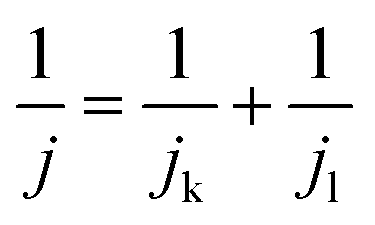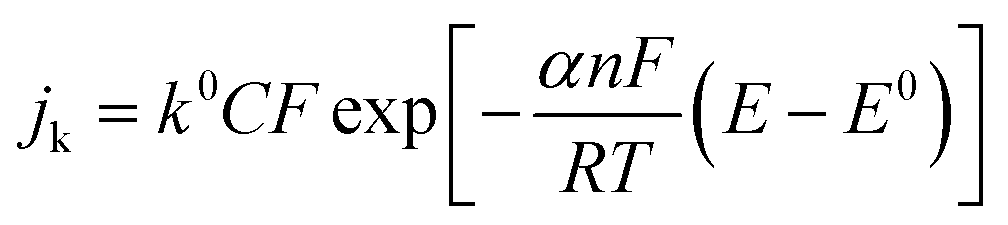A vitamin C fuel cell with a non-bonded cathodic interface†
Received
14th May 2018
, Accepted 18th June 2018
First published on 19th June 2018
Abstract
Vitamin C is a naturally occurring molecule with antioxidant properties, and it often plays a pivotal role in many chemical and biochemical processes. We show that a cobalt-based molecular electrocatalyst can mediate the electron donation from vitamin C which upon coupling with a non-bonded and reversible electron acceptor; the electron flow between the half cells can be channeled in a precious-metal-free configuration. The non-bonded nature of the electron acceptor allows fast interfacial kinetics even on simple carbon particles and arrests the cathode-derived parasitic chemistry often encountered in oxygen breathing fuel cells. Consequently, a vitamin C fuel cell driven by the non-bonded cathodic interface demonstrates ∼18 times higher performance metrics compared to the precious-metal-based vitamin C–O2 configuration. Due to the renewable nature of the fuel and the precious metal-free configuration in the proposed non-bonded architecture, the cell can noticeably reduce the cost of electricity per kW with potential practical applications in powering commercial electrical appliances.
1. Introduction
Electrochemical energy storage and conversion devices have great potential to address the climate change and the adverse effects of anthropogenic pollution.1–9 Fuel cells have received significant attention in this regard as they are potential zero emission energy conversion devices.6–9 The applications of small molecule-based fuel cells such as direct alcohol fuel cells, direct borohydride fuel cells, and direct hydrazine fuel cells are hampered by the safety and storage issues associated with the hydrogen fuel.10–17 Vitamin C is another potential small molecule used in fuel cells because of its abundance in natural resources and low reduction potential.18–24 Vitamin C is often used in many synthetic strategies as a reducing agent owing to its low reduction potential;25–29 it belongs to the category of essential nutrients with antioxidant properties and is used as a medication for many diseases such as scurvy, drug poisoning, liver disease, atherosclerosis, and schizophrenia.30–32 Herein, we show a vitamin C fuel cell where its catalysis is mediated by a