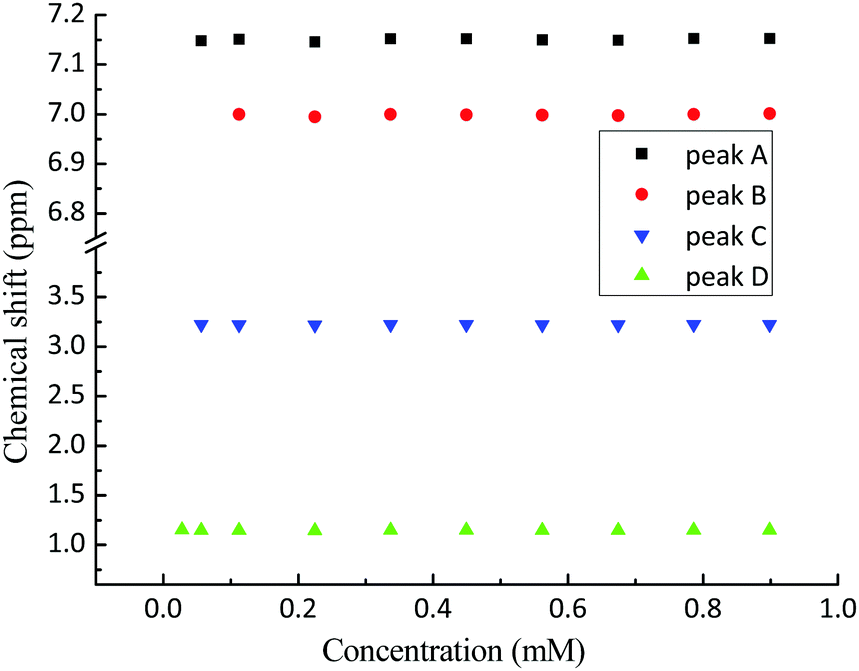 | ||
| Fig. 2 The chemical shifts (see Fig. 1 for peak assignment) recorded in solution of propofol at different concentrations between 0.02 mM and 0.89 mM. A – aromatic H3 and H5, B – aromatic H4, C – CH(CH3)2, and D – CH3. | ||
The absence of such effects points to the absence of any significant aggregation/dimerisation phenomena. This is in contradiction to what has been inferred in gas-phase where “nano-micelles” containing several multimers of propofol were found.55 In addition, the NMR intensity was proportional to the concentration in the whole range which also excludes the presence of any large aggregates (for which the NMR signal could possibly be lost).
Propofol adsorption isotherm
In order to gain insight into the adsorption of propofol at the air/water interface we have combined information from surface tensiometry, NR and VSFS.Fig. 3a shows the surface tension isotherm of aqueous propofol solutions. The decrease in the surface tension is due to adsorption of propofol at the air/water interface. The surface tension drops smoothly with concentration up to a concentration of about 1 mM, above which it remains constant. The limiting area per molecule is about 42 Å2, as seen in Fig. 3b. A discussion regarding the structure of the adsorbed propofol layer will follow in the light of the NR results presented next. Details on the area per molecule calculation are given in ESI.†
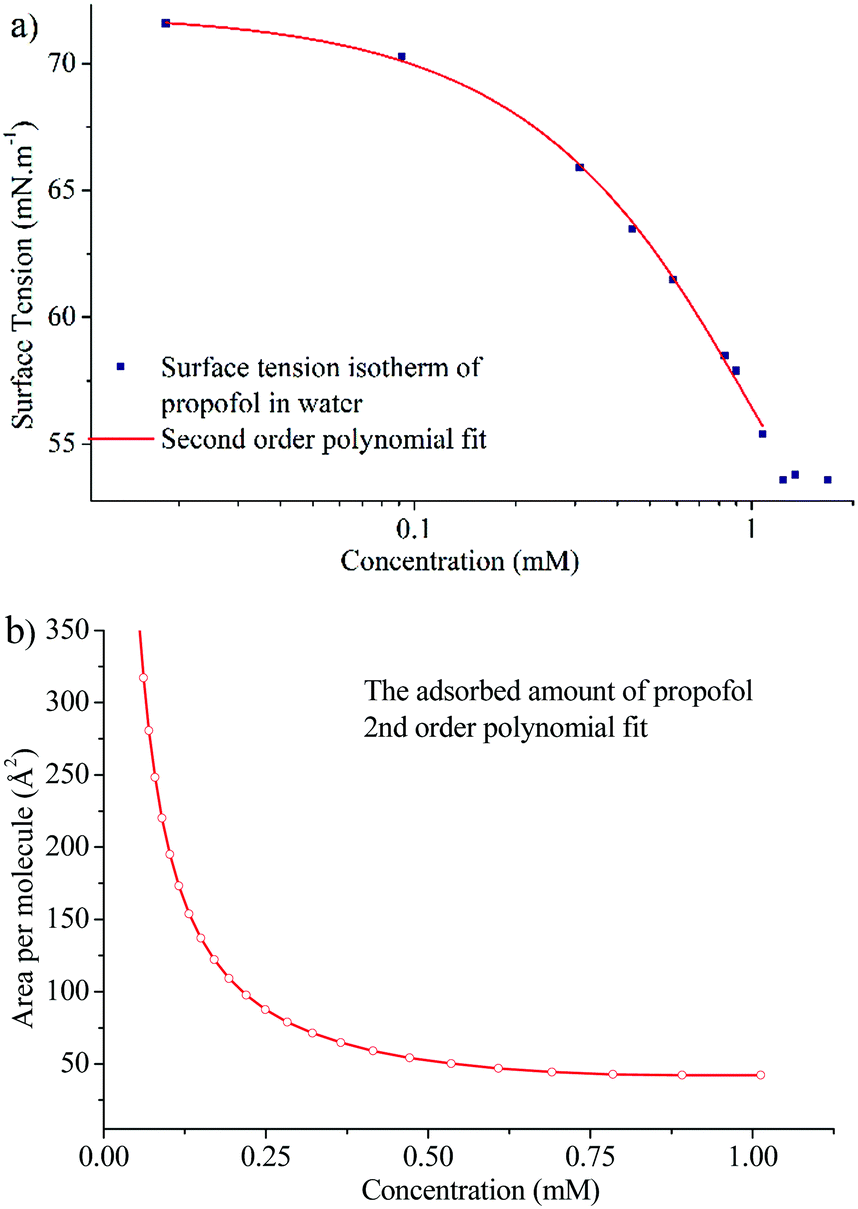 | ||
