
Flexible and non-volatile redox active quasi-solid state ionic liquid based electrolytes for thermal energy harvesting†
Abuzar
Taheri
 a,
Douglas R.
MacFarlane
b,
Cristina
Pozo-Gonzalo
a and
Jennifer M.
Pringle
*a
a,
Douglas R.
MacFarlane
b,
Cristina
Pozo-Gonzalo
a and
Jennifer M.
Pringle
*a
aARC Centre of Excellence for Electromaterials Science, Deakin University, 221 Burwood Highway, Burwood, VIC 3125, Australia. E-mail: j.pringle@deakin.edu.au
bSchool of Chemistry, Monash University, Wellington Road, Clayton, VIC 3800, Australia
First published on 28th June 2018
Abstract
The conversion of low-grade thermal energy to electricity using thermoelectrochemical cells (TECs) is a promising route to sustainable electricity production. Developing a leak-free and flexible electrolyte is an important requirement for increasing the safety of these energy harvesting devices. Here, the addition of polymers to non-volatile redox active ionic liquid electrolytes has been investigated. Polyvinylidene difluoride (PVDF) or poly(vinylidene fluoride-co-hexafluoropropene) (PVDF-HFP) was added to an ionic liquid electrolyte, consisting of a [Co(bpy)3]2+/3+ redox couple in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2mim][NTf2]), to produce either soft gels or free-standing films depending on the polymer content. These quasi-solid state electrolytes have a Seebeck coefficient as high as the liquid electrolyte. The effect of gelation, with different amounts of polymer, on the diffusion coefficient of the redox species is reported. Finally, the first thermal energy harvesting performance of a non-volatile, flexible redox active quasi-solid state electrolyte is demonstrated. The quasi-solid state electrolytes allow use of a much thinner electrolyte layer while still maintaining more than half of the power output of the best ionic liquid electrolyte TEC for the same temperature gradient.
Introduction
The harvesting of low-grade thermal energy is one approach to meeting the increasing need for clean energy, but this approach is still underutilised compared to, for example, solar or wind technologies. An increasingly promising device under development is the thermoelectrochemical cell (TEC), also known as a thermocell, which converts thermal energy directly to electricity by utilising the temperature dependence of a redox couple within an electrolyte.1–4 A potential difference (ΔV) is produced across the cell by holding the two electrodes at different temperatures, creating ΔT. The open-circuit potential is dependent on the Seebeck coefficient, Se, of the redox couple, which is determined by the entropy change (ΔS) of the redox reaction: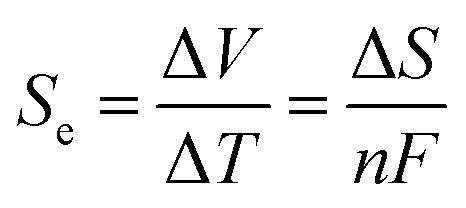 | (1) |
The choice of solvent in the TEC is important not only because of the impact on Se, as introduced above, and power output, but also because it affects its safety and applicability. For these reasons, a non-flammable, non-volatile and leak-free electrolyte is highly desirable. While the volatility can be overcome, to some extent, by using high boiling point organic solvents, flammability can remain a problem. Ionic liquids (ILs) have been extensively investigated for use in electrochemical devices because they can possess high thermal stability, non-flammability and non-volatility.11–14 The application of ILs in TECs therefore provides an opportunity to increase the operating temperature of the device to access higher temperature heat sources in the 100–150 °C range.13,15–18
In addition to improving the thermal stability, using redox couples in ILs instead of molecular solvents can affect the entropy change of the redox reaction and, thus, Se. For example, study of a range of iron, ruthenium and nickel couples in different ILs achieved Se as large as −1.47 mV K−1 and showed that the Se can depend on the charge of the redox couple and the charge density of the IL ions.18 Measurement of Se of various chromium and iron complexes in N-butyl-N-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ([C4mpyr][NTf2]) also showed a dependence on the size and charge of the redox species, and Se as large as 0.43 mV K−1 and −1.49 mV K−1, respectively.19 Note that the sign of Se depends on if the more oxidised species of the redox couple has a larger absolute charge than the more reduced species, or vice versa. More recently, using a cobalt-based redox couple, [Co(bpy)3]2+/3+[NTf2−]2/3 (where bpy = 2,2′-bipyridyl, NTf2 = bis(trifluoromethylsulfonyl)amide) achieved a higher Se (up to 2.2 and 1.9 mV K−1 in organic solvents or IL, respectively), attributed to additional effects from the change in spin state of the Co2+/3+ species during the redox reaction.16,20–22 A further advantage to the [Co(bpy)3]2+/3+[NTf2]2/3 redox couple is that it provides a positive Se, allowing it to be used in TEC arrays in combination with a negative Se redox couple such as K3[Fe(CN)6]/K4[Fe(CN)6],23,24 in a design analogous to that of p-type and n-type semiconductors in thermoelectric generators.25
However, despite these recent advances and the advantages of IL electrolytes, liquid electrolytes have an inherent potential leakage problem that limits their application in devices such as TECs. The solidification of liquid electrolytes by gelation is a viable method for preparing a safer and leak-free electrolyte. Further, the use of quasi-solid state electrolytes in TECs can reduce the problem of heat transfer within the cell, due to convection, which is detrimental to the ΔT being harvested and thus to the power output. To-date this approach has only been explored using hydrogels, where aqueous electrolytes have been gelled using polymers such as cellulose,26 polyvinyl alcohol (PVA),23 and poly (sodium acrylate),27 or ion exchange membranes such as Nafion,28 or Nepton (a condensation product of phenol sulphonic acid/formaldehyde).29