cobalt-based molecular electrocatalyst, leading to Pt-free anodic half-cell chemistry. To design a Pt-free fuel cell, we have replaced the conventional electron acceptor O2 by an outer sphere or a non-bonded redox species that can undergo facile electron transfer even on carbon-based electrodes.33–36 The vitamin C fuel cell driven by the non-bonded cathodic interface delivers ∼18 times higher performance metrics compared to precious metal-based vitamin C–O2 fuel cells due to fast anode as well as cathode interfacial kinetics and the absence of parasitic chemistry involving the cathodic interface. Interestingly, the catholyte in the proposed configuration is electrochemically reversible, leading to a hybrid fuel cell redox flow battery system. We further demonstrate the use of a precious-metal-free vitamin C fuel cell to power commercial electrical appliances, suggesting its feasibility for practical applications.
2. Results and discussion
2.1. Anodic half cell chemistry
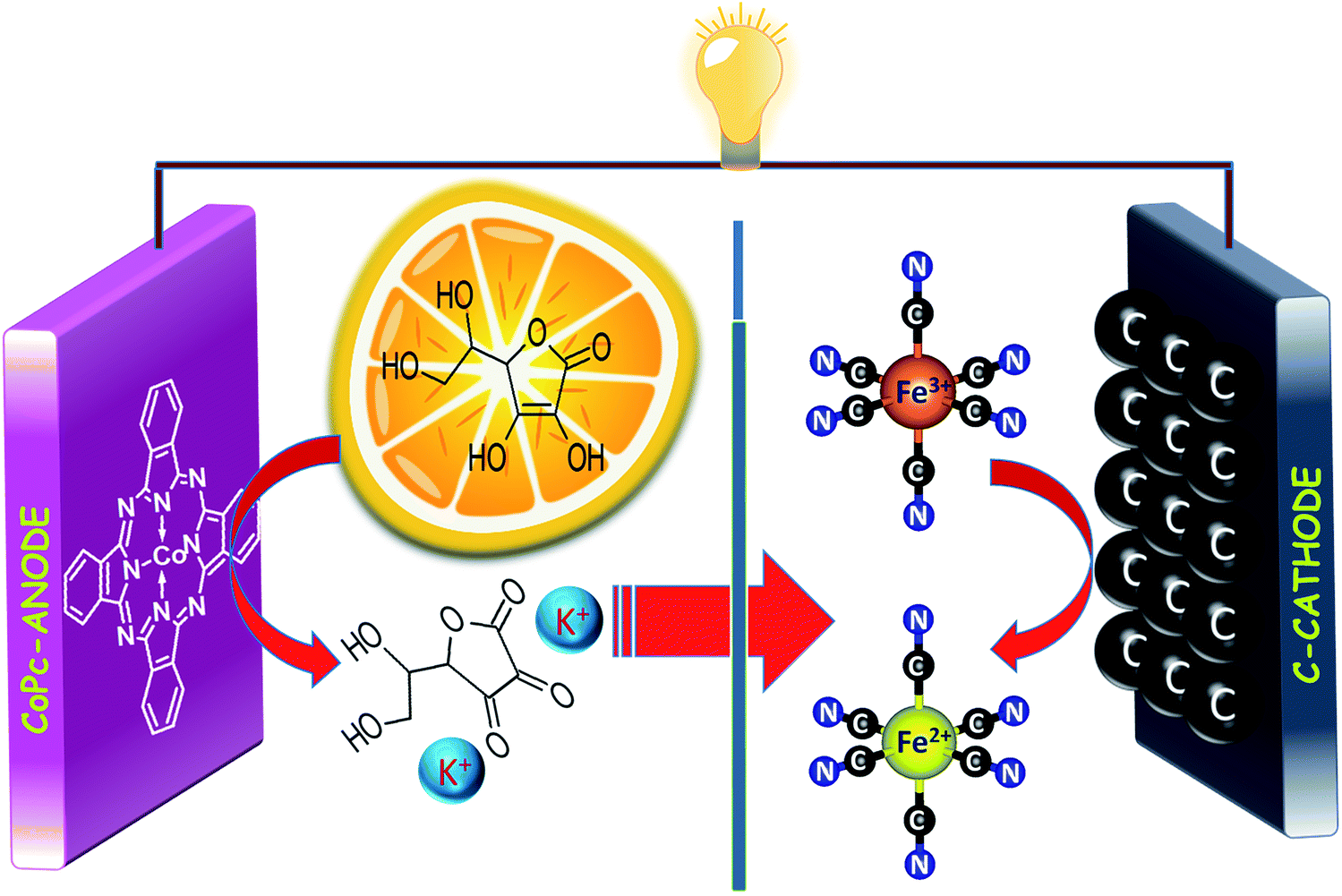
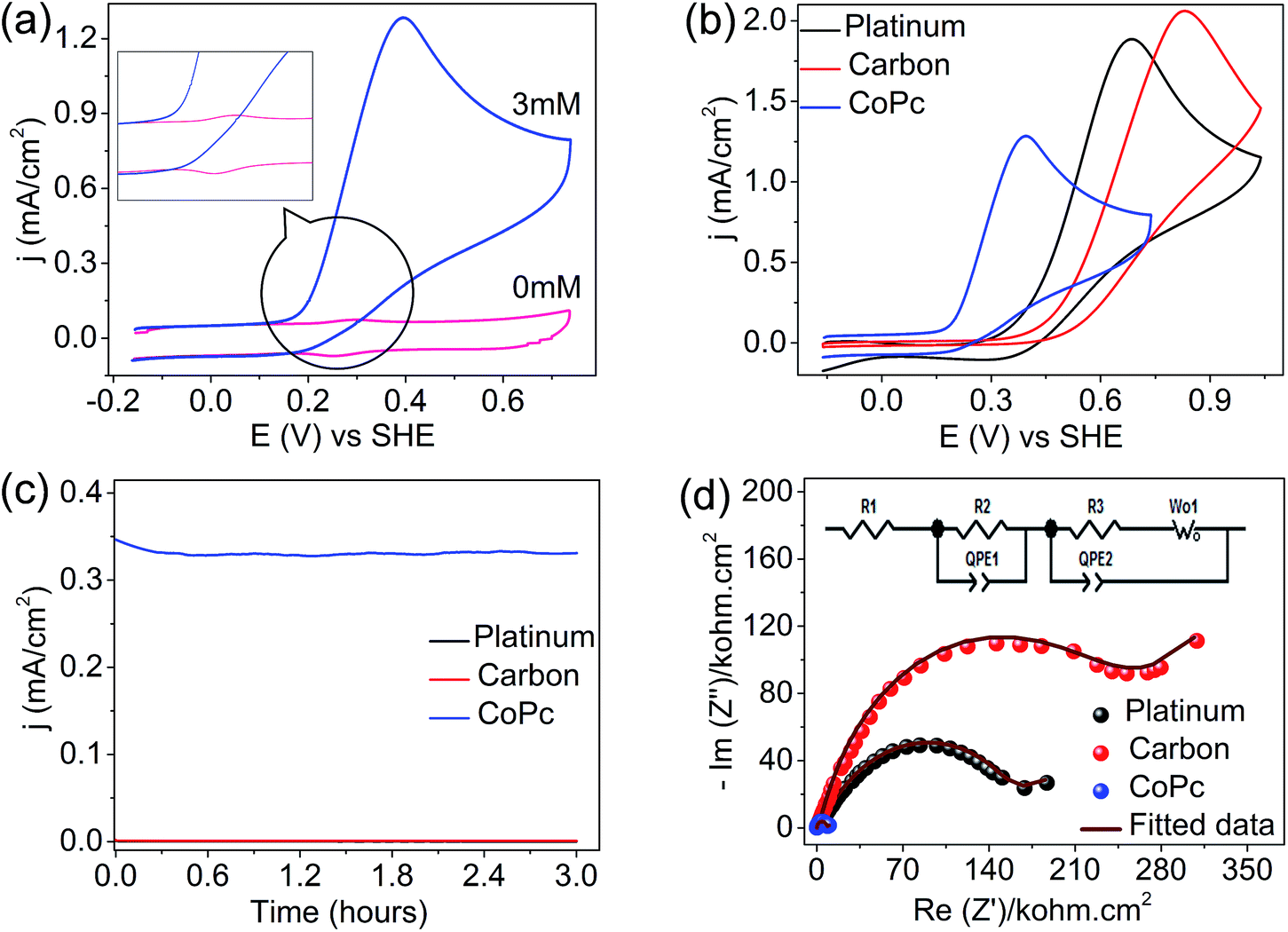
The precious-metal-free vitamin C fuel cell is shown in Scheme 1; it consists of an anodic half-cell with a cobalt-based phthalocyanine (CoPc) molecule as the anodic electrocatalyst for vitamin C oxidation and a cathodic half-cell containing an outer sphere electron acceptor (potassium ferricyanide) that can undergo fast and reversible redox reactions on carbon-based electrodes. The anodic and cathodic cells were separated by a cation-conducting Nafion 117 membrane to prevent internal short circuits. The mediation of vitamin C oxidation by the cobalt-based molecular electrocatalyst is visible in the cyclic voltammogram (Fig. 1a), wherein the addition of vitamin C resulted in the disappearance of the reduction peak of Co3+ (inset of Fig. 1a); the fuel oxidation commenced at Co2+ to Co3+ oxidation, suggesting an ECcat mechanism.37–39 The onset and peak potentials significantly shifted to negative values in comparison to those of Pt-based and carbon-based electrodes, clearly demonstrating the electrocatalytic effect of CoPc towards vitamin C oxidation (Fig. 1b and Table S1, ESI†). The investigation of long-term stabilities of all three electrodes by chronoamperometry at the same electrode potential (Fig. 1c) clearly demonstrated a higher current for a longer duration for the cobalt-based molecular electrocatalyst. Almost similar results were obtained