| Fig. 3 (a) Surface tension isotherm of propofol and a second order polynomial fit to the data (line), and (b) area per molecule calculated using the second order polynomial fit and eqn (1). | ||
The surface excess was measured directly at six different bulk concentrations at low qz values in ACMW, as shown in Fig. 4a. Also shown is the measurement of pure ACMW used to determine the background level. The resulting adsorption isotherm is shown in Fig. 4b, where it is compared to the surface excess obtained from surface tension measurements. We regard the agreement as satisfactory, particularly at high propofol concentrations, considering the different evaluation methods.
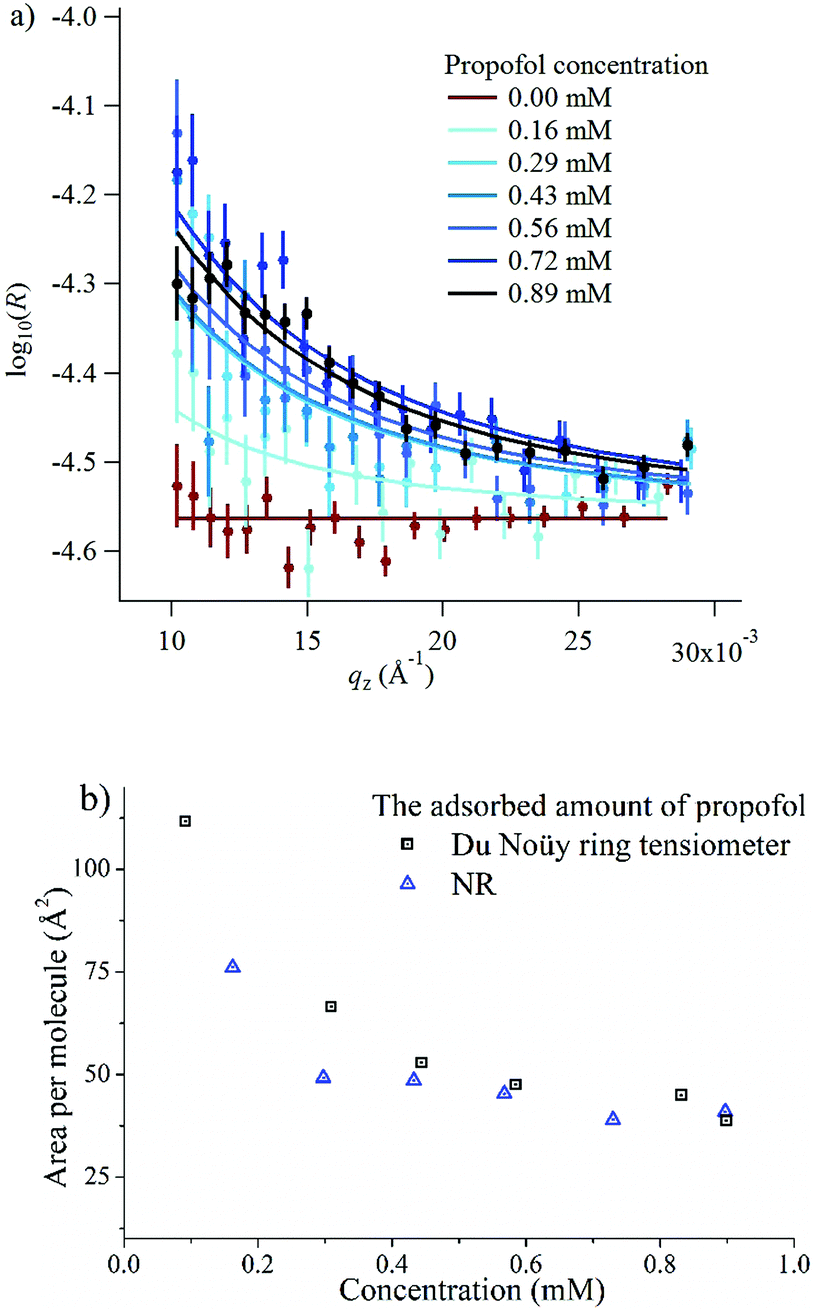 | ||
| Fig. 4 (a) Neutron reflectivity, R, and model fits for propofol solutions at the air/water interface, where the six bulk concentrations indicated are drawn progressively darker in colour; the pure ACMW background data are shown in red, and model fits are shown as lines; (b) a comparison of the area per molecule obtained from surface tension measurements and NR. | ||
Propofol interfacial organisation
In Fig. 5, the NR data and model fits for a 0.89 mM propofol solution at the air/water interface are presented. The inter-layer roughness values were constrained to the capillary wave value of 3.1 Å, the residual background was fitted to 2 × 10−7 (a.u.), and the thickness of the propofol monolayer and its volume fraction were fitted. The fit result using a generic algorithm converged to a dense propofol layer of 8.4 Å thicknesses with a volume fraction of 0.86. These values demonstrate that propofol forms a uniform fluid monolayer of high coverage and that propofol does not form islands at the interface.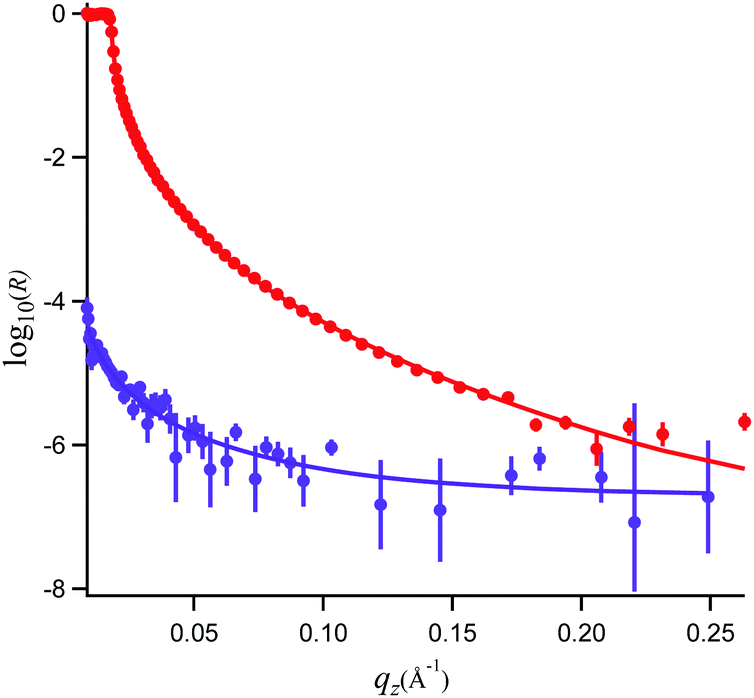 | ||
| Fig. 5 Neutron reflectivity, R, and model fits for propofol solutions at the air/water interface recorded in D2O (red, top) and air contrast matched water (purple, bottom). | ||
Propofol is considered to be of cylinder-like shape. The total volume of such cylinder, calculated from the inverse bulk density, is 285 Å3. Close packing of such cylinder-like shapes accounts for the van der Waals volume of propofol56 which is 198 Å3, 11% water (from the NR analysis) and about 22% air of the total volume. Let's consider the two extreme cases. If the molecules (thought as cylinders) are considered to stand on their ends, and if the cylinders are considered to have a cross section area of 25 Å2, slightly larger than for straight chain alkanes, then its length would be about 11 Å. The other extreme case would be a cylinder lying down giving an area per molecule of about 60 Å2 and a thickness of about 5–6 Å. Clearly, NR and tensiometry data are consistent with an intermediate situation (area per molecule 40–42 Å2, thickness 8.4 Å), suggesting a tilted orientation of propofol, and consistent at least (see below) with the OH-group towards the aqueous phase (the latter supported by the VSFS data discussed next). We are reluctant to go further and report a mean tilt angle as it would require a specific assumption about the packing of the molecules, which is not known a priori.
