
Ex situ solid electrolyte interphase synthesis via radiolysis of Li-ion battery anode–electrolyte system for improved coulombic efficiency†
Fanny
Varenne
a,
John P.
Alper
a,
Frédéric
Miserque
b,
Chandra Sekhar
Bongu
ac,
Adrien
Boulineau
d,
Jean-Frédéric
Martin
e,
Vincent
Dauvois
f,
Alexandre
Demarque
g,
Mickaël
Bouhier
a,
Florent
Boismain
a,
Sylvain
Franger
c,
Nathalie
Herlin-Boime
a and
Sophie
Le Caër
 *a
*a
aNIMBE, UMR 3685 CEA, CNRS, Université Paris-Saclay, CEA Saclay, F-91191 Gif-sur-Yvette Cedex, France. E-mail: sophie.le-caer@cea.fr
bDEN, Service de Corrosion et d'étude du Comportement des Matériaux dans leur Environnement (SCCME), CEA, Université Paris-Saclay, F-91191 Gif-sur-Yvette Cedex, France
cEquipe de Recherche et Innovation en Electrochimie pour l'Energie, ICMMO, UMR 8182, CNRS, Université Paris Sud, Université Paris-Saclay, F-91405 Orsay Cedex, France
dCEA, LITEN, F-38054 Grenoble Cedex 9, France
eCEA, LITEN, DEHT, STB, LM, F-38054 Grenoble Cedex 9, France
fCEA, DEN, DANS, DPC, SECR, LRMO, F-91191 Gif-sur-Yvette Cedex, France
gLaboratoire de Chimie-Physique, ELYSE, UMR 8000 CNRS, UPS Université Paris Sud, F-91405 Orsay Cedex, France
First published on 17th July 2018
Abstract
The radiolysis of a mixed solvent electrolyte–carbon anode material is investigated for the first time. The present work demonstrates the radiolytic growth of an SEI with a chemical composition similar to that formed during electrochemical cycling, as determined by XPS. The quantity of the SEI increases with increasing irradiation dose. Degradation products formed in the liquid and gas phase are also identified as matching those formed during electrochemical cycling. TEM results support the XPS results of increasing SEI content with increasing irradiation dose. Electrochemical characterization by galvanostatic cycling of test cells indicates that the radiolysis generated SEI greatly improves first cycle efficiency of the materials assembled in half cells, and impedance spectroscopy supports the result with an increase in resistivity observed for irradiated samples. This first study opens the door to the use of irradiation tools for the artificial generation of an SEI and for producing LIB anode materials with improved performance.
1. Introduction
Widespread implementation of renewable energy solutions, an imperative for human society, requires advanced energy storage devices to compensate for the intermittent nature of many renewables. Lithium-ion batteries (LIB) are attractive for this application1,2 as they display high energy density and long cycle life in comparison to other rechargeable systems.3,4 In general, LIBs are composed of a carbonaceous anode, a lithium metal oxide cathode, an organic liquid electrolyte and a separator.5 Although these devices have been commercialized since 1991, important issues related to electrolyte–electrode interactions (affecting post-production processing requirements, cell capacity, and safety) remain.Commercial electrolytes generally consist of a mixture of linear (low viscosity and low dielectric constant) and cyclical (high viscosity and high dielectric constant) carbonates in which a lithium salt such as lithium hexafluorophosphate (LiPF6) is dissolved. The selection of these electrolytes is based on cost, stable working potential, and Li-ion solvation/transport properties.6 None of these electrolytes however, are stable at the working potential of Li-ion anode materials. Thus, during the first cycles of the battery, the electrolyte decomposes at the anode surface producing a solid-electrolyte interphase (SEI).7 This interphase is vital to device performance and safety, passivating the electrode surface to further reactions. However, its formation also irreversibly traps lithium ions and decreases cell capacity (around 10% in the first cycle for typical graphite anode materials).8,9 It is now well known that the SEI contains both solid inorganic and organic compounds. Gaseous products, such as H2 and HF, may also be formed in the packaged cell during electrolyte degradation, leading to safety and performance concerns.6,10 Ideally once a stable, high quality SEI is formed on the electrode surface, further solvent degradation reactions are prevented, while Li cations can still be intercalated in the electrode material.11,12 Due to the importance of SEI formation, battery producers run formation protocols on packaged cells prior to distribution, requiring days to weeks on battery cycling devices. Besides the initial capacity loss, these formation protocols impart significant energy and production costs to the final product.9 Processes which passivate anode materials prior to cell fabrication could thus provide safer cells with increased capacity and reduced production costs. Indeed, much work has gone into developing “artificial” SEIs based on various polymers and oxides, summarized in the recent review by Peled and Menkin.13 Here we present a new and promising method to generate a chemically similar SEI via radiolysis of anode materials in the presence of an electrolyte.