at an overpotential of 340 mV (Eappl − Eonset) (Fig. S1, ESI†), suggesting that CoPc was highly catalytic towards vitamin C oxidation. This was further supported by electrochemical impedance spectroscopy, wherein at the open circuit voltage (OCV), the charge transfer resistance on CoPc (inset of Fig. 1d) was found to be ∼11 times smaller than that on Pt (Table S2, ESI†), suggesting facile charge transfer kinetics on the cobalt-based molecular electrocatalyst compared to those on the other electrocatalysts (Fig. 1d). All these results demonstrated that the cobalt-based molecular catalyst is a better electrocatalyst than Pt towards vitamin C, and it can efficiently mediate the electron transfer from vitamin C.
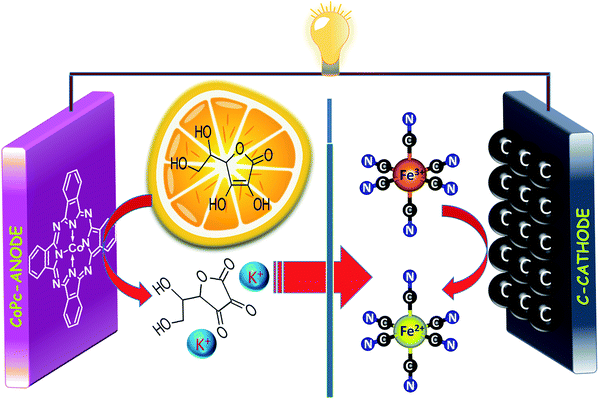 |
| | Scheme 1 Schematics of the precious-metal-free vitamin C fuel cell driven by the non-bonded cathodic interface. | |
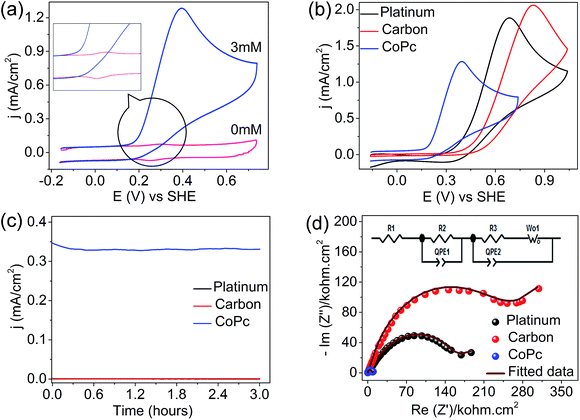 |
| | Fig. 1 (a) Cyclic voltammogram for vitamin C (3 mM in phosphate buffer of pH = 7) oxidation on a cobalt-based phthalocyanine electrocatalyst (CoPc) at a scan rate of 100 mV s−1, (b) comparative cyclic voltammograms on CoPc, Pt and carbon-based electrodes for vitamin C oxidation (3 mM in phosphate buffer of pH = 7) at a scan rate of 100 mV s−1, (c) chronoamperometry curves on Pt, GC, and CoPc at an electrode potential of 0.25 V vs. SHE and (d) electrochemical impedance spectra on CoPc, Pt and carbon electrodes at the open circuit voltage. Frequency range was 100 kHz to 1 mHz and the AC amplitude was 10 mV peak to peak. Inset shows the fitted circuit. | |
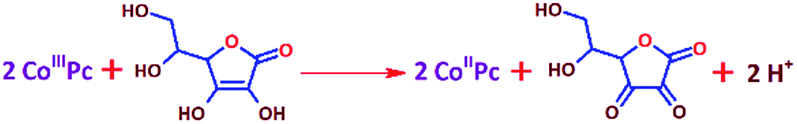
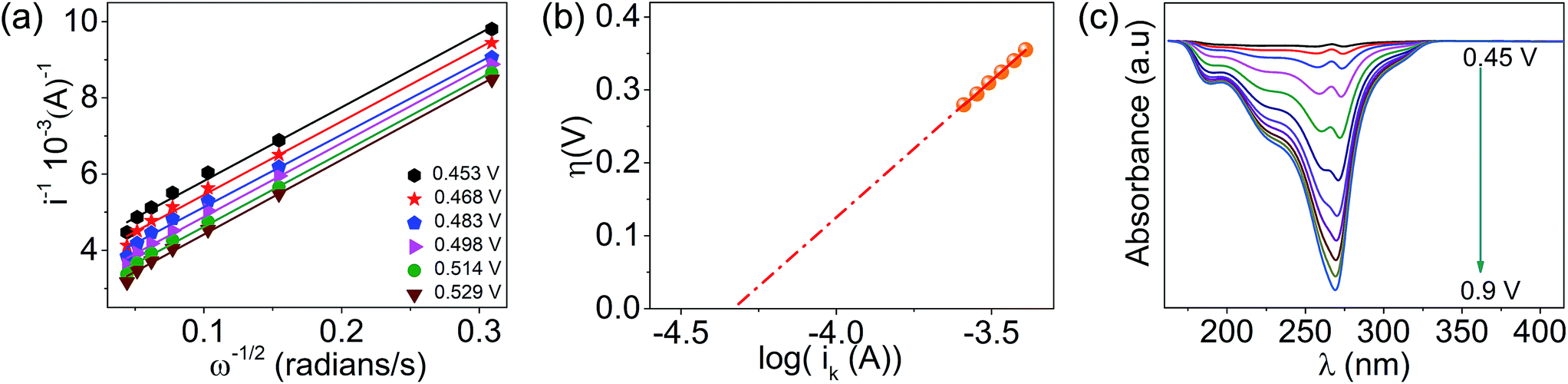
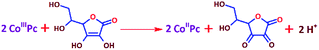
The lower activity on Pt may be due to the irreversible adsorption of vitamin C molecules on the electrode surfaces. The oxidation peak current on CoPc was found to scale up with the concentration and the square root of the scan rate, and a logarithmic plot of current vs. scan rate yielded a slope of ∼0.5, indicating a diffusion-controlled process (Fig. S2a–e, ESI†). The number of electrons involved in the vitamin C oxidation on the cobalt-based molecular electrocatalyst was extracted using the rotating disk electrode (RDE) technique (Fig. S3, ESI†) at different rotations per minute, and the Koutecky–Levich plot (K–L plot) suggested that the number of electrons was close to 2 (Fig. 2a and Table S3, ESI†). The