Iron(III)-based metal–organic frameworks as oxygen-evolving photocatalysts for water oxidation
Received
29th June 2018
, Accepted 18th July 2018
First published on 20th July 2018
Abstract
Development of economical and highly efficient catalysts to achieve visible light-induced water oxidation is a potential approach to mitigate energy shortage and global warming. Herein, we report Fe-based MOFs (MIL-53 and NH2-MIL-53) which show photocatalytic activity for water oxidation under visible light. Compared with MIL-53 (Fe), NH2-MIL-53 (Fe) shows higher photocatalytic efficiency for water oxidation due to the narrower band gap and more efficient separation of the photo-excited electron–hole pairs. Furthermore, through active species trapping experiments and mechanism studies, we find that the photogenerated holes and hydroxyl radicals are possibly responsible for the photocatalytic water oxidation activity of the Fe-based MOFs.
Introduction
Photocatalytic water splitting, an efficient strategy for storing solar energy, consists of two half-reactions: the oxidation of water to O2 and the reduction of protons to H2. Compared with water reduction, water oxidation is much less advanced due to its implicit complexity which involves the transfer of four electrons and the formation of oxygen–oxygen bonds.1 To date, some materials, such as metal oxides,2 metal oxynitrides,3 metal oxysulfides4 and molecular catalysts,5 have been explored for water oxidation. In particular, 3d transition metal oxide materials have received more attention as potential photocatalysts for water oxidation, because they are inexpensive and non-toxic. To enhance the catalytic activity of oxide materials, the use of porous support materials has been demonstrated. Those reports show a significant improvement in stability and catalytic activity compared with unsupported or bulk metal oxides due to a high concentration of surface active sites and the promotion of charge transfer.6
Metal–organic frameworks (MOFs) are a new class of porous hybrid materials composed of metal (oxide) clusters and multidentate organic linkers. Recently, it has been reported that some MOFs show attractive semiconducting performance, absorbing visible light and photocatalytically driving various useful chemical reactions.7 For example, some Fe-based MOFs can be viewed as semiconductor quantum dots (Fe(III)-oxide clusters) held in a three-dimensional structure by a polycarboxylic acid. Several Fe-based MOFs have recently been used as heterogeneous photocatalysts for pollutant degradation, CO2 reduction, hydrogen production and so on.8 Compared with classical semiconductors, the advantages of MOFs are their desirable topology and high surface area, which are beneficial for fast transport and good accommodation of small molecules.9 To achieve efficient utilization of solar energy, their light absorption ability could be more easily tuned by the modification of the metal clusters or the organic linkers.10
Recently, Fe-based MOFs have been reported as heterogeneous catalysts for oxidation reactions.11 To develop environmentally friendly catalysts for water oxidation, we investigate the photocatalytic water oxidation performance of Fe-based MOFs MIL-53 and NH2-MIL-53. The photocatalytic results show that the Fe-based MOFs (MIL-53 and NH2-MIL-53) are promising photocatalysts for water oxidation, and photo-generated holes and hydroxyl radicals are the main active species for the photocatalytic water oxidation. A photocatalytic performance comparison between MIL-53 and NH2-MIL-53 indicates that the amine-substituted linker can improve the photocatalytic water oxidation performance due to the narrower band gap and more efficient separation of the photo-excited electron–hole pairs.
Experimental section
Materials
All reagents were of analytical grade and were used without further purification. Iron(III) chloride hexahydrate (FeCl3·6H2O), terephthalic acid (H2BDC), 2-aminoterephthalic acid and N,N′-dimethylformamide (DMF) were purchased from Sinopharm Chemical Reagent Co. Ltd., China. De-ionized water was obtained from a Millipore Milli-Q system.
Synthesis of MIL-53 (Fe) and NH2-MIL-53 (Fe)
Metal–organic framework MIL-53 (Fe) was synthesized according to a reported method.12 Typically, a mixture of FeCl3·6H2O, H2BDC and DMF with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:280 is heated at 150 °C for 15 h in a Teflon-lined autoclave. The product is isolated by filtration and washed with DMF, ethanol and water in turn; the resulting solid is dried at 150 °C in a vacuum oven. The NH2-functionalized MIL-53 (Fe) was prepared in a similar manner to the parent MIL-53 (Fe) except that terephthalic acid was replaced by 2-aminoterephthalic acid.