Recently we demonstrated that the reactive species generated by radiolysis (the chemical reactivity induced by the interaction between matter and ionizing radiation) of typical LIB solvents, and single solvent electrolytes are the same as those generated during the cycling of LIBs.14–16 Furthermore, radiolysis generated species in measurable amounts at time scales significantly shorter than electrochemical cycling in a battery cell (i.e., minutes vs. days).14–17 These results inspired the work presented here, which aims to extend the radiolysis approach to both study the chemical processes at the anode–electrolyte interface in mixed solvent electrolytes and investigate ex situ generation of SEI. We selected a model anode material consisting of carbon nanoparticles,18 which are known to have a high reactivity towards LIB electrolytes when cycled in half cells, resulting in large irreversible capacity during the first charge/discharge cycle. The composition and the morphology of the SEI formed on carbonaceous anodes during battery cycling with a variety of electrolytes have been intensively investigated, enabling comparison between the degradation products formed via radiolysis and those in the literature obtained via cycling, and validation of the radiolysis approach.9,11,13,19 Major advantages of our process are that no battery assembly or cycling is needed to synthesize the degradation products and the process is in comparison shorter to electrochemical formation. These advantages in turn have important implications in terms of time and cost for both battery production and fundamental chemical studies of new anode/electrolyte combinations.
Herein we present the evolution of the nanoparticle surface chemistry and morphology with increasing irradiation dose, as followed by X-ray Photoelectron Spectroscopy (XPS) and Transmission Electron Microscopy (TEM) respectively. The results of the electrochemical analysis of the materials in test cells, conducted through galvanostatic cycling and Electrochemical impedance spectroscopy (EIS) experiments are correlated to the observed evolution of the surface. The identification and quantification of the degradation products generated in the gas phase from the irradiated mixed solvent electrolyte is performed to give a full picture of the phenomena occurring in different phases. We show that this radiolysis process enables generation of an SEI, whose composition is similar to that formed in cycling experiments, on the surface of the carbon nanoparticles. We show also that while electrochemical formation of SEI still occurs in all samples during galvanostatic cycling, there is a significant decrease in the total current consumed by this process for irradiated samples, with ∼50% decrease in irreversibility on the first cycle for material irradiated at the highest dose. These results indicate that irradiation is a powerful tool not only in the study of electrolyte degradation, but also has potential to reduce cost and improve performance of Li-ion batteries.
2. Results and discussion
2.1 Physico-chemical characterizations
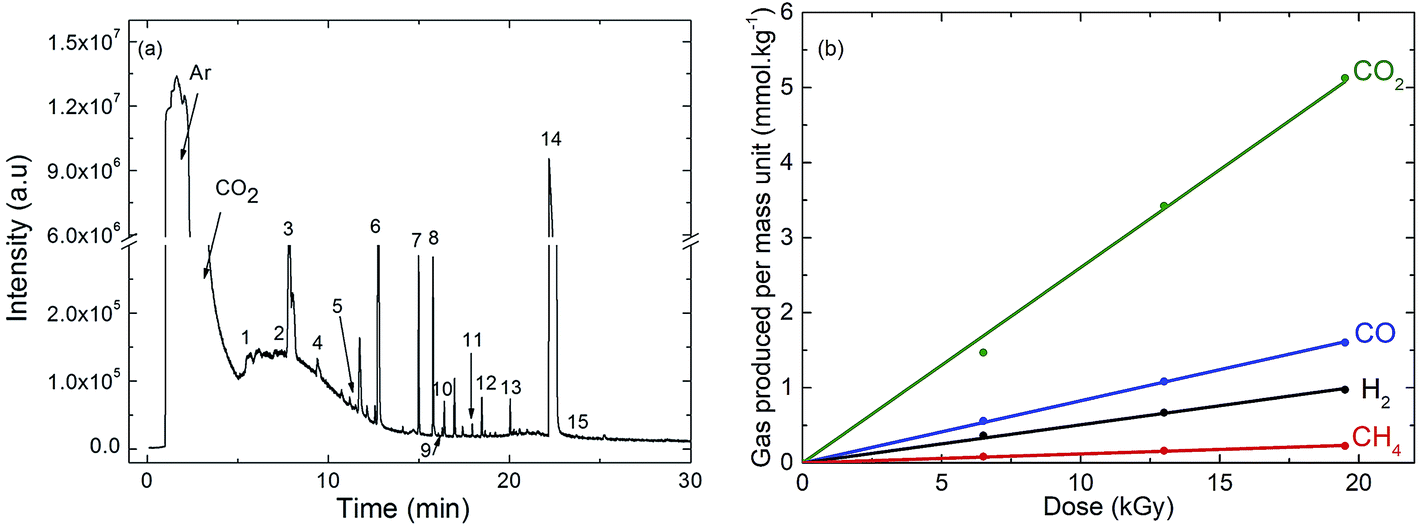 | ||
| Fig. 1 (a) Gas phase chromatogram (GC-EI/MS) obtained after γ-irradiation at a dose of 19 kGy of EC (ethylene carbonate)/DEC (diethyl carbonate)/LiPF6 1 M containing 1% of amorphous carbon nanoparticles. Except Ar and CO2, the identified species are numbered. The corresponding assignments are given in Table 1. (b) Evolution of the main decomposition products formed in the gas phase and measured by μ-GC after γ-irradiation of EC/DEC/LiPF6 1 M containing 1% of amorphous carbon nanoparticles as a function of the dose. | ||
| Identification of species by gas phase chromatography | |
|---|---|
| Number | Assignment |
| 1 | C2H5F |
| 2 | CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
| 3 | C3H8 |
| 4 | CH3CHO |
| 5 | CH3CH2OH |
| 6 | C4H10 |
| 7 | CH3CH2OCHO |
| 8 | C2H5OC2H5 |
| 9 | C5H12 |
| 10 |
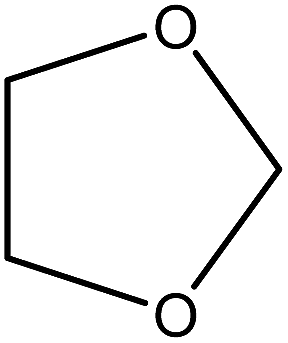
|
| 11 | CH3CH2OCOCH3 |
| 12 |

|
| 13 | CH3CH2OCOOCH3 |
| 14 | DEC |
| 15 | EC |
The same experiments were also performed in the absence of nanoparticles (Fig. S1 and S2†). The compounds observed are the same in the presence or in the absence of nanoparticles and are produced with the same yields (Table S2, ESI†).