rate constant of the reaction was found to be ∼5 × 10−4 cm s−1 from the plot of kinetic currents (Ik) vs. overpotential (η) (Fig. 2b and Table S3, ESI†). Spectroelectrochemistry data (Fig. 2c) indicated that the characteristic peak for vitamin C at ∼265 nm decreased in intensity when the potential was positively switched, suggesting the formation of the oxidized form (dehydro) of vitamin C.40 This indicated the following reaction as the anodic half-cell reaction (eqn (1)).
| |  | (1) |
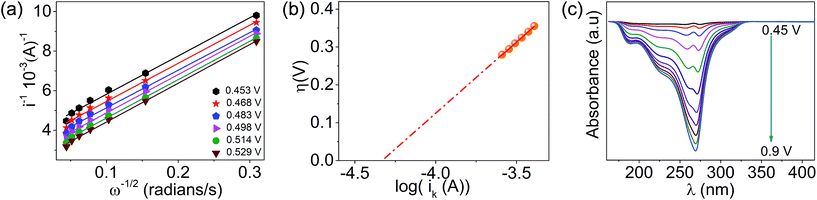 |
| | Fig. 2 (a) Koutecky–Levich plot obtained on a CoPc-modified glassy carbon disk electrode for the oxidation of vitamin C in phosphate buffer (pH = 7) at different potentials, (b) plot of overpotential versus the log of kinetic current and (c) spectroelectrochemistry data during the oxidation of vitamin C. | |
2.2 Cathodic half cell chemistry
The cathodic half-cell chemistry of the outer sphere electron acceptor ferricyanide was investigated by electrochemistry and spectroelectrochemistry on carbon-based electrodes (Fig. S4–S6, ESI†). On a glassy carbon electrode (GC), the redox reaction of the outer sphere species was clearly visible with a peak separation of ∼61 mV, which was close to the value of 59 mV expected for a fast and reversible one-electron transfer reaction33–36 (Fig. S4a, ESI†). The peak current was found to scale up as a function of the square root of the scan rate (Fig. S5a and b, ESI†), and the log of peak current vs. log of scan rate plot demonstrated a slope of nearly 0.5, indicating a diffusion-controlled process. Furthermore, the peak current was found to be proportional to the concentration of the outer sphere species (Fig. S5c and d, ESI†). The rate constant of the reaction was independently extracted from Klinger and Kochi method and Nicolson's method, and the values were found to be in the order of 6.7 × 10−2 cm s−1 and 2.8 × 10−2 cm s−1, suggesting a fast redox reaction (Table S4, ESI†). RDE and the Koutecky–Levich plot (Fig. S4b and c†) demonstrated that the number of electrons was close to 1, and the plot of overpotential vs. log(kinetic currents) (Fig. S4d, ESI†) suggested that the rate constants matched the values extracted from Klinger and Kochi and Nicholson's methods (Table S4, ESI†). These results suggested that ferricyanide was an outer sphere or a non-bonded electron acceptor, which can undergo fast and reversible electron transfer even on carbon-based electrodes. The spectroelectrochemistry data (Fig. S6, ESI†) suggested the formation of ferricyanide during oxidative scans and the generation of ferrocyanide during reductive scans (eqn (2)).| | | [Fe(CN)6]3− + e− → [Fe(CN)6]4− | (2) |