The application of ILs in TECs has provided an opportunity to increase the operating temperature of TECs and achieve a higher power output.2 The gelation of IL-based redox electrolytes using polymers could provide the combined benefits of non-volatility, reduced thermal convection and no solvent leakage. However, this requires the use of alternative polymers to those suitable for hydrogel formation. For the preparation of polymer-based electrolytes, achieving good flexibility and miscibility between the IL and polymer is important and it has been shown that PVDF and PVDF-HFP, in combination with [C2mim][NTf2], can form free-standing membranes with a good mechanical properties.30–35
Much of the prior work looking at the combination of ILs with PVDF or PVDF-HFP has focussed on developing electrolytes for actuators,30,33,36 or lithium-conducting electrolyte materials for batteries.35,37 The combination of [C2mim][NTf2], PVDF-HFP and zinc triflate has also been investigated for application in zinc batteries.38 One of the challenges in developing a quasi-solid state electrolyte for TECs is achieving the mechanical properties sufficient to prevent solvent leakage while also maintaining sufficient transport of the redox couple, which in this case is a relatively large size. However, there is a paucity of studies that report the gelation of ILs containing redox couples. The formation of quasi-solid state IL electrolytes containing the I−/I3− redox couple, for dye-sensitised solar cell (DSSC) applications, has been reported.39–41 Prior research in the DSSC field has also shown that PVDF or PVDF-HFP can be used to gel [Co(bpy)3]2+/3+[NTf2−]2/3 in methoxypropionitrile (MPN) or acetonitrile.42,43
We have recently investigated the use of these polymers to form soft gels with this redox couple in MPN for thermal energy harvesting.44 However, while this approach successfully eliminates leakage concerns, the volatility associated with the use of MPN remain, and the gels in this case were still too soft to form free-standing films. Here, we advance this electrolyte development in two ways, by developing two different kinds of quasi-solid state electrolytes with negligible vapour pressure (i) ionic liquid-based electrolyte gels, and (ii) ionic liquid-based freestanding solid-state electrolyte films. These two different kinds of electrolyte were developed in each case using the minimum amount of polymer within the IL, to achieve either a gel or a freestanding film. The ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2mim][NTf2]) was used as it provides among the highest power outputs of the different ILs tested to-date and is also widely available commercially. This IL was used in combination with the redox couple and polymers polyvinylidene difluoride (PVDF) or poly(vinylidene fluoride-co-hexafluoropropene) (PVDF-HFP) to form the quasi-solid state electrolytes. The effect of polymer type and content on the mechanical properties, diffusion coefficient, Se and TEC performance of the quasi-solid state electrolytes is reported.
Experimental
Materials
Poly(vinylidene fluoride-co-hexafluoropropene) (PVDF-HFP) powder (Mw = 3.13 × 105 from Solvay, Belgium), and polyvinylidene difluoride (PVDF) powder (KF850, Mw = 3 × 105 from Kureha Chemicals, Japan) were used as received. 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2mim][NTf2]) was purchased from Merck (purity ≥ 98%), and used as received. The redox couple [Co(bpy)3][NTf2]2/[Co(bpy)3][NTf2]3 was synthesised following our previously published procedure.16To prepare the redox active electrolytes, equimolar amounts of [Co(bpy)3][NTf2]2 and [Co(bpy)3][NTf2]3 were dissolved in [C2mim][NTf2]. For example, to prepare 5 ml of 0.05 M solution of [Co(bpy)3]2+/3+[NTf2−]2/3 in [C2mim][NTf2], [Co(bpy)3][NTf2]2 (271 mg, 0.25 mmol) and [Co(bpy)3][NTf2]3 (341 mg, 0.25 mmol) were mixed and dissolved in the minimum amount of [C2mim][NTf2] by sonication, then the total volume of solution was taken to 5 ml by addition of [C2mim][NTf2].
For the two different kinds of quasi-solid state electrolyte developed here, the following procedures were followed. A minimum 2.5 wt% of polymer was needed to gel the ionic liquid electrolyte. For gelation of the ionic liquid electrolyte, 25 mg of polymer powder (PVDF or PVDF-HFP) was added to 1 gram of liquid electrolyte, and then the resulting mixture was heated at 120 °C and stirred in air at this temperature for 30 minutes to dissolve the powder. The clear solution was left at room temperature for 1 hour to get a polymer-based gel.
The free standing polymer film was prepared by solvent casting using the minimum amount of polymer (18 wt%). PVDF or PVFD-HFP (180 mg) was added to 1 gram of liquid electrolyte (0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2]), following by addition of 3 ml of acetone. The resulting mixture was stirred in air at 50–55 °C to get a clear solution. The solution was cast in a Petri dish, left overnight to evaporate the solvent, and then dried under vacuum for 2 hours at 50 °C. Using this method, 0.2 and 1 mm thick PVDF films, and 0.2 mm thick PVDF-HFP films, measured by callipers, were prepared.
Thermoelectrochemistry
The Seebeck coefficient of [Co(bpy)3]2+/3+[NTf2]2/3 redox couple was measured using a non-isothermal cell. To measure the Seebeck coefficient of the redox couple in the liquid or gelled electrolyte, a non-isothermal H-cell setup equipped with two platinum wires as electrodes was used. A heating foil and a water bath were used to control the temperature of two legs of the H-cell. A UNI-T UT803 TRMS bench voltameter was used to measure the potential difference.To measure the Seebeck coefficient of the redox couple in the freestanding polymer films, two platinum disks were fixed on a heating block and a Peltier cooler and the polymer-film placed in-between. The temperature of cold side was set to 20 °C, while the other side was increased by 5 °C increments and the potential difference recorded using the same potentiometer as above.