 | ||
| Fig. 2 Comparison of C-1s core level spectra (left side) measured on carbon nanoparticles: not irradiated (a) and irradiated at increasing doses of 28 (b), 58 (c), 92 (d) and 202 (e) kGy. The C nanoparticle line shape is presented by the red peak contribution. Other contributions used for the spectra deconvolution are showed on the right side. Peak assignments are summarized in Table 2. | ||
| Core level | Binding energy (eV) | References for LIBs data, with corresponding binding energy (eV) | Non-irradiated nanoparticles | Irradiated nanoparticles | Assignment | |||
|---|---|---|---|---|---|---|---|---|
| 28 kGy | 58 kGy | 92 kGy | 202 kGy | |||||

|
282.3 ± 0.3 | 282.5 (ref. 11) | ● | Li2–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) 2 2 |
||||

|
283.8 ± 0.3 | 283.7 (ref. 11) | ● | ● | ● | ● | ● | Nanoparticles, ––H, sp2 carbon |

|
285.0 ± 0.3 | 285.0 (ref. 22) | ● | ● | ● | ● | ––C– |
|

|
286.1 ± 0.3 | 286.5 (ref. 11 and 22) | ● | ● | ● | ● | (–H2CH2O–)n and/or ROLi |
|

|
286.8 ± 0.3 | 286.8 (ref. 21) | ● | ● | ● | ● | R-H2-OCO2Li |
|

|
288.5 ± 0.3 | 288.6 (ref. 34) | ● | ● | ● | ● | Oxalates (Li22O4) |
|

|
290.0 ± 0.3 | 290.0,22 290.6 (ref. 11) | ● | ● | ● | ● | ROO2Li and/or Li2O3 |
|
The C-1s spectrum of non-irradiated nanoparticles exhibits a single asymmetrical peak at 283.8 ± 0.3 eV, which corresponds to sp2 C–C bonding in pristine material (Fig. 2, left side). The global shape of the C 1s spectra obtained after irradiation is similar to spectra obtained on graphite LIB anodes after cycling.21 XPS C-1s spectra before and after irradiation at a dose of 1.5 MGy were also recorded in the case of micrometric material (SLP30 graphite in the present case). These spectra are displayed in Fig. S3 (ESI†) and demonstrate that SEI contributions formed upon radiolysis are chemically similar to what is observed upon electrochemical cycling of graphite. The C-1s spectra of all irradiated samples present additional peaks at 285.0 ± 0.3 eV, 286.1 ± 0.3 eV, 286.8 ± 0.3 eV, 288.5 ± 0.3 eV, and 290.0 ± 0.3 eV (Fig. 2, right side). Based on literature data, the contributions are assigned as follows. The peak at 285.0 ± 0.3 eV corresponds to carbon atoms bound to carbon atoms, as in hydrocarbons.21 The second contribution, at 286.1 ± 0.3 eV, indicates the presence of oligomeric species of polyethylene oxide (–CH2CH2O–)n or ROLi species.11,22 This assignment is additionally supported by findings that radiolysis of DEC/LiPF6 solutions generates precursors of polyethylene oxide.15 These two species are in good agreement with the work of Shkrob et al., who provided evidence that one electron reactions in cycling LIB cells containing EC, result in radical formation and subsequent polymerization of EC.23 The peak at 286.8 ± 0.3 eV is assigned to carbon atoms in a one-oxygen environment, as in lithium alkyl carbonates (R-CH2-OCO2Li).21 From a mechanistic point of view, we have previously observed by picosecond pulse radiolysis the formation of the EC˙− radical anion in EC/DEC mixtures.17 This radical anion EC˙− dimerizes into the ethylene dicarbonate dianion (−OCOOCH2CH2OCOO−) which precipitates, in the presence of lithium as solid lithium ethylene dicarbonate (Li2EDC).9 Alkyl carbonate compounds are furthermore, known to be major components formed in the SEI on graphite anodes.24 The contribution at 288.5 ± 0.3 eV is assigned to oxalates (Li2C2O4),25 which have been proposed to result from CO2 formation during electrolyte degradation, consistent with our gas phase product data (see above).25,26 Finally, the highest binding energy contribution at 290.0 ± 0.3 eV is assigned to carbon atoms in a three-oxygen environment such as Li2CO3, which is normally present in the SEI formed during LIB cycling in EC-based electrolytes,11,27 and/or ROCO2Li.21,28 Globally, all contributions are present, even at the lowest dose used. Contrary to the other contributions that increase with the irradiation dose, the peak at 282.3 ± 0.3 eV is observed only at 92 kGy. It may be attributed to lithium intercalated in the structure of amorphous carbon (Li2–C2) or to a complex charge effect.29–31 We can conclude that all the different carbon chemical environments