2.3 Fuel cell performance
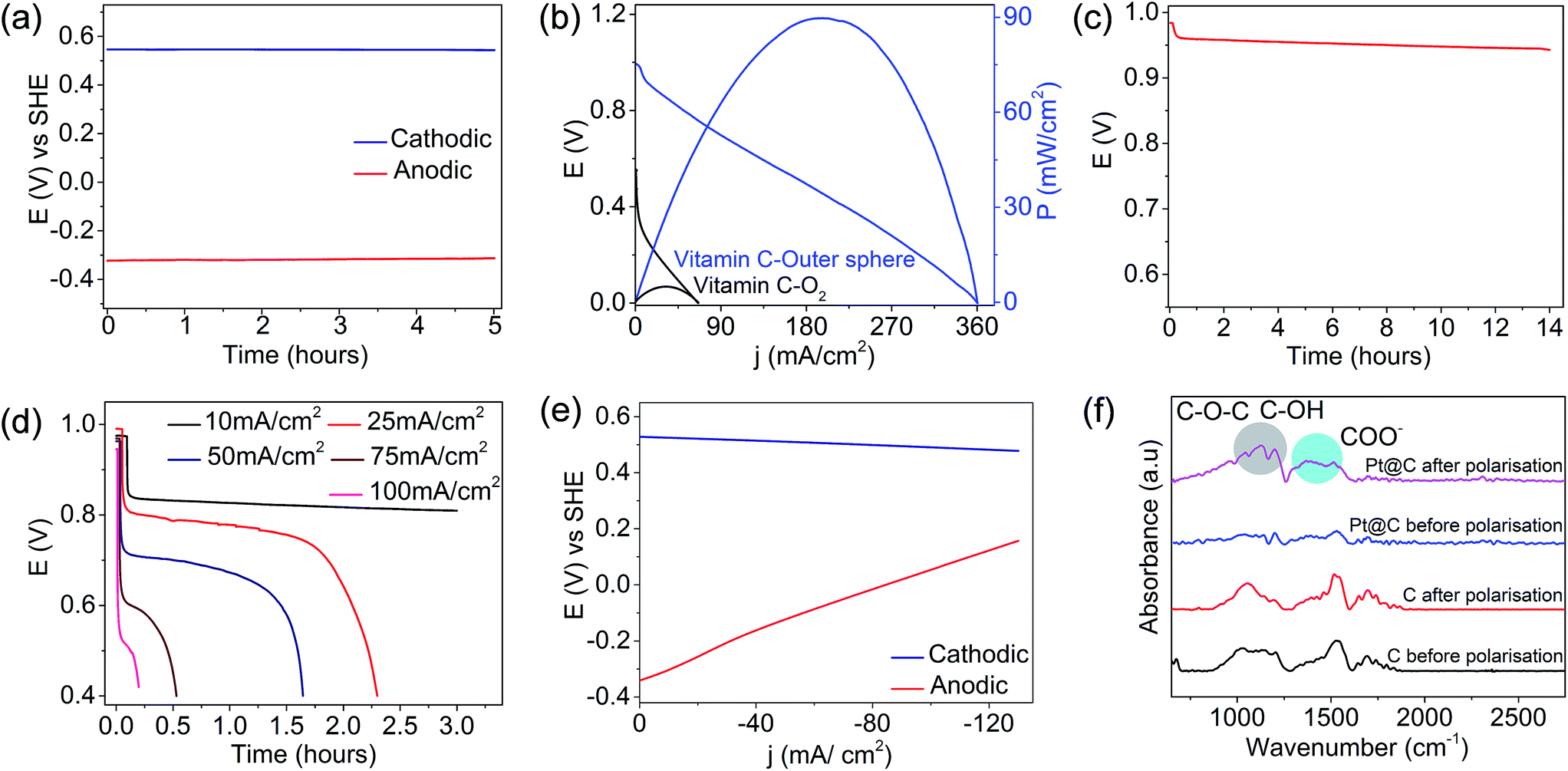
The analysis of single electrode potentials suggested that the anodic half-cell (vitamin C) possessed a voltage of ∼−0.35 V vs. SHE on the CoPc electrode, and the cathodic half-cell with the non-bonded electron acceptor possessed a voltage of ∼0.6 V vs. SHE on the carbon-based electrode (Fig. 3a), suggesting that vitamin C can be an electron donor and ferricyanide can be an electron acceptor in the proposed precious-metal-free configuration. The performance of the precious-metal-free fuel cell (blue trace, Fig. 3b) demonstrated an OCV of ∼1.0 V and a peak power density of ∼90 mW cm−2 at a peak current density of ∼195 mA cm−2. The performance metrics based on the peak power density were ∼18 times higher than those of Pt-based vitamin C–O2 fuel cells (black trace, Fig. 3b). The sufficiently amplified performance metrics in the proposed precious-metal-free fuel cell compared to those of Pt-based vitamin C–O2 fuel cells were due to the enhanced electrode kinetics at either interfaces (as noted above) and the absence of parasitic chemistry involving the cathodic interface (as illustrated below). This should be read in line with the fact that the oxygen reduction reaction on Pt-based electrocatalysts is the performance-limiting interface in state-of-the-art air breathing fuel cells.41–44 However, we noted that the performance of the O2-based fuel cells can be further amplified by increasing the precious metal loading and the O2 partial pressure but with substantial cost implications. The open circuit voltage vs. time plot for vitamin C outer sphere fuel cell (Fig. 3c) suggested a fairly stable profile for several hours, demonstrating the stability at either interfaces. The cell voltages at different discharge current densities are shown in Fig. S7a (ESI†), and the results indicated a decline in the cell voltage when the rate was increased and its reversal on decreasing the rate. The long term stability by galvanostatic polarization at different rates (Fig. 3d) suggested a decent rate capability for the proposed vitamin C fuel cell in a Pt-free configuration.