A platinum working electrode (1.6 mm diameter, ASL Japan) and two platinum wires (XRF, Australia) in a three-electrode cell were used for cyclic voltammetry (CV) scans between −1 V and +1 V at different scan rates (25 to 125 mV s−1). The Cottrell equation45 was used to calculate the diffusion coefficient of the [Co(bpy)3]2+/3+ ions in the liquid or polymer-based electrolytes: a potential step (−1 or +1 V) was applied for 10 seconds, after which the oxidative or reductive process was under diffusion control, and the current was recorded.
The performance of the TEC was studied using a Teflon cell with a 9 mm internal diameter representing the active electrode area. The Teflon cell was sandwiched between two platinum disks (18 mm diameter) with a cell orientation that had both electrodes vertical. A TE Technology cold plate cooler CP- 031, and a cartridge heater inside a copper block, connected to a Manson NP-9613 DC regulated power supply were used to control the temperature of the cold and hot electrodes, respectively. The temperatures were measured using sensors connected to Novous NI020 temperature controllers, with an accuracy of ± 0.1 °C. The CVs and chronoamperometry were performed using BioLogic VMP3 multichannel potentiostat driven by EC-Lab software. The performance of the cell was measured using a Bio-Logic SP-200 and EC-Lab software. Constant Load Discharge (CLD) technique was used to apply different resistances and measure the power and current output. Enough time (60 minutes) was given at each resistance to get a constant voltage and power output. To calculate the power and current density, the averaged final 60 seconds power and current data at each resistance were divided by the working area of one electrode. Longer term testing (Fig. 5) was performed at Pmax by applying the external resistance that corresponds to the maximum power of the electrolyte (determined by the maximum in Fig. 4). The power vs. time recording was started (t = 0) after the standard initial equilibration time was applied (60 minutes).
Results and discussion
Preparation of the quasi-solid state electrolytes
A gelled electrolyte was produced by addition of a minimum of 2.5 wt% PVDF or PVDF-HFP to the 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2mim][NTf2]) electrolyte (Fig. 1a and b). Gelation of the electrolyte was verified through rheology measurements, which showed a higher storage modulus (G′) compared to loss modulus (G′′) in the strain sweep (Fig. S1†). The temperature sweep, which showed nearly constantly higher storage modulus than loss modulus, indicates maintenance of the leak-free gel with both PVDF and PVDF-HFP from 25 to 70 °C, which is promising for application in the TEC operating over this temperature range. | ||
| Fig. 1 Quasi-solid state electrolytes of 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2], with: (a) 2.5 wt% PVDF, (b) 2.5 wt% PVDF-HFP, (c) 18 wt% PVDF and (d) 18 wt% PVDF-HFP, and the mechanical properties of the polymer films with: (e and f) 18 wt% PVDF, (g and h) 18 wt% PVDF-HFP. | ||
To get a flexible and free standing polymer-based film electrolyte, 18 wt% of PVDF or PVDF-HFP was required (Fig. 1c and d). Both PVDF and PVDF-HFP based films were free standing and flexible, but the PVDF-HFP film showed more elasticity (Fig. 1e–h). This is supported by the dynamic mechanical analysis (DMA) measurements, which show that the PVDF-HFP film with lower Young's modulus (5.19 ± 0.86 kPa) than PVDF film (7.98 ± 0.31 kPa) has elastic behaviour, and even at higher strain does not break (Fig. S2†).
Thermal analysis (Fig. S3†) shows that the PVDF based electrolytes, in either gel or film form, have higher melting points than the PVDF-HFP electrolyte. The PVDF gel and PVDF film melt at temperatures above 110 °C, which is promising for TEC applications.
Seebeck coefficient and electrochemical behaviour
The interaction between the redox couple and the surrounding solvent affects the entropy change of the redox reaction and thus the Seebeck coefficient. Thus, it is interesting to investigate the effect of the addition of polymer on Se. A prior report on the development of hydrogels with the Fe(CN)63−/4− redox couple showed a slight decrease in the absolute magnitude of Se, from −1.4 mV K−1 for the liquid (0 wt% cellulose) to −1.32 mV K−1 for 20 wt% cellulose gel.26The effect of PVDF and PVDF-HFP on the Se of the [Co(bpy)3]2+/3+[NTf2−]2/3 in [C2mim][NTf2] is shown in Table 1. The absence of any significant change in Se upon gelation of the IL-based electrolytes in this study suggests that the interaction between the redox couple and the IL is sufficiently strong that the solvation shell is largely unaffected by polymer addition. Thus, the high Se of this redox couple is maintained upon gelation, which is highly advantageous for the development of TECs with the quasi-solid state electrolytes.
| Electrolyte with 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 | Seebeck coefficient (mV K−1) | Diffusion coefficient (D × 107 cm2 s−1) | |
|---|---|---|---|
| [Co(bpy)3]2+ | [Co(bpy)3]3+ | ||
| [C2mim][NTf2] | 1.50 ± 0.01 | 1.81 ± 0.07 | 1.63 ± 0.10 |
| 2.5 wt% PVDF/[C2mim][NTf2] | 1.51 ± 0.02 | 1.65 ± 0.06 | 1.66 ± 0.07 |
| 18 wt% PVDF/[C2mim][NTf2] | 1.53 ± 0.02 | 0.87 ± 0.06 | 0.67 ± 0.03 |
| 2.5 wt% PVDF-HFP/[C2mim][NTf2] | 1.52 ± 0.02 | 1.75 ± 0.06 | 1.56 ± 0.07 |
| 18 wt% PVDF-HFP/[C2mim][NTf2] | 1.56 ± 0.01 | 0.61 ± 0.05 | 0.57 ± 0.06 |
Cyclic voltammetry (CV) of the polymer-based electrolytes (Fig. 2), using a three-electrode cell, was performed at different scan rates to assess the impact of polymer on the electrochemical properties of the quasi-solid state electrolytes, prior to full thermoelectrochemical cell testing. This shows that the cobalt redox couple in the quasi-solid state electrolytes has stable cycling performance and quasi-reversible behaviour. However, there is a wider peak-to-peak separation in the freestanding films, which have a higher polymer content than the gels. This reflects a significant increase in resistance, primarily attributed to a larger mass transfer resistance in the electrolyte films. In addition, a lower current was measured in PVDF-HFP systems compared to the PVDF systems, which is consistent with the lower diffusion coefficients (Table 1).