identified by XPS on the irradiated particles' surfaces are also observed in electrochemically formed SEI.32,33 The ratio between the integrated intensity of the various SEI compound contributions to the C-1s spectra (at the exception of the 283.8 eV peak) and the total integrated intensity increases with irradiation dose up to 92 kGy, indicating that the proportion of degradation products near the surface of the particles increases in this dose range. At higher doses, this ratio remains relatively unchanged (Fig. S4, ESI†). Degradation products can also contribute to the 283.8 eV peak, attributed to the carbon nanoparticles in the non-irradiated sample, through sp2 carbons and C–H bonds as in hydrocarbons.11 Therefore, even if the ratio (Fig. S4†) mentioned above remains relatively unchanged at doses above 92 kGy, the degradation product quantity may still increase at the highest doses, as is implied by the electrochemical results below. It is worth mentioning that other XPS spectra were measured. In the case of Li-1s however, the signals are too weak and noisy to allow for a relevant interpretation. In the case of P-2p and O-1s, the signals are very complex, preventing a careful interpretation. We have provided in the ESI† the F-1s spectra (Fig. S5 and Table S3†). These are however not informative. Indeed, the fluorine species formed upon are the same as those arising from our drying procedure (Fig. S5† and corresponding discussion).
The XPS C-1s spectra suggest a surface chemistry on the irradiated carbon nanoparticles which matches that of anode materials coated with electrochemically formed SEI. All the classes of compounds identified here (see Table 2) are the same as those reported in typical LIBs experiments.11,21,30–32,34,35 Furthermore, with increased irradiation dose the relative quantity of SEI increases up to ∼100 kGy. We conclude that irradiation of the anode material–electrolyte system is a promising ex situ technique for mimicking the surface chemistry during LIB operation.
 | ||
| Fig. 3 TEM images of the nanoparticles exposed to electrolyte (a) not irradiated, (b) after irradiation at a dose of 55 kGy, and (c) at a dose of 93 kGy. (d) Is a zoom of the blue boxed area in (c). Areas outlined in red indicate agglomerates of degradation products. | ||
2.2 Electrochemical characterization
As described above, the first cycle of a battery, often called the “formation step”,9 is used to generate a stable SEI which passivates the electrode surfaces and is important for performance and safety of the cell during its lifetime. As the step often involves a low current, it adds a significant cost to LIB production in terms of time and capital.9 SEI formation ex situ at a higher rate could thus reduce these costs and provide useful information in investigating new electrolytes and their decomposition. To investigate the functionality of the SEI generated during irradiation, electrodes were made from three samples of carbon nanoparticles (CNPs): CNPs irradiated as described above at a 92 kGy dose (moderate irradiation dose), 202 kGy dose (high irradiation dose), and reference CNPs which had been exposed to the electrolyte but not irradiated. These three samples were used to fabricate test coin cells and cycled as described in the Experimental section. The first cycle charge and discharge potential vs. specific integrated current curves are presented in Fig. 4. The 60 h time limit was reached by the reference and 92 kGy dose samples while the 202 kGy dose samples reached the 0 V low potential limit. The total “specific integrated current” values obtained from these tests are provided in Table 3. It is important to note that the specific integrated current includes the lithiation/delithiation currents as well as those resulting from SEI formation. Both irradiated samples exhibit electrochemical activity starting at ∼2 V during the lithiation segment, indicated by the inflection of the curves. The reference sample electrochemical activity onset occurs at a lower voltage, ∼1.5 V. This matches well with the onset of reduction for these electrolytes in contact with micro-graphitic carbons, which is typically observed ∼1.4 V, although there is a