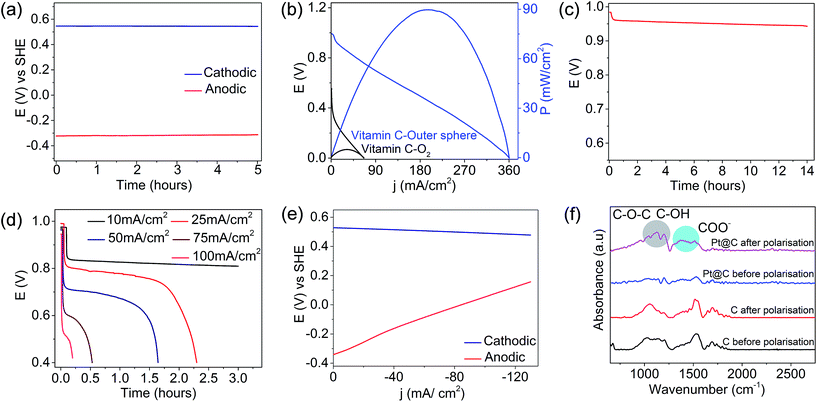 |
| | Fig. 3 (a) Single electrode potentials of anodic and cathodic half cells vs. SHE and (b) polarization curves at 25 °C for the vitamin C fuel cell driven by the non-bonded electron acceptor (blue trace) (anode CoPc catalyst loading and cathode carbon loading were 10 mg cm−2 and the flow rates of the anolyte and the catholyte were 25 mL per minute) in comparison to Pt-based vitamin C–O2 fuel cells (black trace) (anode Pt catalyst loading and cathode Pt loading were 0.5 mg cm−2; the flow rate of the anolyte was 25 mL per minute and the flow rate of O2 was 100 mL per minute). (c) OCV of the Pt-free vitamin C fuel cell as a function of time, (d) galvanostatic discharge curves at different current densities, (e) half-cell polarization curves and (f) the stability of cathodic interfaces after the long-term stability tests in either configurations. All the currents were normalised with respect to the geometrical area. | |
Half-cell polarization suggested that the anodic half-cell chemistry limited the overall fuel cell performance (Fig. 3e), and this could be due to the lower electrode kinetics of vitamin C oxidation compared to that of ferricyanide, as demonstrated before. The stabilities of the electrodes were investigated before and after the long-term stability tests in either configurations, and the results suggested that the cathode in the vitamin C–O2 fuel cell was prone to carbon corrosion (FTIR spectrum, Fig. 3f). The vibrations observed in the FTIR spectrum of the Pt/C electrode after the stability tests at ∼1350–1500 cm−1 corresponded to COO− and those at ∼1100–1250 cm−1 corresponded to COC and COH, suggesting the oxidation of carbon. Carbon corrosion on the cathodic interface is often catalyzed by the Pt catalyst with serious cost and performance implications in the long run.45–47 This sort of parasitic chemistry was not encountered in the vitamin C fuel cell driven by the non-bonded cathodic interface (Fig. 3f), possibly due to the absence of the Pt catalyst and oxygen on the cathode side. The CoPc anode was found to be quite durable (FTIR spectra, Fig. S7b, ESI†). The vibrations at 1500–1600 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the phenyl ring, at 1348 cm−1 due to the C–N stretching vibration, the intense peak at 1139 cm−1 corresponding to the in plane bending vibration, and the 745–800 cm−1 bands due to the C–H out of plane bending vibration of the ring remained largely unaffected even after long-term galvanostatic polarization (Fig. S7b, ESI†).48,49 Overall, the proposed vitamin C outer sphere fuel cell is a type of hybrid fuel cell redox flow battery since the catholyte is electrochemically reversible (Fig. S4, ESI†), and it has potential for practical applications. The practical advantage of the proposed vitamin C outer sphere fuel cell is demonstrated by powering a table lamp (Videos 1 and 2†).
C stretching of the phenyl ring, at 1348 cm−1 due to the C–N stretching vibration, the intense peak at 1139 cm−1 corresponding to the in plane bending vibration, and the 745–800 cm−1 bands due to the C–H out of plane bending vibration of the ring remained largely unaffected even after long-term galvanostatic polarization (Fig. S7b, ESI†).48,49 Overall, the proposed vitamin C outer sphere fuel cell is a type of hybrid fuel cell redox flow battery since the catholyte is electrochemically reversible (Fig. S4, ESI†), and it has potential for practical applications. The practical advantage of the proposed vitamin C outer sphere fuel cell is demonstrated by powering a table lamp (Videos 1 and 2†).
3. Conclusion
In conclusion, we have demonstrated a precious-metal-free vitamin C fuel cell by exploiting the mediated electron donation of vitamin C by a cobalt-based molecular electrocatalyst and the non-bonded nature of the cathodic interface. The resulting hybrid fuel cell redox flow battery in a precious-metal-free configuration arrests the parasitic chemistry driven by molecular oxygen and delivers ∼18 times higher performance metrics compared to Pt-based vitamin C–O2 fuel cells.
4. Experimental section
4.1. Materials and methods
The chemicals such as cobalt phthalocyanine (CoPc), KH2PO4, K2HPO4, tetrahydro furan, dimethyl sulfoxide, ethanol, isopropyl alcohol, Nafion solution and sulphuric acid were procured from Sigma Aldrich, India. Toray carbon, Ketjen black (KB) and Nafion membranes were purchased from the Fuel cell store and used as received. Super P conducting additive carbon was obtained from Alfa Aesar, India.
4.2. Experimental procedure
The CoPc-modified glassy carbon electrode was used for the electrochemical oxidation of vitamin C. Prior to the use of the glassy carbon electrode (GC), the electrode was manually polished using 0.05 micron alumina powder and washed thoroughly with distilled water. Then, GC was electrochemically cleaned using a 0.1 M H2SO4 solution by cycling between the potentials of −0.2 V and 1.1 V using platinum as the counter electrode and Ag/AgCl (saturated KCl) as the reference electrode. For the modification of the GC electrode, the CoPc catalyst was prepared by mixing 5.0 mg of CoPc with 10% of super P (conducting carbon additive) and 5 wt% of PTFE solution dispersed in isopropyl alcohol. The mixture was sonicated for about 30 minutes, and the catalyst was drop-casted on the pre-cleaned glassy carbon electrode and dried in an oven for about 1 h prior to use.