VSFS spectra: CH region
The VSFS data in Fig. 6a show well-defined peaks, which testify that propofol adsorbs at the air/water interface in a non-random fashion. In Fig. 6b the attenuated total reflection (ATR)-IR spectrum of propofol in the CH region is presented, showing that the bulk IR peaks are reproduced in the surface VSFS spectra. The assignments are based on published IR data31,57 and VSFS spectroscopy work on similar molecules.13,58–62 It is clear from Fig. 6a that the SSP polarisation combination exhibits most of the vibrational features. The peak centred at 2873 cm−1 is assigned to the symmetric CH3 stretch (sCH3), and is similar to the sCH3 peak found for alkyl chains. The small peak at 2910 cm−1, which is present only at high concentrations, is assigned to the CH of the isopropyl unit. The peak at 2965 cm−1 dominates the entire SSP spectrum and is assigned to the antisymmetric CH3 stretch (aCH3). This peak is also observed in the PPP polarisation, but barely distinguishable in the SPS polarisation. There are two additional peaks in the SSP polarisation: the aromatic CH stretches at 3035 and 3071 cm−1, which are clearly distinguished at high concentrations.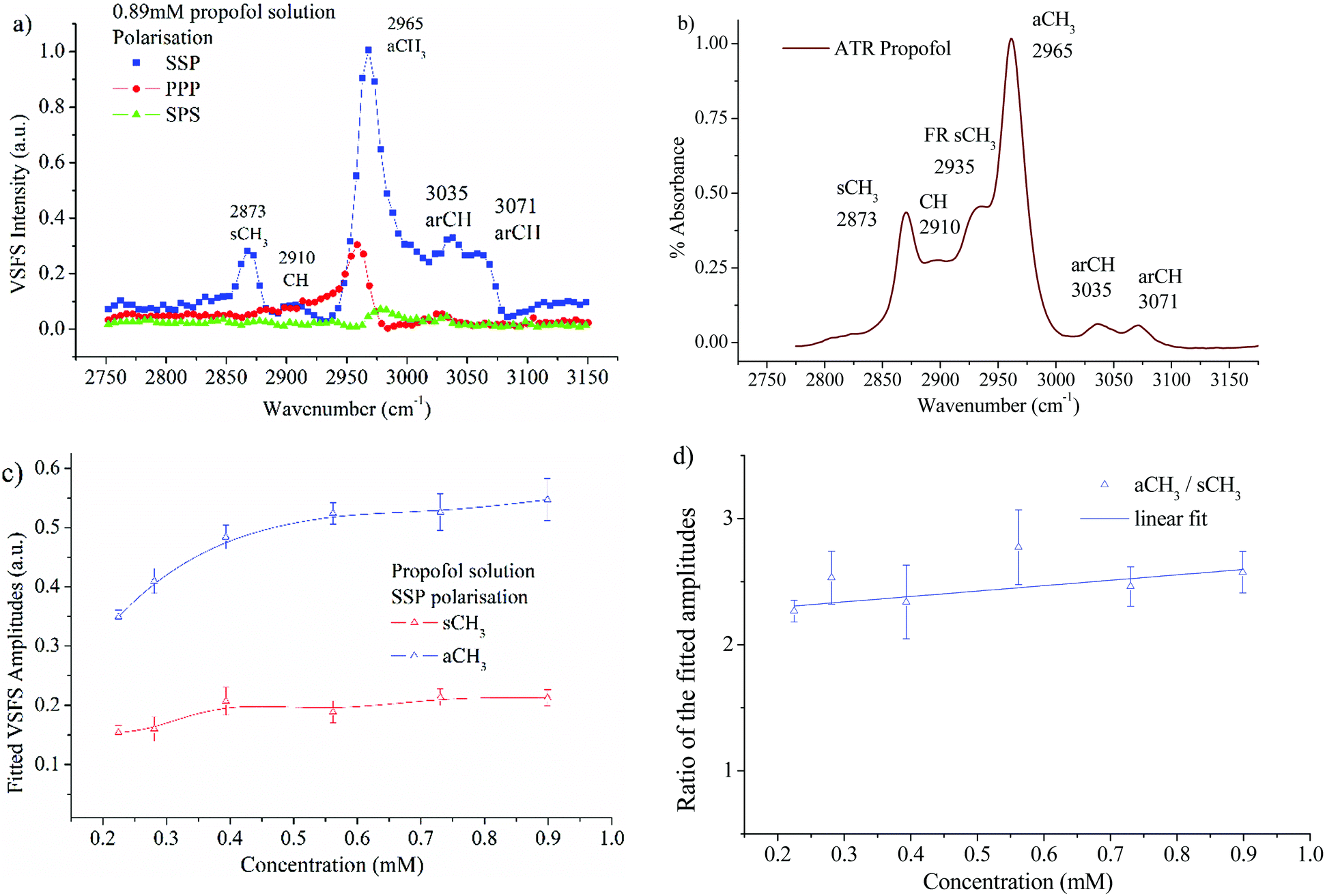 | ||
| Fig. 6 (a) VSF spectra of a 0.89 mM propofol solution in the SSP, PPP, and SPS polarisations. All peaks are normalised to the highest intensity peak – SSP aCH3; (b) ATR-IR spectrum of pure propofol in the CH stretching region; (c) fitted amplitudes for the sCH3 stretch and the aCH3 stretch as a function of propofol concentration in the SSP polarisation. (d) The ratio of the aCH3 to sCH3 amplitudes as a function of concentration. The line is a linear fit to the data. | ||
For alkyl chains, the aCH3 peak is normally significantly weaker than the sCH3 peak in the SSP polarisation combination due to the chain orientation.44,63,64 However, for propofol the opposite is found. The reason is that the total VSFS signal has contributions from all four closely spaced methyl groups (see Fig. 1), and due to different directions of the transition dipole moments for the symmetric and antisymmetric methyl stretches, their signal strength will be differently enhanced. The same argument rationalises that the antisymmetric methyl stretch is stronger in SSP than PPP, which normally is not observed for alkyl chains. It should be pointed out that in different environments (e.g. without water as solvent) the propofol SSP spectrum (not shown) resembles a typical spectrum from an alkyl chain, which emphasises that the unusual SSP spectrum for propofol at the air/water interface is due to the propofol orientation.