broadening of the reactivity window for nanometric and amorphous carbons such as those used here.9 The higher voltage onset for the irradiated samples may be due to reactive surface species generated during the irradiation (for instance surface oxygen functionality on carbon materials have been associated with LIB electrolyte breakdown above 2 V).12 More interestingly however is that the main plateau between 0.6 and 0.8 V, where the majority of SEI formation is expected to take place,9 has the lowest total current consumption for the 202 kGy material, followed by the 92 kGy. The reference material (non-irradiated) consumes the largest total current with SEI formation.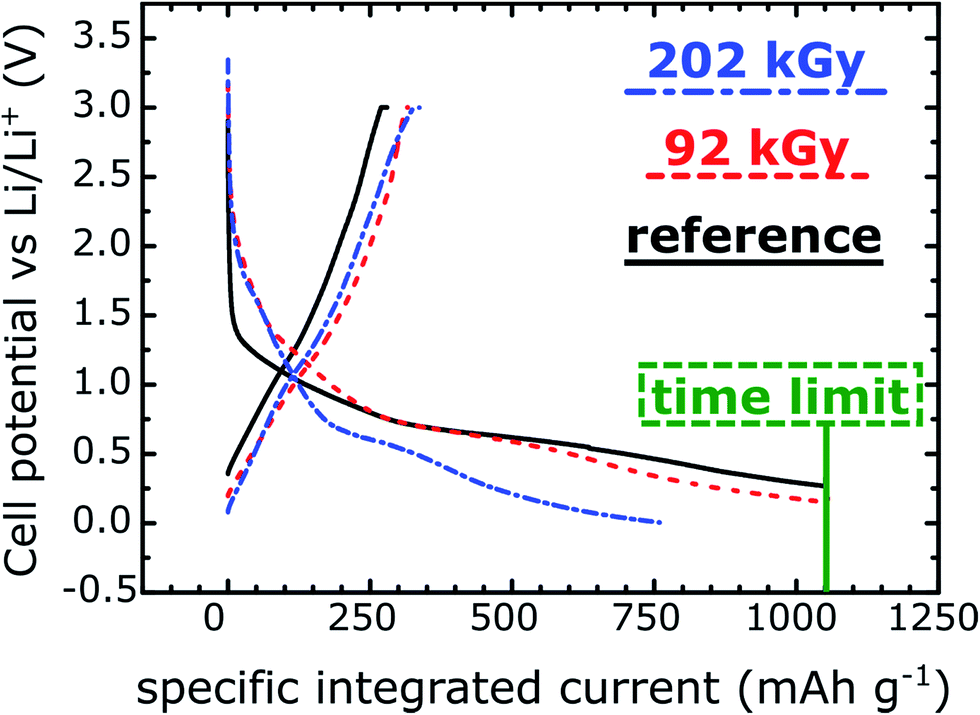 | ||
| Fig. 4 First galvanostatic cycle (“formation step”) of the three tested carbon nanoparticles. | ||
| Specific integrated current – lithiation (mA h g−1) | Specific integrated current – delithiation (mA h g−1) | Coulombic efficiency (%) | |
|---|---|---|---|
| Reference | 1050 ± 1 | 279 ± 1 | 27% |
| 92 kGy irradiation dose | 1050 ± 1 | 327 ± 3 | 31% |
| 202 kGy irradiation dose | 746 ± 14 | 342 ± 8 | 46 ± 2% |
Towards the end of the initial lithiation process, the lithiation and SEI formation appear to proceed concurrently, with Li-intercalation being complete at the voltage cut-off of 0 V, as expected for disordered carbons.36 At the time limit set for the test, only the 202 kGy material cell reached the low voltage cutoff whereas the 92 kGy material cell reached ∼0.15 V, and the exposed material cell only reached ∼0.27 V. From the delithiation integrated current, we observe that the 202 kGy cell intercalated the most lithium, exhibiting a delithiation integrated specific current of 342 mA h g−1. The 92 kGy cell followed with an integrated specific current of 327 mA h g−1 and the reference cell exhibited an integrated specific current of only 279 mA h g−1. Furthermore all the materials present similar profiles during this portion of the cycle and no difference in polarization is observed.
Electrochemical formation of SEI still occurred in all samples, albeit less with increasing radiation dose, and the first cycle efficiency increased from 27% for the reference to 46% for the 202 kGy dose, as shown in Table 3. These efficiencies are inherently low due to the nanometric size of the materials and large surface area to volume ratio as compared to typical micrometric graphitic materials used in LIBs. For this proof of concept study however, the large surface area materials were selected specifically for increased SEI growth, as explained above.
The Nyquist plots, obtained by electrochemical impedance spectroscopy, are presented in Fig. 5a and b. Analysis of the results was performed using the electrical equivalent circuit (Fig. 5). A more detailed description of the model, the raw parameters, and their definition is provided in the (Table S4, ESI†). Within the practical limitations of applying such a model to this complex system, the general conclusions which may be drawn from the fitted data support the formation of a resistive phase on the surface, in coherence with the results discussed above. The charge transfer resistance element (Rct, extracted from the lower frequency response) increases from 271 Ω to 457 Ω between the reference and particles irradiated at 106 kGy. The surface layer resistance element measured at high frequencies (Rf: in the case of the reference particles the surface layer can be considered as the mix of residues from salt decomposition under vacuum as observed with XPS, not to be confused with the irradiation generated decomposition products, and adsorbed solvent molecules which are on the particle side of the electrochemical double layer), is approximately two times that of the reference sample, 73 Ω and 35 Ω respectively. As well, the surfaces of both the irradiated and reference samples present spectra which indicate inhomogeneous surfaces, as the factor α associated with the film pseudocapacitance (ZCPE-f = 1/Cf(jω)α) is calculated to be 0.8, deviating significantly from the ideal value of 1 for a homogeneous film37 in agreement with the irregular aggregates seen by TEM in the vicinity of carbon nanoparticles.