A single glass compartment in the three-electrode configuration containing 0.1 M phosphate buffer (pH = 7.0) electrolyte was used for the study of the oxidation of vitamin C. The CoPc-modified glassy carbon electrode was used as the working electrode, Ag/AgCl (saturated KCl solution) was used as the reference electrode and a platinum foil was used as the counter electrode. The voltammograms were recorded using the CoPc-modified glassy carbon electrode in the presence and absence of vitamin C in an N2-saturated electrolyte.
The RDE experiment was performed using an RDE glassy carbon electrode (area 0.192 cm2). A previously cleaned electrode was modified by drop casting the CoPc catalyst, and it was dried well. All the RDE electrochemical measurements were conducted in a nitrogen-purged 5 mM vitamin C solution at a scan rate of 5 mV s−1 with a three-electrode system containing CoPc-modified electrode as the working electrode, Ag/AgCl (saturated KCl) as the reference electrode and a platinum electrode as the counter electrode by varying the rotation speed (100, 400, 900, 1600, 2500, 3600, and 4900). For the calculation of the number of electrons involved in the reaction, Koutecky–Levich (K–L) equation (eqn (3)) was used:
| | 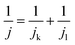 | (3) |
here,
j is the total current density,
jk is the kinetic current density and
jl is the limiting current density.
| jl = 0.62nFD2/3ν−1/6Cω1/2 |
here,
n is the number of electrons,
F is the Faraday constant,
D (cm
2 s
−1) is the diffusion coefficient,
ν (cm s
−1) is the kinematic viscosity,
C (moles per cm
3) is the bulk concentration and
ω (rad s
−1) is the rotation rate.
The plot of reciprocal of current density and reciprocal of rotation rate at different overpotentials gave a slope equal to 1/0.62nFD2/3ν−1/6C. From this, the number of electrons was calculated.
The intercept of the plot gives the reciprocal of the kinetic current density. The kinetic current is related to the overpotential by the following equation (eqn (4)):
| | 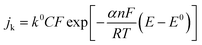 | (4) |
here,
k0 is the rate constant and
α is the transfer coefficient.
k0 and
α were calculated from the plot of overpotential
vs. log(kinetic current density). The obtained slope is equal to 2.303
RT/
αnF and the intercept on the independent variable axis gives the exchange current density equal to
FCk0.
50
4.3. Characterization
In situ spectroelectrochemistry data of potassium ferricyanide were collected using an ALS Electrochemistry and Spectroelectrochemistry SEC2000 spectrometer, with 10 mM ferricyanide in phosphate buffer solution at a 5 mV s−1 scan rate in a three electrode system containing CoPc-coated indium tin oxide (ITO) as the working electrode, Ag/AgCl (saturated KCl) as the reference electrode and a platinum wire as the counter electrode. Potassium ferricyanide signal was taken as a background. The electrochemical measurements and impedance spectra (100 kHz to 1 mHz at open circuit voltage) of vitamin C oxidation were obtained using a Biologic potentiostat (VMP-300) in a three-electrode configuration, as discussed above. The RDE experiment was performed using the Princeton Applied Research Ametek (model-636A) R-D electrode system in the three-electrode configuration in 0.1 M phosphate buffer containing 5 mM vitamin C. ATIR-FTIR (Bruker Alpha) was carried out for the characterization of electrodes before and after the stability tests.
4.4. Fuel cell fabrication
The vitamin C fuel cell was constructed by using CoPc as the anode and Ketjen black carbon (KB) as the cathode separated by a Nafion 117 membrane. The anode was prepared by spraying the catalyst ink prepared by dispersing CoPc, 10% Super P carbon and 20 wt% PTFE binder in isopropyl alcohol; it was then spray coated on a Toray carbon paper with a loading of 10 mg cm−2. In the same way, for the cathode preparation, KB nanoparticles and 10 wt% of PTFE were dispersed in isopropyl alcohol by sonication for about 30 minutes and spray coated on a Toray carbon paper at a loading of 10 mg cm−2. A pre-treated Nafion 117 membrane was sandwiched between the cathode and the anode. The sandwiched setup was kept in between two graphite plates with serpentine channels with an inlet and an outlet for the flow of solutions. Finally, stainless steel end plates were used as backing plates. A 3 M vitamin C solution in KOH solution of pH 14 was used as the anolyte, and 1 M potassium ferricyanide in KOH solution of pH 14 was used as the catholyte. The flow rate was 25 mL min−1. For vitamin C–O2 fuel cells, the anode Pt catalyst loading and the cathode Pt loading were 0.5 mg cm−2. The flow rate of the anolyte was 25 mL per minute, and the flow rate of O2 was 100 mL per minute. The operating temperature for all fuel cell studies was 25 °C. The geometric area of the electrodes was 4 cm2 (2 cm × 2 cm). All the currents were normalised with respect to the geometrical area.
Conflicts of interest
There are no conflicts to declare.
References
- E. Mourad, L. Coustan, P. Lannelongue, D. Zigah, A. Mehdi, A. Vioux, S. A. Freunberger, F. Favier and O. Fontaine, Nat. Mater., 2016, 16(4), 446–453 CrossRef PubMed.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 17036 CrossRef.
- Y. Nabil, S. Cavaliere, I. A. Harkness, J. D. B. Sharman, D. J. Jones and J. Rozière, J. Power Sources, 2017, 363, 20–26 CrossRef.
- W. Gao, N. Singh, L. Song, Z. Liu, A. L. M. Reddy, L. Ci, R. Vajtai, Q. Zhang, B. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6(8), 496–500 CrossRef PubMed.
- T. Du, C. H. Burgess, J. Kim, J. Zhang, R. Durrant and M. A. Mclachlan, Sustainable Energy Fuels, 2017, 1, 119–126 RSC.
- M. del Cueto, P. Ocón and J. M. L. Poyato, J. Phys. Chem. C, 2015, 119(4), 2004–2009 CrossRef.