It is interesting that the IR spectrum shown in Fig. 6b shows the Fermi resonance CH3 at 2935 cm−1 which is not clearly observed in the VSFS spectra. The fitted amplitudes of the VSFS peaks are provided in the ESI.† In Fig. 6c the fitted amplitudes of the sCH3 and the aCH3 stretches are presented as a function of propofol concentration. Above 0.2 mM the fitted amplitudes of both peaks approach a plateau, a result that is consistent with the findings from surface tension isotherms, as discussed earlier (Fig. 3). The sCH3/aCH3 fitted amplitude ratio as a function of concentration is presented in Fig. 6d. The fitted amplitude depends both on the average orientation and the number of molecules probed, whereas the sCH3/aCH3 ratio is only affected by the orientation, since the number density is cancelled by taking this ratio.
We note that this ratio is almost constant over the concentration range 0.2–0.89 mM, meaning that the average orientation of the adsorbed propofol is almost constant. The SPS polarisation did not show any CH feature that could be used reliably in the fitting procedure. This, and the fact that there are four methyl groups pointing in different directions that contribute to that signal, prohibits a more detailed orientational analysis. The assignments of vibrational features of propofol in the CH stretching region are presented in Table 1.
| Peak (cm−1) | Polarisation | Observed | Assignments | Ref. |
|---|---|---|---|---|
| 2873 | SSP | VSFS/IR | sCH3 | 13, 57 and 65 |
| 2910 | SSP, PPP | VSFS/IR | CH isopropyl | 66 |
| 2965 | SSP, PPP, SPS | VSFS/IR | aCH3 | 13, 57, 61 and 62 |
| 3035 | SSP, PPP | VSFS/IR | arCH | 13 and 57 |
| 3071 | SSP | VSFS/IR | arCH | 13 and 57 |
VSFS spectra: water region
In order to have a reference between different measurement sessions, spectra of the free OH vibration of a pure water surface were recorded before all measurements. In Fig. 7 spectra of a 0.89 mM propofol solution together with the pure water spectra in the SSP and PPP polarisations are shown. | ||
| Fig. 7 Water region spectra of a 0.89 mM propofol solution together with a pure water spectrum in (a) SSP, and (b) PPP polarisation combination. These two spectra are normalised to the intensity of the free OH vibration at the pristine water surface. | ||
Both the SSP and the PPP spectra of water agree with the spectra recorded before,17,44,67 where the most prominent features exhibited in the SSP polarisation are the broad band spanning over several hundred wavenumbers and centred at around 3300 cm−1 as well as the sharp peak at 3704 cm−1.
The latter peak, which is present in both the SSP and PPP polarisation, is assigned to the OH stretching mode of the topmost surface OH bonds of water molecules protruding into air and vibrating free from hydrogen bonding.68 The assignment of the broad band was for a long time a source of debate.69–71 However, it is broadly agreed now that the broad band is assigned to hydrogen bonded water molecules with varying strength and coordination meaning that the OH signal is a signature of a collective vibration of several water molecules.72–75
The SSP polarisation spectrum of a 0.89 mM propofol solution, shown in Fig. 7a, is considerably different from the pure water spectrum. At this propofol concentration, the free OH peak at 3704 cm−1 has essentially vanished, suggesting that none or very few free OH bonds remain. However, a new broad band, centred around 3670 cm−1, appears. A similar band has been observed for several amphiphilic molecules (e.g., decanol, sugar surfactants and alkyl polyethylene glycol surfactants) at the air/water interface, and has been assigned to the OH stretch of ordered water molecules weakly interacting with hydrocarbon moieties.17 This is consistent with the relatively large hydrophobic part of propofol, and suggests direct contact between water and this region of propofol. The small red shift of around 30 cm−1 compared to the free OH vibration indicates that the interaction is weak. We note that since propofol is non-ionic, any significant enhancement of the water signal as observed for ionic amphiphiles76,77 is neither expected nor seen.
In the PPP spectrum in Fig. 7b a band covering nearly the whole OH stretching region is observed, with the maximum intensity around 3600 cm−1. Thus, the maximum is red shifted with around 70 cm−1 in comparison with the SSP spectrum, which exhibits nearly zero intensity at 3600 cm−1. The OH bond populations responsible for the peaks at 3600 cm−1 and 3670 cm−1 possess weak interactions, due to proximity to the hydrophobic moieties of propofol. However, since the two peaks have their maximum at different wavenumbers, the two populations experience dissimilar environments.
An interesting observation is that the right hand side of the broad band, towards 3400 cm−1 is almost completely suppressed in the SSP polarisation combination (Fig. 7a). Instead, a new band centred at about 3175 cm−1 clearly shows up in the spectrum. The fact that this band appears at such low frequency, close to the centre frequency for ice,78,79 suggests that it is associated with interfacial water that experience strong interactions with the hydroxyl part of propofol, similar to what was found for long chain alcohols.20 A similar band, centred at 3150 cm−1 has further been observed for interfacial sugar surfactants possessing –OH groups (C10 maltoside and C10 glucoside) and assigned to OH stretching vibrations of the hydroxyl groups in the sugar rings and their hydration shells.17 In contrast, 1-decanol, which, like propofol, contains a single –OH group and a large hydrophobic moiety, exhibited a band centred at 3250 cm−1, thus indicating relatively weaker hydrogen bonds.