 | ||
| Fig. 5 Electrochemical impedance spectroscopy (EIS) of the carbon nanoparticles before (a) and after (b) irradiation at a dose of 106 kGy. The electrical equivalent circuit is shown above. | ||
3. Conclusion
In the present work, we have irradiated suspensions of carbon nanoparticles, as a representative anode material for LIBs, in a typical electrolyte and we have followed the evolution of the surface of the nanoparticles with increasing irradiation dose. In summary, the radiolysis treatment generates electrolyte decomposition products on the particles which are found by XPS to be chemically similar to electrochemically formed SEI, passivate the surface electrochemically, and indeed create a solid interphase between the electrode materials and the electrolyte. Thus we conclude that by irradiation the ex situ generation of synthetic SEI is achieved. Degradation products in the gas and solid phases of the irradiated system were also identified as having the same chemical signatures as those in cycling batteries. Electrochemical formation of SEI still occurred in all samples during the galvanostatic cycling, albeit less with increasing irradiation dose, and the first cycle efficiency was significantly increased with increasing irradiation. As a proof of concept these results are exciting.Radiolysis is thus a powerful tool to rapidly provide a full picture of phenomena taking place in different phases in LIBs. Furthermore, irradiation treatment is a promising method for ex situ conditioning of electrode materials. Indeed, irradiation of electrode materials prior to their implementation in LIBs could significantly reduce manufacturing costs associated with electrochemical formation steps. It should also be noted that such radiolysis results can be obtained using gamma ray generators or electron accelerators such as those that are commonly used in hospitals, by the food industry for sterilization, or for water treatment. Additionally it should be noted that the number of such facilities is steadily growing in number.38 Slurries of carbon material/binder/electrolyte could also be continuously irradiated in order to streamline the process. More work is needed to optimize the irradiation configuration to enable larger concentrations of particles, and the irradiation conditions such as dose to ensure homogenous surface passivation and avoid embrittlement of the binder. The present work however paves the way to the use of radiolysis for the synthesis of materials with an artificial SEI. Application of these methods to next generation LIB anodes for improved energy storage, such as silicon nanoparticles where the first cycle efficiency is especially important, is currently ongoing.
4. Materials and methods
4.1 Preparation and characterization of carbon nanoparticles
Carbon nanoparticles were prepared by laser pyrolysis as described previously.18 In summary, ethylene gas at ∼40 mol% in helium (at ∼1 standard liter per minute) was pyrolysed by a CO2 laser, PRC 2200, operating at 10.6 μm wavelength with 870 watts of power.After collection under air, the powders were stored in a glove box under argon atmosphere. The nanoparticles consist of highly disordered carbon, as evidenced by Raman measurement (Fig. S6, ESI†) with an average diameter around 80 nm (see TEM images in Fig. S7, ESI†).
4.2 Irradiation treatment
The nanoparticles were heated at 400 °C for 2 h under argon to eliminate residual water as well as any organic residual material generated during pyrolysis of C2H4. Thermally treated nanoparticles (1% w) were then dispersed in the electrolyte, 50/50 (v/v) ethylene carbonate (EC)/diethyl carbonate (DEC) mixture containing LiPF6 1 M (provided by Sigma-Aldrich, anhydrous grade, purity >99%), in a glove box under argon atmosphere. No higher amount of nanoparticles was studied, as nanoparticles are no longer properly dispersed in the solution for concentrations higher than 2.5% w, which prevents then to perform reproducible experiments. The electrolyte mix for irradiation was selected to have a large proportion of EC, as it is known to undergo decomposition reactions resulting in more robust, polymeric SEI as compared to linear carbonates such as DEC, dimethyl carbonate (DMC), or ethyl methyl carbonate (EMC).9 About 1.3 mL of dispersion was added to a glass ampoule and subsequently degassed by argon bubbling for 30 min. The ampoules were degassed three times and filled with argon 6.0 (99.9999%) at 1.5 bar. Water concentration of the prepared dispersion of carbon nanoparticles was determined to be less than 50 ppm by a coulometric Karl Fischer titrator (Mettler Toledo).The irradiation of the samples was carried out with a panoramic 60Co γ source. During irradiation experiments, the samples were stirred using a specially designed sample holder to avoid sedimentation or agglomeration of nanoparticles. The samples were irradiated at a dose rate ranging between 42 and 56 Gy min−1 (1 Gy = 1 J kg−1) depending on the irradiation position on the sample holder. The dose rate was determined by using the aqueous Fricke dosimeter.39 For the XPS, TEM and electrochemical experiments, the nanoparticles were separated after irradiation from the supernatant by centrifugation (3000 g, 5 min, 20 °C), rinsed three times with DEC, dried under vacuum for 16 h, and stored in Ar before subsequent analysis.