:1:280 is heated at 150 °C for 15 h in a Teflon-lined autoclave. The product is isolated by filtration and washed with DMF, ethanol and water in turn; the resulting solid is dried at 150 °C in a vacuum oven. The NH2-functionalized MIL-53 (Fe) was prepared in a similar manner to the parent MIL-53 (Fe) except that terephthalic acid was replaced by 2-aminoterephthalic acid.
Characterization of the photocatalysts
X-ray powder diffraction (XRD) measurements of MIL-53 (Fe) and NH2-MIL-53 (Fe) were performed on a Bruker D8 diffractometer operated at 40 V and 40 mA using a Cu-Kα radiation source (λ = 1.54056 Å). Their electronic absorption spectra were recorded using a UV-2450 UV-vis spectrophotometer with BaSO4 as the reflectance standard reference at room temperature. FT-IR spectra of the photocatalysts were recorded on a Nicolet Nexus 470 FT-IR spectrophotometer. The N2 adsorption isotherms and BJH pore size distributions were measured at 77 K by using a Micromeritics ASAP 2020M volumetric adsorption analyzer. Scanning electron microscopy (SEM) images were obtained on a MERLIN Compact instrument. Cyclic voltammetry (CV) was performed on a CHI-730 electrochemical analyser with FTO paste and Fe-based MOF, Ag/AgCl and Pt wire electrodes as the working, reference and auxiliary electrodes, respectively, at room temperature with a scan rate of 100 mV s−1. The photocurrent response was determined using a CHI-852C electrochemical workstation with complex-pasted FTO as the working electrode, a piece of platinum wire as the counter electrode, a saturated Ag/AgCl electrode as the reference, and a 20 mM phosphate buffer solution (pH = 8.5) as the electrolyte. Oxygen evolution was monitored by gas chromatography using a thermal conductivity detector (GC-TCD).
Photocatalytic water oxidation
Light-driven water-oxidation reactions were performed in a glass photolysis vessel containing 0.70 mM catalyst, 1.0 mM [Ru(bpy)3]Cl2 (sensitizer), 0.08 M Na2S2O8 (electron acceptor) and 20 mM buffered water (pH = 8.5, Na2HPO4/NaH2PO4). After purging with N2, the vessel was illuminated by the light source using an LED lamp (450–550 nm, 4 W). The aqueous suspension was maintained under stirring and illumination for 120 min. Oxygen was analysed with a gas chromatograph equipped with a thermal conductivity detector.
Results and discussion
Characterization of MIL-53 (Fe) and NH2-MIL-53 (Fe)
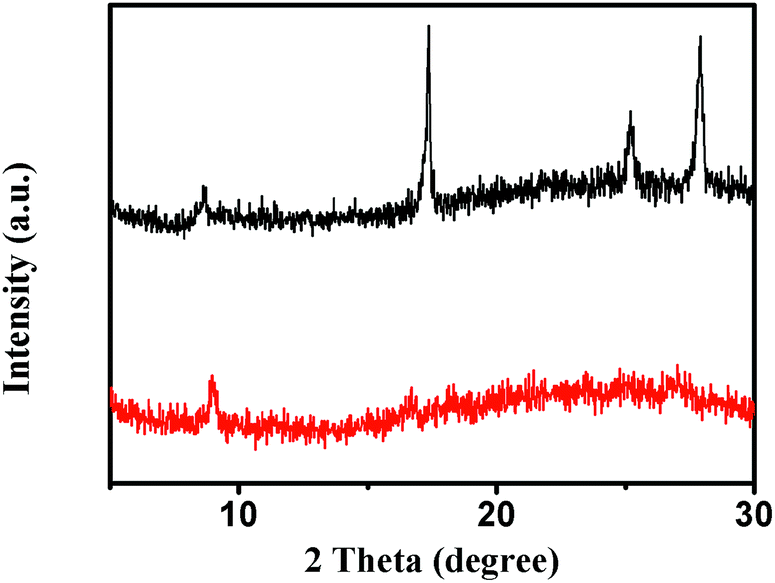
The crystallographic structures of MIL-53 (Fe) and NH2-MIL-53 (Fe) were examined by XRD (X-ray diffraction). Apparently, MIL-53 (Fe) and NH2-MIL-53 (Fe) exhibit similar diffraction patterns, which are consistent with a previous report on the MIL-53 framework.12 Amine functionalization has no effect on the crystal phase structure (Fig. 1).