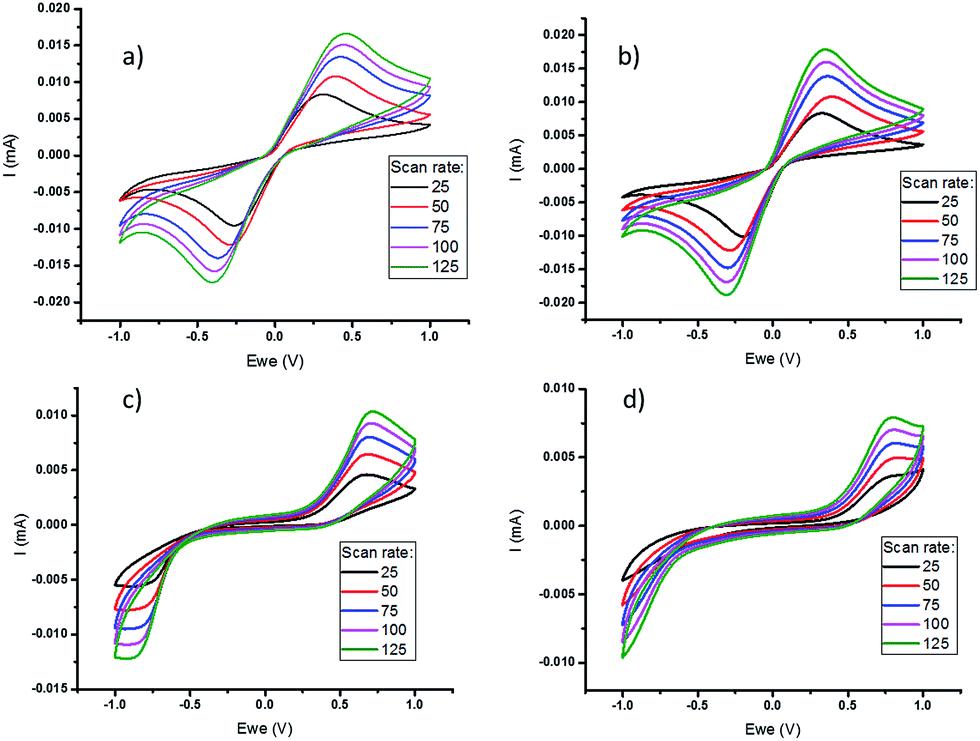 | ||
Fig. 2 Cyclic voltammetry of 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2] with (a) 2.5 wt% PVDF, (b) 2.5 wt% PVDF-HFP, (c) 18 wt% PVDF and (d) 18 wt% PVDF-HFP at different scan rates ( 25, 25,  50, 50,  75, 75,  100 and 100 and  125 mV s−1), using a three-electrode cell equipped with a platinum working electrode (1.6 mm diameter) and two platinum wires as the counter and reference electrodes. 125 mV s−1), using a three-electrode cell equipped with a platinum working electrode (1.6 mm diameter) and two platinum wires as the counter and reference electrodes. | ||
Measurement of the diffusion coefficient of the cobalt redox species by chronoamperometry shows that the formation of a soft gel with either PVDF or PVDF-HFP does not significantly decrease the diffusivity (Table 1). However, after formation of the free standing polymer film, upon increasing the amount of polymer, there is a notable decrease in diffusion coefficient. The diffusion coefficient is also slightly lower in the PVDF-HFP systems compared to the PVDF, possibly indicating some degree of interaction between the cationic redox couple and the HFP chains in the PVDF-HFP.46
Thermoelectrochemical cell (TEC) performance
During TEC operation, the two electrodes are maintained at different temperatures, creating a potential difference across the cell as a result of the temperature dependence of the electrochemical potential of the redox couple. Before current is drawn, this potential difference is represented by the open circuit voltage. The power and current output of the cell is determined by applying a sequence of external resistances across the cell and measuring the voltage. The highest power output is produced when the applied external resistance is equal to the internal resistance of the cell. The total resistance of the cell is the sum of the activation, ohmic and mass transport resistances and, as discussed below, the latter can be the most significant resistance when viscous electrolytes are used. The performance of the TEC with the polymer-based electrolytes was measured using cold and hot electrode temperatures of Tcold = 20 °C and Thot = 60 °C, respectively, and the power and current output is shown in Fig. 3 and 4.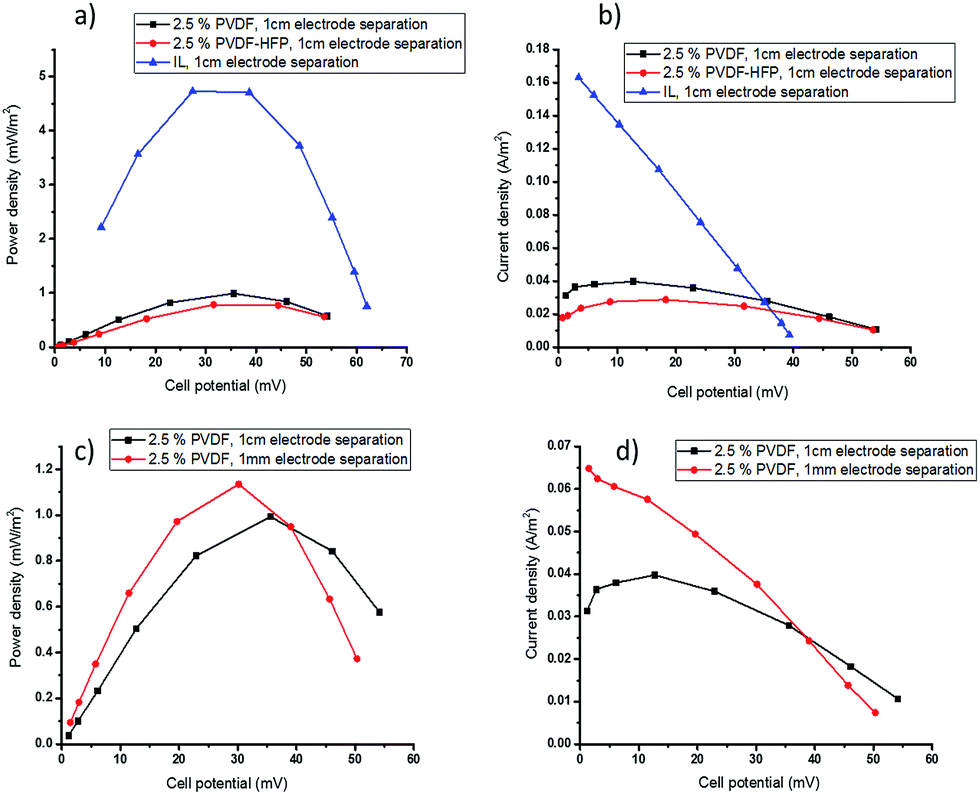 | ||