4.3 Characterization of products formed in the gas phase
The products formed in the gas phase were identified by means of hyphenated gas chromatography (Agilent, 6890 GC) with a mass spectrometer (Agilent, 5973 MS). The separation was performed with a CP-PorabondQ column (Varian, 25 m × 0.32 mm) operated in splitless mode. The flow rate of helium (gas vector) was fixed at 2 mL min−1. The temperature of the injector was set at 110 °C. The separated products were fragmented using an electron impact source of 70 eV and detected by a quadrupole mass analyser within a mass range from 4 to 160 m/z. The products were identified by comparing experimental spectra to the mass spectra library from NIST. The quantification of the main products (H2, CH4, CO and CO2) was performed by gas chromatography (μGC R3000, SRA Instruments) with argon as carrier gas.14 Prior to the experiments, calibration was performed by using a standard bottle containing 1000 ppm of each studied gas (CO, CO2, H2 and CH4). Various dilutions of these gases with ultra-pure argon were also accomplished. For each gas, a line linking the area of the peak to the concentration could then be plotted.4.4 Characterization of the surface of the nanoparticles by X-ray photoelectron spectroscopy (XPS)
XPS measurements were performed using a Thermofisher Escalab 250 XI spectrometer equipped with a monochromatic X-ray Al-Kα source (hν = 1486.6 eV). A constant pass energy of 20 eV was used for high resolution spectra. The energy resolution of the instrument has been found to be 0.3 eV. To control the charging of the samples a charge neutralizer flood gun was used. All spectra were calibrated using the C–C contribution from the nanoparticles fixed at 283.8 eV. No degradation of the samples under X-ray beam was observed. Data acquisition and processing were carried out using Avantage data processing software (Thermofisher Scientific). After calibration, the background from each spectrum was subtracted using a Shirley-type background. The experimental spectra were fitted using mixed Lorentzian–Gaussian contributions.4.5 Transmission electron microscopy (TEM) characterization
Carbon particles were analysed by the means of a FEI TECNAI OSIRIS microscope in TEM mode and operated at 200 kV. Care was taken to control the electron dose and avoid damaging materials. The particles were deposited onto lacey carbon grids in a glove box and transferred to the microscope under argon atmosphere with a dedicated sample holder.4.6 Electrochemical characterization
Electrode inks were prepared in an argon filled glovebox by mixing carbon nanoparticles (before and after irradiation treatment), SuperP carbon black (Timcal), and PVdF binder (polyvinylidenedifluoride – Solvay) in N-methyl-pyrrolidone (NMP) at a weight ratio of 80/10/10. The obtained ink was spread on copper foil using a doctor blade at 100 μm thickness. Electrodes were dried under vacuum at 80 °C prior to assembly in test cells, and mass loadings were ∼3–4.5 mg cm−2.Galvanostatic tests were performed on 2032 coin cells in a half cell configuration with a Celgard 2400 polypropylene separator. EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC:EMC 1:1:1 with 1 M LiPF6 was used as the electrolyte in these tests, as it is the typical electrolyte utilized in our labs for cycling carbon based anodes. Galvanostatic tests were performed using a BCS-805 cycler (Biologic) working with BT Lab software. Cells were cycled at C/20 between 0 V and 3 V vs. Li+/Li, with a 60 hour limit imposed on the test. The capacity of the material was estimated to be 350 mA h g−1, and the C-rate was calculated accordingly.
:DMC:EMC 1:1:1 with 1 M LiPF6 was used as the electrolyte in these tests, as it is the typical electrolyte utilized in our labs for cycling carbon based anodes. Galvanostatic tests were performed using a BCS-805 cycler (Biologic) working with BT Lab software. Cells were cycled at C/20 between 0 V and 3 V vs. Li+/Li, with a 60 hour limit imposed on the test. The capacity of the material was estimated to be 350 mA h g−1, and the C-rate was calculated accordingly.
EIS experiments were performed in symmetrical two electrode Swagelok type cells with a microporous polypropylene separator (Celgard® 2400). The electrolyte was the same as that used in irradiation experiments. EIS was performed in the frequency range 83 kHz to 33 mHz, with a VMP3 (BioLogic) frequency response analyzer. The excitation signal was 10 mV peak to peak. The equilibrium potential was considered to be reached when the drift in open circuit voltage remained less than 0.1 mV for 1 hour. Fitting of the impedance spectra was performed with the Zplot® software from Scribner Associates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by “DSM-Energie” program from CEA (Project SOLEIL). The MOMENTOM (Initiative de Recherche Stratégique, Université Paris Saclay) project is also gratefully acknowledged for financial support. J. P. A. gratefully acknowledges CEA Eurotalents fellowship support. The authors thank Dr Céline Cannes and Dr Véronika Zinovyeva from Institut de Physique Nucléaire of Orsay (France) for Karl Fischer measurements.References
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 RSC.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef PubMed.
- C. L. Campion, W. T. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327 CrossRef.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503 CrossRef PubMed.