 |
| | Fig. 1 XRD patterns of MIL-53 (black line) and NH2-MIL-53 (red line). | |
The presence of the free NH2 group was confirmed from the FT-IR spectra (Fig. 2) by peaks at around 3327 and 3458 cm−1, which can be ascribed to the asymmetrical and symmetrical stretching vibrations of free primary amines without coordination. The characteristic absorption peaks of MIL-53 (Fe) and NH2-MIL-53 (Fe) are identical to the data reported in the literature.12 The bands at around 1576 and 1394 cm−1 are assigned to the asymmetric and symmetric vibrations of carboxyl groups, respectively. The stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O appears at around 1668 cm−1. The peak at around 764 cm−1 corresponds to C–H bending vibrations of the benzene in the ligand. Furthermore, the stretching vibration of Fe–O bonds appears at around 538 cm−1, implying the formation of an Fe-oxo bond between Fe3+ and the carboxylic group. From the results, together with the XRD data, it is clear that NH2-MIL-53 (Fe) possesses a similar structure to MIL-53 (Fe) and NH2 groups do not coordinate with Fe3+, which do not affect the structure of the framework.
O appears at around 1668 cm−1. The peak at around 764 cm−1 corresponds to C–H bending vibrations of the benzene in the ligand. Furthermore, the stretching vibration of Fe–O bonds appears at around 538 cm−1, implying the formation of an Fe-oxo bond between Fe3+ and the carboxylic group. From the results, together with the XRD data, it is clear that NH2-MIL-53 (Fe) possesses a similar structure to MIL-53 (Fe) and NH2 groups do not coordinate with Fe3+, which do not affect the structure of the framework.
 |
| | Fig. 2 FT-IR spectra of MIL-53 (Fe) and NH2-MIL-53 (Fe). | |
The surface area and porous structure of MIL-53 (Fe) and NH2-MIL-53 (Fe) were investigated using N2 adsorption–desorption isotherms. From Fig. 3, we can see that both of them exhibit a typical type I isotherm characteristic of microporous solids with high adsorption in the low pressure region. The BET surface area of NH2-MIL-53 is smaller than that of MIL-53 due to a partial blocking of the pores by the uncoordinated amino group (–NH2).13
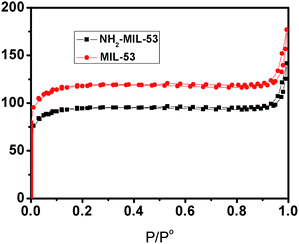 |
| | Fig. 3 Nitrogen adsorption isotherms of the Fe-based MOFs (MIL-53 and NH2-MIL-53). | |
The morphological features and particle size of the Fe-based MOFs (MIL-53 (Fe) and NH2-MIL-53 (Fe)) were investigated by scanning electron microscopy (SEM) and the results are shown in Fig. 4. Fig. 4(a) shows that MIL-53 (Fe) has a sheet structure with an average size of 0.5 μm width. NH2-MIL-53 (Fe) is spindle-shaped with an average size of 0.7 μm length and 0.4 μm width.