| Fig. 3 TEC performance with 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2] electrolytes, Tcold = 20 °C, Thot = 60 °C, (a) power density and (b) current density of TEC gelled with 2.5 wt% PVDF or PVDF-HFP (electrode separation = 1 cm), (c) power density and (d) current density with electrode separation 1 cm or 1 mm. | ||
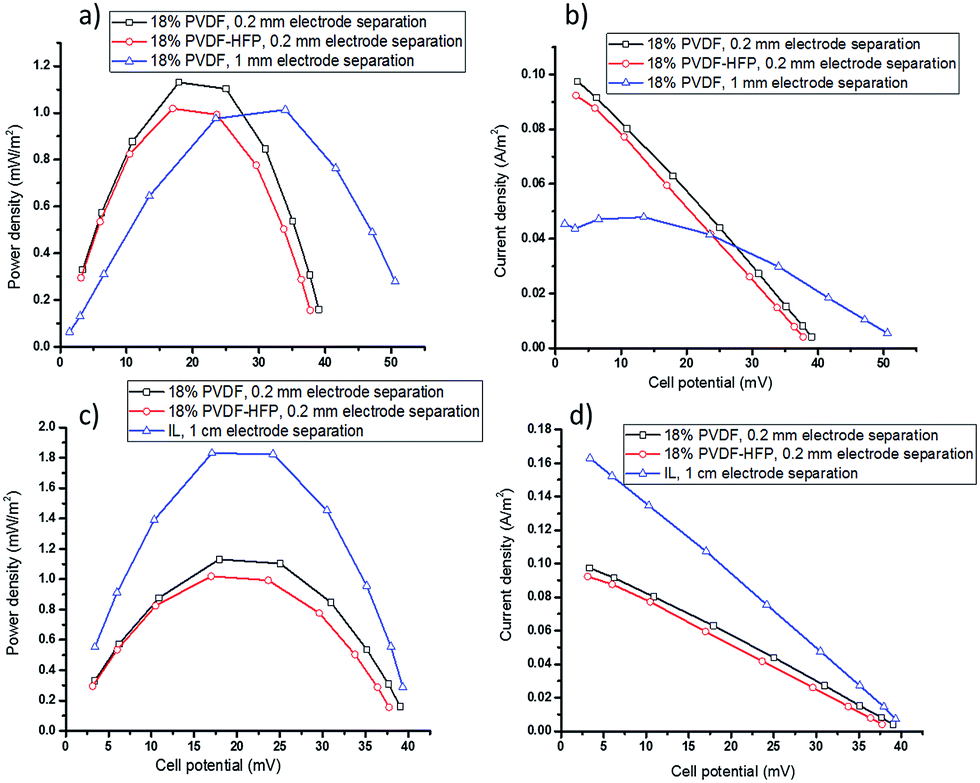 | ||
| Fig. 4 TEC performance with 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2] electrolytes, (a) power density and (b) current density of TEC gelled with 18 wt% PVDF or PVDF-HFP, Tcold = 20 °C, Thot = 60 °C, electrode separation = 0.2 mm or 1 mm; (c) power density and (d) current density of IL electrolyte with applied Tcold = 20 °C, Thot = 46 °C, and polymer film electrolyte with applied Tcold = 20 °C, Thot = 60 °C, giving actual ΔT = 26 °C. | ||
Gelation with 2.5 wt% polymer reduces the Pmax from 4.80 ± 0.12 mW m−2 for the ionic liquid electrolyte to 0.98 ± 0.05 and 0.78 ± 0.06 mW m−2 (Fig. 3a) for PVDF or PVDF-HFP respectively. This drop is attributed to a significant decrease in mass transport rates of the redox couple. The observed decrease in current density at low potentials in the current density–potential plots (Fig. 3b) also indicates mass transfer limitations in the gelled electrolyte. In TECs, the transport rate of a redox couple in the ionic liquid electrolyte is the sum of convection, diffusion and migration effects. However, in the quasi-solid state electrolytes it is limited to transfer by diffusion and migration; the decreased convection upon solidification of the electrolyte reduces the overall mass transport of the redox couple and can significantly decrease the power output.47 The results also show that using PVDF gel electrolyte gives better performance compared to the PVDF-HFP. This is consistent with the slightly slower diffusivity of the redox species in the latter gel (Table 1).