Moreover, the PPP spectrum of 1-decanol showed a broad band extending over the region 3000–3800 cm−1, obviously different from that obtained in presence of propofol. Accordingly, the strength of the hydrogen bonds of water that are hydrating propofol (SSP spectra) is in between those observed with the sugar surfactants and decanol, which indicates that the hydrogen bond strength depends not only on the hydrophilic group, which is the same for decanol and propofol, but also the surrounding hydrophobic environment.20
The freedom of the molecules to rotate or move around may also influence the strength of the hydrogen bond. Propofol has a larger area per molecule then decanol17 and therefore has less constraints in adopting a position that allows for stronger hydrogen bonds to be formed.
Summary and conclusion
Propofol does not form dimers or multimers in bulk solution up to the solubility limit (0.89 mM). It adsorbs at the air/water interface forming a dense (volume fraction 0.86) uniform film (area/molecule ≈40–42 Å2, thickness ≈8.4 Å) close to the solubility limit. The propofol molecule is tilted relative the surface normal and oriented with the OH-group towards water. Its orientation at the interface is almost constant in the concentration range 0.2–0.89 mM.We have identified different water populations that hydrate propofol at the air/aqueous solution interface. Strong hydrogen bonds, similar to those found for the tetrahedrally-coordinated hydrogen bonding in ice, are formed between water and the OH-group of propofol. These hydrogen bonds are stronger than those found between water and decanol, but weaker than those found next to sugar surfactants. Thus, the hydrogen bond strength does not only depend on the hydrophilic group, which is the same for decanol and propofol, but also on the surrounding hydrophobic environment and the ability to adopt conformations that allow formation of strong hydrogen bonds.
Effective molecular information regarding the arrangement and hydration of propofol at the air/water interface opens the door for a more extensive examination of the interfacial properties of propofol, especially at the buried membrane/water interfaces where this may have important implications for understanding the driving forces for the underlying interactions of drugs in model systems, and they set the stage for us and others to progress with studies of the interactions of propofol and other small model drugs with more complex interfacial morphologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was kindly supported by Omya International AG. We thank the Institut Laue-Langevin for an allocation of beam time on FIGARO (DOI: 10.5291/ILL-DATA.TEST-2589) as well as access to complementary instruments in the Partnership for Soft Condensed Matter. JD and PC thank the NanoS3-290251 ITN and the Swedish Research Council VR for support.References
- F. Franks and J. E. Desnoyers, Alcohol-water mixture revisited, in Water Science Reviews, ed. F. Franks, Cambridge University Press, Cambridge, 1989, vol. 1, pp. 171–232 Search PubMed.
- Water: A Comprehensive Treatise: The Physics and Physical Chemistry of Water, ed. F. Franks, Plenum, New York, 1972 Search PubMed.
- A. P. G. Arthur and W. Adamson, Physical Chemistry of Surfaces, John Wiley & Sons, Inc., 1997 Search PubMed.
- J. Grimshaw, in Electrochemical Reactions and Mechanisms in Organic Chemistry, ed. J. Grimshaw, Elsevier Science B.V., Amsterdam, 2000, pp. 261–299 Search PubMed.
- M. Patra, E. Salonen, E. Terama, I. Vattulainen, R. Faller, B. W. Lee, J. Holopainen and M. Karttunen, Biophys. J., 2006, 90, 1121–1135 CrossRef CAS PubMed.
- A. R. Mazzeo, J. Nandi and R. A. Levine, Am. J. Physiol.: Gastrointest. Liver Physiol., 1988, 254, G57–G64 CrossRef CAS PubMed.
- G. Banfalvi, in Permeability of Biological Membranes, Springer International Publishing, Cham, 2016, pp. 1–71, DOI:10.1007/978-3-319-28098-1_1.
- A. K. Lugli, C. S. Yost and C. H. Kindler, Eur. J. Anaesthesiol., 2009, 26, 807–820 CrossRef CAS.
- F. H. Frimmel, Angew. Chem., 1993, 105, 800 CrossRef.
- J. W. Whalen, J. Chem. Educ., 1983, 60, A322 CrossRef.
- C. M. Johnson and S. Baldelli, Chem. Rev., 2014, 114, 8416–8446 CrossRef CAS.
- J. Sung, K. Park and D. Kim, J. Phys. Chem. B, 2005, 109, 18507–18514 CrossRef CAS PubMed.
- R. Lu, W. Gan, B.-h. Wu, Z. Zhang, Y. Guo and H.-f. Wang, J. Phys. Chem. B, 2005, 109, 14118–14129 CrossRef CAS.
- G. Ma and H. C. Allen, J. Phys. Chem. B, 2003, 107, 6343–6349 CrossRef CAS.
- Y. Rao, M. Subir, E. A. McArthur, N. J. Turro and K. B. Eisenthal, Chem. Phys. Lett., 2009, 477, 241–244 CrossRef CAS.
- W.-T. Liu, L. Zhang and Y. R. Shen, J. Chem. Phys., 2006, 125, 144711 CrossRef.