 |
| | Fig. 4 SEM images of (a) MIL-53 (Fe) and (b) NH2-MIL-53 (Fe). | |
Photophysical properties
The light absorption properties of the photocatalytic materials were analyzed by diffuse-reflectance UV-vis spectroscopy. As shown in Fig. 5, MIL-53 (Fe) and NH2-MIL-53 (Fe) have a clear absorption in the visible light region. For MIL-53 (Fe), the characteristic absorption at about 360 nm can be assigned to the ligand-to-metal charge transfer (LMCT) of carboxylate oxygen to Fe(III).14 In addition, a peak centered at 445 nm may be attributed to the transition 6A1g ⇒ 4A1g + 4Eg(G) in Fe(III).15 Although there are some similarities between MIL-53 (Fe) and NH2-MIL-53 (Fe), the absorption properties of NH2-MIL-53 (Fe) have been significantly enhanced compared with MIL-53 (Fe). Combined with the band gap energy estimated from UV-vis absorption spectra (Fig. 4 inset), the calculated optical bandgaps of MIL-53 (Fe) and NH2-MIL-53 (Fe) are 2.72 eV and 2.59 eV, respectively.16 These results indicate that introducing the amino group (–NH2) into the Fe-based MOF can enhance its absorption capacity in the visible light region and decrease the band gap energy, which benefit its photocatalytic activity.17
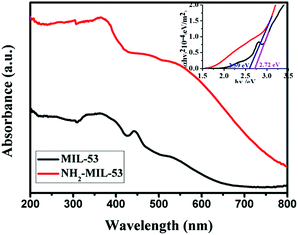 |
| | Fig. 5 UV-vis diffuse reflectance spectra of MIL-53 (Fe) and NH2-MIL-53 (Fe) (inset: optical bandgaps of MIL-53 (Fe) and NH2-MIL-53 (Fe)). | |
Electrochemical analysis
To better understand the water oxidation activity of the Fe-based MOFs, electrochemical measurements were carried out in phosphate buffer (pH = 8.5) using the different Fe-based MOF coated FTO working electrodes. As shown in Fig. 6(a), the cathodic wave of MIL-53 (Fe) and NH2-MIL-53 (Fe), respectively, appears at −0.63 V (vs. SCE) and −0.60 V, assigned to the reduction of dioxygen formed at the working electrode. The results confirm that Fe-based MOFs MIL-53 (Fe) and NH2-MIL-53 (Fe) possess the ability of photocatalysis for water oxidation.
 |
| | Fig. 6 (a) Cyclic voltammograms (CVs) of MIL-53 (Fe) and NH2-MIL-53 (Fe) coated FTO working electrodes (black line, blank; red line, MIL-53; blue line, NH2-MIL-53 (Fe)). (b) The transient photocurrent responses of MIL-53 (Fe) and NH2-MIL-53 (Fe). | |
Fig. 6(b) shows the photocurrent–time curves of MIL-53 (Fe) and NH2-MIL-53 (Fe) under intermittent visible light illumination. From Fig. 6(b), we can see that NH2-MIL-53 (Fe) exhibits a higher photocurrent density than MIL-53 (Fe). This suggests a more efficient separation of the photo-excited electron–hole pairs of NH2-MIL-53 (Fe), which indicates that the photocatalytic performance of NH2-MIL-53 (Fe) is higher than that of MIL-53 (Fe).
The electrochemical results indicate that the water oxidation activity of NH2-MIL-53 (Fe) is higher than that of MIL-53 (Fe) and the incorporation of the amine group (–NH2) into the Fe-based MOF can be beneficial for enhancing the photocatalytic water oxidation performance.
Photocatalytic properties
The photocatalytic water oxidation tests of the Fe-based MOFs were carried out in a glass photolysis vessel containing [Ru(bpy)3]Cl2 (sensitizer), Na2S2O8 (electron acceptor) and water at pH = 8.5 (Na2HPO4/NaH2PO4). Furthermore, it is known that in the [Ru(bpy)3]2+–Na2S2O8 system, water oxidation is highly dependent on solution pH (Fig. 7). At lower pH values (pH = 7), the activity of MIL-53 dropped remarkably. From Fig. 8, it can be deduced that the Fe-based MOFs (MIL-53 and NH2-MIL-53) are active water oxidation catalysts. Compared with MIL-53, amino-functionalized NH2-MIL-53 shows improved catalytic activity for water oxidation. The maximum amount of O2 evolved over NH2-MIL-53 after 140 min is 120 μmol (TON: 51.1), which is remarkably larger than that obtained with MIL-53 (TON: 35.4). Considering that MIL-53 and NH2-MIL-53 possess similar structures, it may be concluded that the amine group from the ligands contributes to the enhanced photocatalytic performance for water oxidation.