To ameliorate the mass transport limitations in the gelled electrolytes, the effect of decreasing the distance between the two electrodes was investigated. The practical limitation to this strategy is that it must remain possible to maintain the same temperature gradient across the thin cell as the thicker cell. The elimination of parasitic heat transfer by convection, by electrolyte solidification, should benefit this approach.
By decreasing the electrode separation from 1 cm to 1 mm, the amount of electrolyte required was significantly decreased, which is an additional benefit of these solid electrolytes. The smaller open circuit voltage (Voc) in the thinner cell (Fig. 3c and d) indicates that using smaller electrode separation did result in a decrease in the temperature gradient across the cell, from ∼40 °C to ∼35 °C. As a result, the power density only increased slightly with the thinner cell, from 0.98 ± 0.05 to 1.18 ± 0.09 mW m−2 (Fig. 3c), even though a significant improvement in current density at low potentials was achieved due to the mass transfer improvement (Fig. 3d).
For further optimisation of the electrolyte and cell design, use of the free-standing polymer film electrolytes to enable thinner cells was investigated. The power and current output of cells with 18 wt% PVDF or PVDF-HFP is shown in Fig. 4a and b. At 1 mm electrode separation, increasing the polymer content from 2.5 wt% to 18 wt% results in a decrease in Pmax from 1.20 ± 0.05 (Fig. 3c) to 1.01 ± 0.04 (Fig. 4a), as expected based on the lower diffusion coefficient of the redox couple in the latter (Table 1). However, increasing the polymer content is beneficial as it allows for further reductions in electrolyte thickness, described below. The formation of flexible, freestanding films will also be very advantageous for development of flexible devices, e.g. for wearable applications or wrapping around hot pipes.
The performance of a TEC containing even thinner (0.2 mm) PVDF film electrolytes with applied Tcold = 20 and Thot = 60 °C is also shown in Fig. 4a and b. This gave higher currents and similar powers compared to the 1 mm cell with either the PVDF gel or film electrolyte. Using 18 wt% PVDF-HFP film with the thickness of 0.2 mm in TEC produces lower power and current density compared to PVDF film electrolyte with the same thickness. This observation is consistent with the above results obtained using PVDF and PVDF-HFP gel electrolytes and with the lower diffusion coefficient. However, the smaller open circuit voltage of the 0.2 mm films indicates that with the smaller electrode separation the temperature gradient had been decreased to around 26 °C (determined by Voc divided by Se: 40/1.53 = 26). Overall, however, this does not result in a decrease in the output power.
To make a definitive comparison between the TEC with the ionic liquid electrolyte and those with the new, thinner polymer film electrolytes, the power and current output of a 1 cm TEC with liquid electrolyte was measured using an applied Thot = 46 °C and Tcold = 20 °C (ΔT = 26 °C), respectively (Fig. 4c and d), thus replicating the actual ΔT in the thin film TEC. The results demonstrate that with the same temperature gradient, 60% of the power generation of the liquid electrolyte TEC can be produced by using the new PVDF films with a thickness of 0.2 mm, but with the significant advantage of much less electrolyte and a leak-free, flexible electrolyte design.
Finally, the long-term performance of the new, thinner TECs was assessed. It should be noted that in a TEC with two electrodes held at constant (different) temperatures, the conversion of thermal energy into electricity is continuous: the cell does not discharge in the way that a battery does. However, any decomposition of the electrolyte or electrodes would affect the longer-term performance. To assess the durability of the TEC with the highest performing quasi-solid state electrolytes, the free standing 0.2 mm films, the cell was operated at Pmax for 13 hours. These longer-term tests show that both thin PVDF and PVDF-HFP films have a promising performance, with less than 4% and 7% decrease, respectively, in power output (Fig. 5). Rather than only reflecting electrolyte decomposition, some of this power drop can likely be attributed to the Soret effect, which is electrolyte migration as a result of thermal diffusion, and hence the decay rate decreases after the first couple of hours.48 It is known that Soret diffusion equilibration in solid electrolytes can take hours to stabilize,28 and this is a more obvious effect in thicker solid electrolytes. Comparing the longer term performance of the 0.2 mm films with that of 1 mm PVDF film also shows that a thicker film takes longer to produce a stable power and current output, because of the larger mass transport limitations. Finally, the TEC with a thin PVDF film yields higher power and current than either the thick PVDF film (1 mm) or the thin PVDF-HFP film (0.2 mm), as result of having superior mass transport properties. These initial results, plus prior studies from the DSSC field that indicate good stability for the [Co(bpy)3]2+/3+ couple (91% efficiency maintained after 2000 hours light soaking using an MPN-based electrolyte),49 are very promising indicators for the long-term stability of the IL-based quasi-solid state TECs.