- E. Tyrode, C. M. Johnson, A. Kumpulainen, M. W. Rutland and P. M. Claesson, J. Am. Chem. Soc., 2005, 127, 16848–16859 CrossRef CAS PubMed.
- S. Baldelli, C. Schnitzer, M. J. Shultz and D. J. Campbell, J. Phys. Chem. B, 1997, 101, 4607–4612 CrossRef CAS.
- Y.-C. Wen, S. Zha, C. Tian and Y. R. Shen, J. Phys. Chem. C, 2016, 120, 15224–15229 CrossRef CAS.
- J. A. Mondal, V. Namboodiri, P. Mathi and A. K. Singh, J. Phys. Lett., 2017, 8, 1637–1644 CAS.
- C. D. Stanners, Q. Du, R. P. Chin, P. Cremer, G. A. Somorjai and Y. R. Shen, Chem. Phys. Lett., 1995, 232, 407–413 CrossRef CAS.
- V. B. Fainerman, R. Miller and H. Möhwald, J. Phys. Chem. B, 2002, 106, 809–819 CrossRef CAS.
- J. A. Killian and G. von Heijne, Trends Biochem. Sci., 2000, 25, 429–434 CrossRef CAS.
- A. F. Miller, R. W. Richards and J. R. P. Webster, Macromolecules, 2000, 33, 7618–7628 CrossRef CAS.
- D. M. Small, L. Wang and M. A. Mitsche, J. Lipid Res., 2009, 50, S329–S334 CrossRef.
- L. Du, X. Liu, W. Huang and E. Wang, Electrochim. Acta, 2006, 51, 5754–5760 CrossRef CAS.
- R. J. Alsop, C. L. Armstrong, A. Maqbool, L. Toppozini, H. Dies and M. C. Rheinstadter, Soft Matter, 2015, 11, 4756–4767 RSC.
- Y. Saaka, D. T. Allen, Y. Luangwitchajaroen, Y. Shao, R. A. Campbell, C. D. Lorenz and M. J. Lawrence, Soft Matter, 2018, 14, 3135–3150 RSC.
- P. Niga, P. M. Hansson-Mille, A. Swerin, P. M. Claesson, J. Schoelkopf, P. A. C. Gane, E. Bergendal, A. Tummino, R. A. Campbell and C. M. Johnson, J. Colloid Interface Sci., 2018, 526, 230–243 CrossRef CAS PubMed.
- I. Vasileiou, T. Xanthos, E. Koudouna, D. Perrea, C. Klonaris, A. Katsargyris and L. Papadimitriou, Eur. J. Pharmacol., 2009, 605, 1–8 CrossRef CAS PubMed.
- A. Mohammad, F. B. Faruqi and J. Mustafa, J. Cancer Ther., 2010, 1, 124–130 CrossRef CAS.
- R. A. Siddiqui, M. Zerouga, M. Wu, A. Castillo, K. Harvey, G. P. Zaloga and W. Stillwell, Breast Cancer Res., 2005, 7, R645–R654 CrossRef CAS.
- G. Fragneto, Eur. Phys. J.: Spec. Top., 2012, 213, 327–342 CAS.
- Y. Gerelli, L. Porcar and G. Fragneto, Langmuir, 2012, 28, 15922–15928 CrossRef CAS.
- T. Soranzo, D. K. Martin, J.-L. Lenormand and E. B. Watkins, Sci. Rep., 2017, 7, 3399 CrossRef.
- S. Isaksson, E. B. Watkins, K. L. Browning, T. Kjellerup Lind, M. Cárdenas, K. Hedfalk, F. Höök and M. Andersson, Nano Lett., 2017, 17, 476–485 CrossRef CAS.
- R. A. Campbell, H. P. Wacklin, I. Sutton, R. Cubitt and G. Fragneto, Eur. Phys. J. Plus, 2011, 126, 107 CrossRef.
- J. R. Lu, R. K. Thomas and J. Penfold, Adv. Colloid Interface Sci., 2000, 84, 143–304 CrossRef CAS.
- J. Hernandez-Pascacio, Á. Piñeiro, J. M. Ruso, N. Hassan, R. A. Campbell, J. Campos-Terán and M. Costas, Langmuir, 2016, 32, 6682–6690 CrossRef CAS PubMed.
- Á. Ábraham, R. A. Campbell and I. Varga, Langmuir, 2013, 29, 11554–11559 CrossRef PubMed.
- R. A. Campbell, A. Tummino, B. A. Noskov and I. Varga, Soft Matter, 2016, 12, 5304–5312 RSC.
- R. A. Campbell, Y. Saaka, Y. Shao, Y. Gerelli, R. Cubitt, E. Nazaruk, D. Matyszewska and M. J. Lawrence, J. Colloid Interface Sci., 2018, 531, 98–108 CrossRef CAS PubMed.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- P. Niga, PhD thesis, Royal Institute of Technology, 2010.
- X. Chen, M. L. Clarke, J. Wang and Z. Chen, Int. J. Mod. Phys. B, 2005, 19, 691–713 CrossRef CAS.
- M. Buck and M. Himmelhaus, J. Vac. Sci. Technol., A, 2001, 19, 2717–2736 CrossRef CAS.