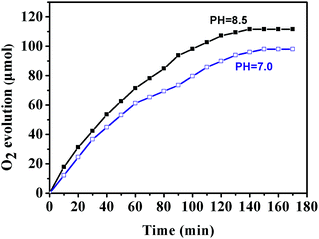 |
| | Fig. 7 Dioxygen (O2) evolution activity of MIL-53 (Fe) in the photocatalytic system under various pH conditions (black line: pH = 8.5, blue line: pH = 7.0). | |
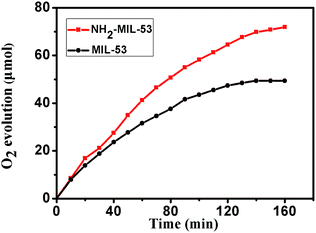 |
| | Fig. 8 O2 evolution activity of the Fe-based MOFs (MIL-53 and NH2-MIL-53) in aqueous suspensions (2 mL). Conditions: LED lamp (λ ≥ 420 nm, 4 W), catalyst concentration 0.70 mM, 1.0 mM [Ru(bpy)3]2+, 0.08 M Na2S2O8, 20 mM sodium phosphate buffer. | |
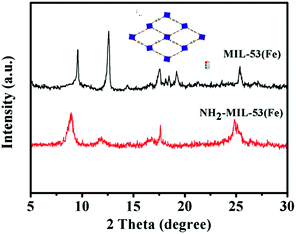
To check the stability and reusability of the photocatalysts, four consecutive catalytic runs were performed. The results show that the Fe-based MOFs (MIL-53 and NH2-MIL-53) can maintain their photocatalytic activity during the whole reaction process (Fig. 9 and 10). From Fig. 9, we can see that MIL-53 (Fe) shows no significant decrease in activity with 90% of the activity retained in the fourth run. In Fig. 10, the yield of NH2-MIL-53 (Fe) decreased to 60% after three additional reaction cycles. As shown in Fig. 11, the XRD pattern of the recovered MIL-53 reveals a reduced peak intensity and the disappearance of some peaks compared with fresh MIL-53. These findings imply that the main framework of MIL-53 remains almost unchanged but a small part of the skeleton is collapsed after the photoreaction, which may lead to a minor drop in activity for photocatalytic water oxidation. Compared to the recovered MIL-53 (Fe), the recovered NH2-MIL-53 (Fe) shows a reduced peak intensity only at peak 2θ = 8.8° and the disappearance of the other peaks. The above XRD results indicate that the framework of MIL-53 (Fe) and NH2-MIL-53 (Fe) collapsed to different degrees after the four consecutive runs, and the collapse of the main framework resulted in a significant drop in the reactivity of NH2-MIL-53 (Fe) (Fig. 12).
 |
| | Fig. 9 O2 evolution activity of MIL-53 (Fe) in four repetitive runs. Other conditions are the same as in Fig. 8. | |
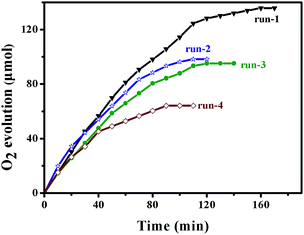 |
| | Fig. 10 O2 evolution activity of NH2-MIL-53 (Fe) in four repetitive runs. Other conditions are the same as in Fig. 8. | |