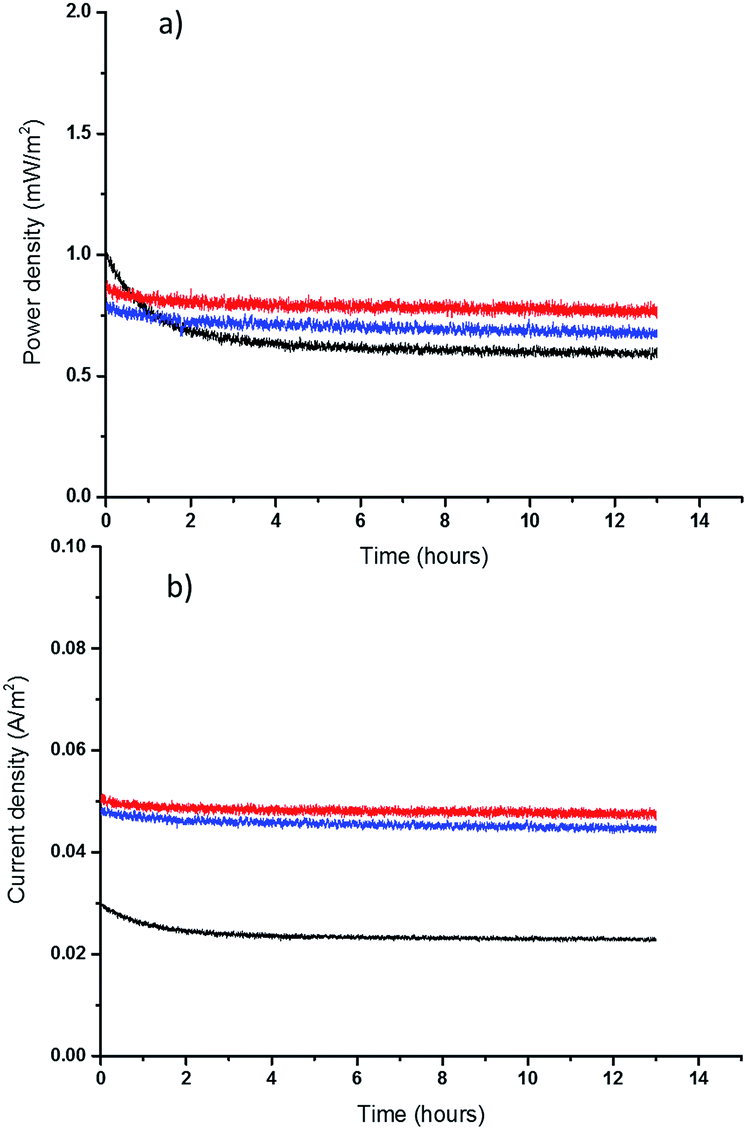 | ||
Fig. 5 (a) Power density and (b) current density of a TEC containing polymer-based film electrolyte; 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2] with: 18 wt% PVDF ( ) or PVDF-HFP ( ) or PVDF-HFP ( ) with the thickness of 0.2 mm, and 18 wt% PVDF ( ) with the thickness of 0.2 mm, and 18 wt% PVDF ( ) with thickness of 1 mm (Tcold = 20 °C, Thot = 60 °C). ) with thickness of 1 mm (Tcold = 20 °C, Thot = 60 °C). | ||
Conclusions
Here we report the first flexible and non-volatile solid-state electrolyte for thermal energy harvesting, achieved by solidification of an ionic liquid electrolyte containing a redox couple. Using PVDF or PVDF-HFP for gelation of the Co(bpy)3 redox couple in an IL electrolyte, leak-free electrolytes that are suitable for use in flexible TEC devices were prepared. The gelation did not significantly affect the Seebeck coefficient, but did decrease the diffusivity of the cobalt redox couple at higher polymer content. The mechanical properties of the PVDF-HFP film electrolyte in terms of flexibility and elasticity were better than with PVDF, but the performance of the TEC with PVDF-based electrolytes was slightly better as a result of the higher redox couple diffusion rate.In the TEC, gelation of the electrolyte caused a decrease in power output, attributed to decreased mass transport. However, this was addressed by using thinner electrolyte films, a strategy aided by the decreased convective heat transfer in the quasi-solid state electrolytes. Using a 0.2 mm thick PVDF polymer electrolyte film, with 0.05 M [Co(bpy)3]2+/3+[NTf2]2/3 in [C2mim][NTf2], the TEC achieved 60% of the power generation of the best 1 cm thick ionic liquid electrolyte cell at the same temperature gradient. Thus, this has the advantage of reduced leakage and electrolyte flexibility as well as requiring much less electrolyte material.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Australian Research Council (ARC) through its Centre of Excellence program (CE140100012) and through the Australian Laureate Fellowship scheme for D. R. M.Notes and references
- B. Burrows, J. Electrochem. Soc., 1976, 123, 154–159 CrossRef.
- M. Dupont, D. MacFarlane and J. Pringle, Chem. Commun., 2017, 53, 6288–6302 RSC.
- A. Gunawan, C.-H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Nanoscale Microscale Thermophys. Eng., 2013, 17, 304–323 CrossRef.
- T. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef.
- A. Debethune, T. Licht and N. Swendeman, J. Electrochem. Soc., 1959, 106, 616–625 CrossRef.
- E. Eastman, J. Am. Chem. Soc., 1928, 50, 292–297 CrossRef.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1981, 122, 155–170 CrossRef.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1981, 124, 35–51 CrossRef.
- E. L. Yee, R. J. Cave, K. L. Guyer, P. D. Tyma and M. J. Weaver, J. Am. Chem. Soc., 1979, 101, 1131–1137 CrossRef.
- C. B. Vining, Nat. Mater., 2009, 8, 83 CrossRef PubMed.
- J. D. Decoppet, S. B. Khan, M. S. Al-Ghamdi, B. G. Alhogbi, A. M. Asiri, S. M. Zakeeruddin and M. Grätzel, Energy Technol., 2017, 5, 321–326 CrossRef.
- A. Eftekhari, Y. Liu and P. Chen, J. Power Sources, 2016, 334, 221–239 CrossRef.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- V. V. Singh, A. K. Nigam, A. Batra, M. Boopathi, B. Singh and R. Vijayaraghavan, Int. J. Electrochem., 2012, 2012, 1–19 Search PubMed.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Chem. Commun., 2011, 47, 6260–6262 RSC.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013, 6, 2639–2645 RSC.
- M. Bonetti, S. Nakamae, B. T. Huang, T. J. Salez, C. Wiertel-Gasquet and M. Roger, J. Chem. Phys., 2015, 142, 244708 CrossRef PubMed.