- J. Yan, J. Zhang, Y.-C. Su, X.-G. Zhang and B.-J. Xia, Electrochim. Acta, 2010, 55, 1785 CrossRef.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, Carbon, 2016, 105, 52 CrossRef.
- D. Aurbach, J. Power Sources, 2000, 89, 206 CrossRef.
- P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55, 6332 CrossRef.
- J. Collins, G. Gourdin, M. Foster and D. Qu, Carbon, 2015, 92, 193 CrossRef.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703 CrossRef.
- D. Ortiz, V. Steinmetz, D. Durand, S. Legand, V. Dauvois, P. Maître and S. Le Caër, Nat. Commun., 2015, 6, 6950 CrossRef PubMed.
- D. Ortiz, I. Jimenez Gordon, J.-P. Baltaze, O. Hernandez-Alba, S. Legand, V. Dauvois, G. Si Larbi, U. Schmidhammer, J. L. Marignier, J.-F. Martin, J. Belloni, M. Mostafavi and S. Le Caër, ChemSusChem, 2015, 8, 3605 CrossRef PubMed.
- D. Ortiz, I. Jimenez Gordon, S. Legand, V. Dauvois, J.-P. Baltaze, J. L. Marignier, J.-F. Martin, J. Belloni, M. Mostafavi and S. Le Caër, J. Power Sources, 2016, 326, 285 CrossRef.
- F. Wang, F. Varenne, D. Ortiz, V. Pinzio, M. Mostafavi and S. Le Caër, ChemPhysChem, 2017, 18, 2799 CrossRef PubMed.
- A. Galvez, N. Herlin-Boime, C. Reynaud, C. Clinard and J.-N. Rouzaud, Carbon, 2002, 40, 2775 CrossRef.
- F. A. Soto, Y. Ma, J. M. Martinez de la Hoz, J. M. Seminario and P. B. Balbuena, Chem. Mater., 2015, 7990–8000 CrossRef.
- H. Yoshida, T. Fukunaga, T. Hazama, M. Terasaki, M. Mizutani and M. Yamachi, J. Power Sources, 1997, 68, 311 CrossRef.
- S. Leroy, F. Blanchard, R. Dedryvére, H. Martinez, B. Carré, D. Lemordant and D. Gonbeau, Surf. Interface Anal., 2005, 37, 773 CrossRef.
- H. Bryngelsson, M. Stjerndahl, T. Gustafsson and K. Edström, J. Power Sources, 2007, 174, 970 CrossRef.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255 CrossRef.
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1987, 134, 1611 CrossRef.
- R. Dedryvère, S. Laruelle, S. Grugeon, L. Gireaud, J.-M. Tarason and D. Gonbeau, J. Electrochem. Soc., 2005, 152, A689 CrossRef.
- D. Aurbach, B. Markovsky, A. Shechter, Y. Ein-Eli and H. Cohen, J. Electrochem. Soc., 1996, 143, 3809 CrossRef.
- D. Aurbach, M. D. Levi, E. Levi and A. Schechter, J. Phys. Chem. B, 1997, 101, 2195 CrossRef.
- R. Dedryvère, L. Gireaud, S. Grugeon, S. Laruelle, J.-M. Tarason and D. Gonbeau, J. Phys. Chem. B, 2005, 109, 15868 CrossRef PubMed.
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824 CrossRef.
- I. A. Profatilova, S.-S. Kim and N.-S. Choi, Electrochim. Acta, 2009, 54, 4445 CrossRef.
- S. Malmgren, K. Ciosek, M. Hahlin, T. Gustafsson, M. Gorgoi, H. Rensmo and K. Edström, Electrochim. Acta, 2013, 97, 23 CrossRef.
- V. Winkler, T. Hanemann and M. Bruns, Surf. Interface Anal., 2017, 49, 361 CrossRef.
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358 CrossRef.
- J.-T. Li, J. Swiatowska, V. Maurice, A. Seyeux, L. Huang, S.-G. Sun and P. Marcus, J. Phys. Chem. C, 2011, 115, 7012 CrossRef.
- P. Niehoff, S. Passerini and M. Winter, Langmuir, 2013, 29, 5806 CrossRef PubMed.
- Y. Ein-Eli, B. Markovsky, D. Aurbach, Y. Carmeli, H. Yamin and S. Luski, Electrochim. Acta, 1994, 39, 2559 CrossRef.
- H. Bouayad, Z. Wang, N. Dupré, R. Dedryvère, D. Foix, S. Franger, J.-F. Martin, L. Boutafa, S. Patoux, D. Gonbeau and D. Guyomard, J. Phys. Chem. C, 2014, 118, 4634 CrossRef.
- A. G. Chmielewski, in Applications of ionizing radiation in materials processing, ed. Y. Sun and A. G. Chmielewski, Institute of nuclear chemistry and technology, Warszawa, 2017, vol. 2, p. 516 Search PubMed.
- H. Fricke and E. J. Hart, in Radiation Dosimetry, ed. F. H. Attix and W. C. Roesch, Academic press, New York and London, 2nd edn, 1966, vol. 2, p. 167 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00257f |
| This journal is © The Royal Society of Chemistry 2018 |