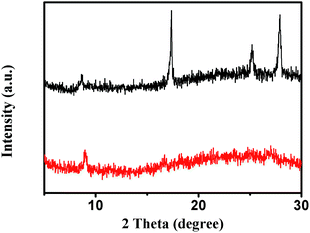 |
| | Fig. 11 XRD patterns of the recovered Fe-based MOFs, MIL-53 (black line) and NH2-MIL-53 (red line). | |
 |
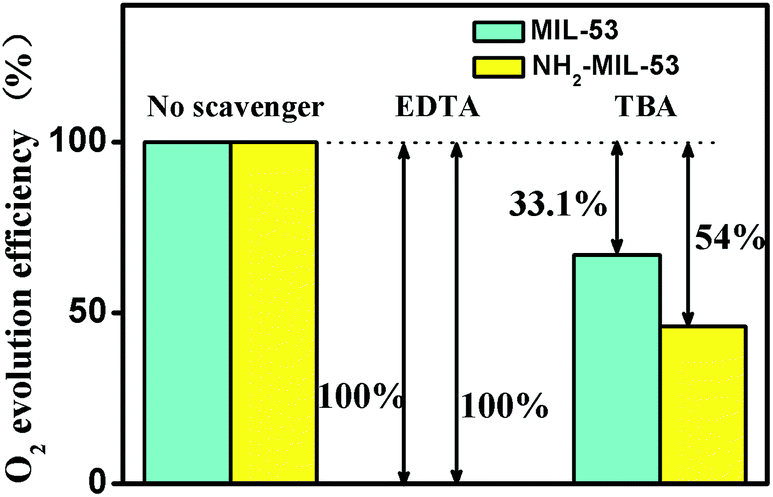
| | Fig. 12 O2 evolution efficiency over MOF-53 (Fe) under visible light irradiation in the presence of radical scavengers (0.1 mmol). | |
On the other hand, we performed active species trapping experiments in the reaction system to determine the role of active species generated during photocatalytic water oxidation using the Fe-based MOFs (MIL-53 and NH2-MIL-53). We found that in the presence of EDTA (a typical hole scavenger), there is nearly no evolution of O2. These results reveal that photogenerated holes are the original active species intermediates in the photocatalytic water oxidation. The addition of tert-butyl alcohol (TBA, a ˙OH scavenger) led to a significant decrease in the photocatalytic water oxidation. The photocatalytic decomposition efficiencies of TBA are about 35% and 41.1% for MIL-53 and NH2-MIL-53, respectively. The results reveal that ˙OH is the other active species.
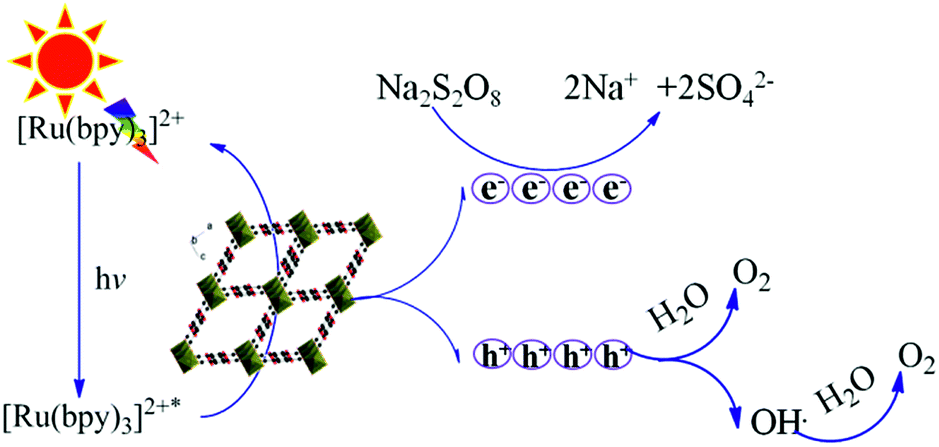
On the basis of the active species trapping experiments and the properties of the Fe-based MOFs, a possible visible-light photocatalytic mechanism of MIL-53 (Fe) and NH2-MIL-53 (Fe) is shown in Fig. 13. [Ru(bpy)3]2+ is the sensitizer, which absorbs visible light and generates [Ru(bpy)3]2+*. [Ru(bpy)3]2+* transfers the energy to the Fe-based MOFs and returns to the [Ru(bpy)3]2+ state, which leads to the formation of photogenerated holes in the VB of the Fe-based MOFs. The photo-generated holes with strong oxidizing ability can oxidise water directly, or react with H2O to generate ˙OH which would produce O2 subsequently. Meanwhile, the photogenerated electrons are accepted by Na2S2O8 (electron acceptor). Generally, the photocatalytic activity increases with decreasing band gaps of Fe-based MOFs. According to UV-vis DRS spectra, the band gap energies of MIL-53 (Fe) and NH2-MIL-53 (Fe) are 2.72 eV and 2.59 eV, respectively. Due to the narrower band gap, NH2-MIL-53 (Fe) exhibits higher activity for water oxidation.