- Y. Yamato, Y. Katayama and T. Miura, J. Electrochem. Soc., 2013, 160, H309–H314 CrossRef.
- T. Migita, N. Tachikawa, Y. Katayama and T. Miura, Electrochemistry, 2009, 77, 639–641 CrossRef.
- J. He, D. Al-Masri, D. R. MacFarlane and J. M. Pringle, Faraday Discuss., 2016, 190, 205–218 RSC.
- N. Jiao, T. J. Abraham, D. R. MacFarlane and J. M. Pringle, J. Electrochem. Soc., 2014, 161, D3061–D3065 CrossRef.
- M. A. Lazar, D. Al-Masri, D. R. MacFarlane and J. M. Pringle, Phys. Chem. Chem. Phys., 2016, 18, 1404–1410 RSC.
- P. Yang, K. Liu, Q. Chen, X. Mo, Y. Zhou, S. Li, G. Feng and J. Zhou, Angew. Chem., 2016, 128, 12229–12232 CrossRef.
- L. Zhang, T. Kim, N. Li, T. J. Kang, J. Chen, J. M. Pringle, M. Zhang, A. H. Kazim, S. Fang and C. Haines, Adv. Mater., 2017, 29, 1605652 CrossRef PubMed.
- M. Hamid Elsheikh, D. A. Shnawah, M. F. M. Sabri, S. B. M. Said, M. Haji Hassan, M. B. Ali Bashir and M. Mohamad, Renewable Sustainable Energy Rev., 2014, 30, 337–355 CrossRef.
- L. Jin, G. W. Greene, D. R. MacFarlane and J. M. Pringle, ACS Energy Lett., 2016, 1, 654–658 CrossRef.
- J. Wu, J. J. Black and L. Aldous, Electrochim. Acta, 2017, 225, 482–492 CrossRef.
- W. B. Chang, C. M. Evans, B. C. Popere, B. M. Russ, J. Liu, J. Newman and R. A. Segalman, ACS Macro Lett., 2015, 5, 94–98 CrossRef.
- W. Grubb, Nature, 1958, 181, 338 CrossRef.
- J. Dias, A. Lopes, B. Magalhaes, G. Botelho, M. M. Silva, J. Esperanca and S. Lanceros-Mendez, Polym. Test., 2015, 48, 199–205 CrossRef.
- K. Hong, Y. K. Kwon, J. Ryu, J. Y. Lee, S. H. Kim and K. H. Lee, Sci. Rep., 2016, 6, 29805 CrossRef PubMed.
- J. C. Jansen, K. Friess, G. Clarizia, J. Schauer and P. Izak, Macromolecules, 2010, 44, 39–45 CrossRef.
- R. Mejri, J. Dias, S. B. Hentati, M. Martins, C. Costa and S. Lanceros-Mendez, J. Non-Cryst. Solids, 2016, 453, 8–15 CrossRef.
- V. Narasimhan and S.-Y. Park, Langmuir, 2015, 31, 8512–8518 CrossRef PubMed.
- H. Ye, J. Huang, J. J. Xu, A. Khalfan and S. G. Greenbaum, J. Electrochem. Soc., 2007, 154, A1048–A1057 CrossRef PubMed.
- Z. Chen, Y. Shen, N. Xi and X. Tan, Smart Mater. Struct., 2007, 16, S262–S271 CrossRef.
- S. Ferrari, E. Quartarone, P. Mustarelli, A. Magistris, M. Fagnoni, S. Protti, C. Gerbaldi and A. Spinella, J. Power Sources, 2010, 195, 559–566 CrossRef.
- J. P. Tafur and A. J. F. Romero, J. Membr. Sci., 2014, 469, 499–506 CrossRef.
- A. Kontos, M. Fardis, M. Prodromidis, T. Stergiopoulos, E. Chatzivasiloglou, G. Papavassiliou and P. Falaras, Phys. Chem. Chem. Phys., 2006, 8, 767–776 RSC.
- P. K. Singh, K. I. Kim, N. G. Park and H. W. Rhee, Macromol. Symp., 2007, 249, 162–166 CrossRef.
- P. Wang, S. M. Zakeeruddin, P. Comte, I. Exnar and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 1166–1167 CrossRef PubMed.
- M. B. Achari, V. Elumalai, N. Vlachopoulos, M. Safdari, J. Gao, J. M. Gardner and L. Kloo, Phys. Chem. Chem. Phys., 2013, 15, 17419–17425 RSC.
- W. Xiang, W. Huang, U. Bach and L. Spiccia, Chem. Commun., 2013, 49, 8997–8999 RSC.
- A. Taheri, D. R. MacFarlane, C. Pozo-Gonzalo and J. M. Pringle, ChemSusChem, 2018 DOI:10.1002/cssc.201800794.
- G. Denuault, M. V. Mirkin and A. J. Bard, J. Electroanal. Chem. Interfacial Electrochem., 1991, 308, 27–38 CrossRef.
- Y. Saito, H. Kataoka, C. Capiglia and H. Yamamoto, J. Phys. Chem. B, 2000, 104, 2189–2192 CrossRef.
- P. F. Salazar, S. Kumar and B. A. Cola, J. Appl. Electrochem., 2014, 44, 325–336 CrossRef.
- J. Goodrich, F. M. Goyan, E. Morse, R. G. Preston and M. Young, J. Am. Chem. Soc., 1950, 72, 4411–4418 CrossRef.
- R. Jiang, A. Anderson, P. R. Barnes, L. Xiaoe, C. Law and B. C. O'Regan, J. Mater. Chem. A, 2014, 2, 4751–4757 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanical and thermal analysis of the electrolytes. See DOI: 10.1039/c8se00224j |
| This journal is © The Royal Society of Chemistry 2018 |