- M. J. Shultz, C. Schnitzer, D. Simonelli and S. Baldelli, Int. Rev. Phys. Chem., 2000, 19, 123–153 Search PubMed.
- X. Zhuang, P. B. Miranda, D. Kim and Y. R. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12632–12640 CrossRef CAS.
- X. D. Zhu, H. Suhr and Y. R. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 3047–3050 CrossRef CAS.
- C. Hirose, N. Akamatsu and K. Domen, Appl. Spectrosc., 1992, 46, 1051–1072 CrossRef.
- L. F. G. Reiner, E. Paula, M. Perillo and D. García, J. Biomater. Nanobiotechnol., 2013, 4, 28–34 CrossRef.
- K. I. Momot, P. W. Kuchel, B. E. Chapman, P. Deo and D. Whittaker, Langmuir, 2003, 19, 2088–2095 CrossRef CAS.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- J. Alsins, M. Björling, I. Furó and V. Egle, J. Phys. Org. Chem., 1999, 12, 171–175 CrossRef CAS.
- I. León, J. Millán, E. J. Cocinero, A. Lesarri and J. A. Fernández, Angew. Chem., Int. Ed., 2014, 53, 12480–12483 Search PubMed.
- K. A. Woll, B. P. Weiser, Q. Liang, T. Meng, A. McKinstry-Wu, B. Pinch, W. P. Dailey, W. D. Gao, M. Covarrubias and R. G. Eckenhoff, ACS Chem. Neurosci., 2015, 6, 927–935 CrossRef CAS PubMed.
- A. Jabłońska, Ł. Ponikiewski, K. Ejsmont, A. Herman and A. Dołęga, J. Mol. Struct., 2013, 1054–1055, 359–366 CrossRef.
- P. Guyot-Sionnest, R. Superfine, J. H. Hunt and Y. R. Shen, Chem. Phys. Lett., 1988, 144, 1–5 CrossRef CAS.
- C.-y. Wang, H. Groenzin and M. J. Shultz, J. Am. Chem. Soc., 2005, 127, 9736–9744 CrossRef CAS.
- T. Ishihara, T. Ishiyama and A. Morita, J. Phys. Chem. C, 2015, 119, 9879–9889 CrossRef CAS.
- C.-y. Wang, H. Groenzin and M. J. Shultz, J. Phys. Chem. B, 2004, 108, 265–272 CrossRef CAS.
- Y. Yu, Y. Wang, K. Lin, N. Hu, X. Zhou and S. Liu, J. Phys. Chem. A, 2013, 117, 4377–4384 CrossRef CAS PubMed.
- J. F. D. Liljeblad, V. Bulone, E. Tyrode, M. W. Rutland and C. M. Johnson, Biophys. J., 2010, 98, L50–L52 CrossRef CAS.
- E. Tyrode, P. Niga, M. Johnson and M. W. Rutland, Langmuir, 2010, 26, 14024–14031 CrossRef CAS.
- H. Chen, W. Gan, R. Lu, Y. Guo and H.-F. Wang, J. Phys. Chem. B, 2005, 109, 8064–8075 CrossRef CAS.
- S. Kataoka and P. S. Cremer, J. Am. Chem. Soc., 2006, 128, 5516–5522 CrossRef CAS PubMed.
- E. Tyrode, C. M. Johnson, M. W. Rutland and P. M. Claesson, J. Phys. Chem. C, 2007, 111, 11642–11652 CrossRef CAS.
- Q. Du, R. Superfine, E. Freysz and Y. R. Shen, Phys. Rev. Lett., 1993, 70, 2313–2316 CrossRef CAS.
- G. L. Richmond, Annu. Rev. Phys. Chem., 2001, 52, 357–389 CrossRef CAS.
- D. F. Liu, G. Ma, L. M. Levering and H. C. Allen, J. Phys. Chem. B, 2004, 108, 2252–2260 CrossRef CAS.
- M. Sovago, R. K. Campen, G. W. H. Wurpel, M. Müller, H. J. Bakker and M. Bonn, Phys. Rev. Lett., 2008, 100, 173901 CrossRef.
- T. Ishiyama and A. Morita, J. Phys. Chem. A, 2007, 111, 9277–9285 CrossRef CAS PubMed.
- A. Morita and J. T. Hynes, Chem. Phys., 2000, 258, 371–390 CrossRef CAS.
- S. Yamaguchi, J. Chem. Phys., 2015, 143, 034202 CrossRef PubMed.
- A. Adhikari, J. Chem. Phys., 2015, 143, 124707 CrossRef PubMed.
- M. R. Watry, T. L. Tarbuck and G. L. Richmond, J. Phys. Chem. B, 2003, 107, 512–518 CrossRef CAS.
- X. Chen, W. Hua, Z. Huang and H. C. Allen, J. Am. Chem. Soc., 2010, 132, 11336–11342 CrossRef CAS PubMed.
- R. Claverie, M. D. Fontana, I. Duričković, P. Bourson, M. Marchetti and J.-M. Chassot, Sensors, 2010, 10, 3815 CrossRef CAS PubMed.
- Q. Sun and H. Zheng, Prog. Nat. Sci., 2009, 19, 1651–1654 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm01677a |
| This journal is © The Royal Society of Chemistry 2019 |