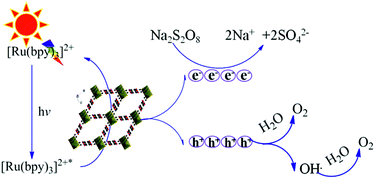 |
| | Fig. 13 The proposed mechanism of the Fe-based MOFs (MIL-53 (Fe) and NH2-MIL-53 (Fe)) for visible-light photocatalytic water oxidation. | |
Conclusions
In summary, Fe-based MOFs (MIL-53 and NH2-MIL-53) display photocatalytic activity for water oxidation under visible-light irradiation. Moreover, the photogenerated holes and hydroxyl radicals (˙OH) are possibly responsible for the photocatalytic water oxidation activity of the Fe-based MOFs. For the first time, we propose that the amine-substituted NH2-MIL-53 (Fe) can decrease the band gap energy and efficiently separate the photo-excited electron–hole pairs, which are beneficial for enhanced photocatalytic performance toward water oxidation. Hence, Fe-based MOFs are promising heterogeneous photocatalysts for water oxidation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21701056 and 21571085), Universities Natural Science Foundation of Jiangsu Province (12KJD150003), and Highly Qualified Professional Initial Funding of Jiangsu University (12JDG052).
References
-
(a) T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC
 ;
(b) S. Berardi, S. Drouet, L. Francas, G. GimbertSurinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC ;
(c) M. Haumann, P. Liebisch, C. Muller, M. Barra, M. Grabolle and H. Dau, Science, 2005, 310, 1019 CrossRef PubMed ;
(d) J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC ;
(e) H. Pan, S. Zhu, X. Lou, L. Mao, J. Lin, F. Tianand and D. Zhang, RSC Adv., 2015, 5, 6543 RSC .
;
(b) S. Berardi, S. Drouet, L. Francas, G. GimbertSurinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC ;
(c) M. Haumann, P. Liebisch, C. Muller, M. Barra, M. Grabolle and H. Dau, Science, 2005, 310, 1019 CrossRef PubMed ;
(d) J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC ;
(e) H. Pan, S. Zhu, X. Lou, L. Mao, J. Lin, F. Tianand and D. Zhang, RSC Adv., 2015, 5, 6543 RSC .
-
(a) D. C. Hong, Y. Yamada, T. Nagatomi, Y. Takaiand and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 19572 CrossRef PubMed ;
(b) H. F. Liu and G. R. Patzke, Chem. Asian J., 2014, 9, 2249 CrossRef PubMed ;
(c) Y. Zhang, J. Rosen, G. S. Hutchings and J. Feng, Catal. Today, 2014, 225, 171 CrossRef .
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CrossRef .
-
(a) A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547 CrossRef PubMed ;
(b) Y. Goto, J. Seo, K. Kumamoto, T. Hisatomi, Y. Mizuguchi, Y. Kamihara, M. Katayama, T. Minegishi and K. Domen, Inorg. Chem., 2016, 55, 3674 CrossRef PubMed .
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef PubMed .
-
(a) Y. Zhang, E. C. Judkins, D. R. McMillin, D. Mehta and T. Ren, ACS Catal., 2013, 3, 2474 CrossRef ;
(b) J. Y. Han, D. P. Wang, Y. H. Du, S. B. Xi, J. D. Hong, S. M. Yin, Z. Chen, T. H. Zhou and R. Xu, J. Mater. Chem. A, 2015, 3, 20607 RSC ;
(c) S. Yusuf and F. Jiao, ACS Catal., 2012, 2, 2753 CrossRef ;
(d) L. L. Qu, J. Cai and Q. Y. Chen, RSC Adv., 2016, 6, 14416 RSC .
-
(a) M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem.–Eur J., 2007, 13, 5106 CrossRef PubMed ;
(b) C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942 CrossRef PubMed ;
(c) L. M. Yang, G. Y. Fang, J. Ma, E. Ganz and S. S. Han, Cryst. Growth Des., 2532, 2014, 14 Search PubMed .
-
(a) K. G. M. Laurier, F. Vermoortele, R. Ameloot, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, J. Am. Chem. Soc., 2013, 135, 14488 CrossRef PubMed ;
(b) D. K. Wang, R. K. Huang, W. J. Liu, D. R. Sun and Z. H. Li, ACS Catal., 2014, 4, 4254, DOI:10.1021/cs501169t ;
(c) C. H. Zhang, L. H. Ai and J. Jiang, J. Mater. Chem. A, 2015, 3, 3074 RSC ;
(d) T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC ;
(e) R. W. Liang, F. F. Jing, L. J. Shen, N. Qin and L. Wu, J. Hazard. Mater., 2015, 287, 364 CrossRef PubMed ;
(f) Z. W. Yang, X. Q. Xu, X. X. Liang, C. Lei, Y. L. Wei, P. Q. He, B. L. Lv, H. C. Ma and Z. Q. Lei, Appl. Catal., B, 2016, 198, 112 CrossRef .
- L. Shi, T. Wang, H. B. Zhang, K. Chang, X. G. Meng, H. M. Liu and J. H. Ye, Adv. Sci., 2015, 2, 1500006 CrossRef PubMed .
-
(a) H. Khajavi, J. Gascon, J. M. Schins, L. D. A. Siebbeles and F. Kapteijn, J. Phys. Chem. C, 2011, 115, 12487 CrossRef ;
(b) Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem. Int. Ed., 2012, 51, 3364 CrossRef PubMed ;
(c) J. H. Choi, Y. J. Choi, J. W. Lee, W. H. Shin and J. K. Kang, Phys. Chem. Chem. Phys., 2009, 11, 628 RSC ;
(d) B. Civalleri, F. Napoli, Y. Noel, C. Roetti and R. Dovesi, CrystEngComm, 2006, 8, 364 RSC ;
(e) L. J. Shen, R. W. Liang, M. B. Luo, F. F. Jing and L. Wu, Phys. Chem. Chem. Phys., 2015, 17, 117 RSC .
-
(a) Y. Horiuchi, T. Toyao, K. Miyahara, L. Zakary, D. D. Van, Y. Kamata, T. Kim, S. W. Lee and M. Matsuoka, Chem. Commun., 2016, 52, 5190 RSC ;
(b) L. Chi, Q. Xu, X. Liang, J. Wang and X. Su, Small, 2016, 10, 1351 CrossRef PubMed .
-
(a) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. A. Vallet-Regí, M. Sebban, F. Taulelle and G. R. Férey, J. Am. Chem. Soc., 2008, 130, 6774 CrossRef PubMed ;
(b) Y. K. Seo, J. W. Yoon, J. S. Lee, U. H. Lee, Y. K. Hwang, C. H. Jun, P. Horcajada, C. Serre and J. S. Chang, Microporous Mesoporous Mater., 2012, 157, 137 CrossRef .
- V. Colombo, C. Montoro, A. Maspero, G. Palmisano, N. Masciocchi, S. Galli, E. Barea and J. A. Navarro, J. Am. Chem. Soc., 2012, 134, 12830 CrossRef PubMed .
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabres i Xamena and H. Garcia, Chem.–Eur J., 2007, 13, 5106 CrossRef PubMed .
- G. T. Vuong, M. H. Pham and T. O. Do, Dalton Trans., 2012, 42, 550 RSC .
- W. Yue, S. L. Suraru, D. Bialas, M. Müller and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 6159 CrossRef PubMed .
-
(a) Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem. Int. Ed., 2012, 51, 3364 CrossRef PubMed ;
(b) L. J. Shen, S. J. Liang, W. M. Wu, R. W. Liang and L. Wu, Dalton Trans., 2013, 42, 13649 RSC .
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jun
Wang
a,
Tian-Yi
Xu
b,
Qiu-Yun
Chen
a,
Jun
Wang
a,
Tian-Yi
Xu
b,
Qiu-Yun
Chen