- S. Goodwin and D. A. Walsh, ACS Appl. Mater. Interfaces, 2017, 9, 23654–23661 CrossRef PubMed.
- Z. Tao, Q. Zhang, X. Xi, G. Hou and L. Bi, Electrochem. Commun., 2016, 72, 19–22 CrossRef.
- T. Ravikumar and K. Scott, Chem. Commun., 2012, 48(45), 5584–5586 RSC.
- D. Sebastián, A. Serov, I. Matanovic, K. Artyushkova, P. Atanassov, A. S. Aricò and V. Baglio, Nano Energy, 2017, 34, 195–204 CrossRef.
- D. Sebastián, A. Serov, K. Artyushkova, J. Gordon, P. Atanassov, A. S. Arico and V. Baglio, ChemSusChem, 2016, 9, 1986–1995 CrossRef PubMed.
- A. S. Gago, Y. G. Ponce, Y. J. Feng, J. P. Esquivel, N. Sabat, J. Santander and N. A. Vante, ChemSusChem, 2012, 5, 1488–1494 CrossRef PubMed.
- K. Scott, A. K. Shukla, C. L. Jackson and W. R. A. Meuleman, J. Power Sources, 2004, 126, 67–75 CrossRef.
- Z. M. Bhat, R. Thimmappa, M. C. Devendrachari, S. P. Shafi, S. Aralekallu, A. R. Kottaichamy, M. Gautam and M. O. Thotiyl, J. Phys. Chem. Lett., 2017, 8(15), 3523–3529 CrossRef PubMed.
- D. K. Ross, Vacuum, 2006, 80(10), 1084–1089 CrossRef.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef.
- R. Borup, J. Meyers, B. S. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, X. J. Boncella, J. E. Mcgrath, O. M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. Kimijima and N. Iwashita, Chem. Rev., 2007, 3904–3951 CrossRef PubMed.
- S. K. Mondal, R. K. Raman, A. K. Shukla and N. Munichandraiah, J. Power Sources, 2005, 145, 16 CrossRef.
- G. Karim-Nezhad, M. Hasanzadeh, L. Saghatforoush, N. Shadjou, B. Khalilzadeh and S. Ershad, J. Serb. Chem. Soc., 2009, 74, 581–593 CrossRef.
- S. S. Kumar, J. Mathiyarasu, K. L. N. Phani and V. Yegnaraman, J. Solid State Electrochem., 2006, 10, 905–913 CrossRef.
- O. Muneeb, E. Do, T. Tran, D. Boyd, M. Huynh, G. Ghosn and J. L. Haan, J. Power Sources, 2017, 351, 74–78 CrossRef.
- N. Fujiwara, K. Yasuda, T. Ioroi, Z. Siroma, Y. Miyazaki and T. Kobayashi, Electrochem. Solid-State Lett., 2003, 6, A257 CrossRef.
- M. L. Chen, S. L. Huang, C. L. Hsieh, J. Y. Lee and T. J. Tsai, Electrochim. Acta, 2014, 141, 241–247 CrossRef.
- J. W. Kim, J. D. Ocon, H. S. Kim and J. Lee, ChemSusChem, 2015, 8(17), 2883–2891 CrossRef PubMed.
- Y. Xiong and Y. Xia, Adv. Mater., 2007, 19(20), 3385–3391 CrossRef.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40(6), 1789–1791 CrossRef.
- L. Lu, I. Sevonkaev, A. Kumar and D. V. Goia, Powder Technol., 2014, 261, 87–97 CrossRef.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126(28), 8648–8649 CrossRef PubMed.
- N. R. Jana, Small, 2005, 1(8–9), 875–882 CrossRef PubMed.
- M. G. Espey, P. Chen, B. Chalmers, J. Drisko, A. Y. Sun, M. Levine and Q. Chen, Free Radical Biol. Med., 2011, 50(11), 1610–1619 CrossRef PubMed.
- J. Verrax and P. B. Calderon, Free Radical Biol. Med., 2009, 47(1), 32–40 CrossRef PubMed.
- S. Harrison, Am. J. Gastroenterol., 2003, 98(11), 2485–2490 CrossRef PubMed.
- J. M. Nugent, K. S. V. Santhanam, A. Rubio and P. M. Ajayan, Nano Lett., 2001, 1(2), 87–91 CrossRef.
- P. A. Henry, A. S. Raut, S. M. Ubnoske, C. B. Parker and J. T. Glass, Electrochem. Commun., 2014, 48, 103–106 CrossRef PubMed.
- M. M. Lounasvuori, M. Rosillo-Lopez, C. G. Salzmann, D. J. Caruana and K. B. Holt, Faraday Discuss., 2014, 172, 293–310 RSC.
- W. Du, B. Zhou and X. Jiang, Chem. Phys. Lett., 2014, 595–596, 1–5 CrossRef.
- S. Fukuzumi, K. Miyamoto, T. Suenobu, E. Van Caemelbecke and K. M. Kadish, J. Am. Chem. Soc., 1998, 120(12), 2880–2889 CrossRef.
- E. V. Kudrik, S. V. Makarov, A. Zahl and R. Van Eldik, Inorg. Chem., 2003, 42(2), 618–624 CrossRef PubMed.
- O. S. A. Aobayama and K. Adachi, Anal. Sci., 1990, 4, 2–6 Search PubMed.
- J. R. Witmer, B. J. Wetherell, B. A. Wagner, J. Du, J. J. Cullen and G. R. Buettner, Redox Biol., 2016, 8, 298–304 CrossRef PubMed.
- D. R. Paul, W. J. Koros, R. Y. F. Liu, Y. S. Hu, E. Baer, A. Hiltner, H. D. Keith, R. Y. F. Liu, A. Hiltner, E. Baer, R. E. Cohen, A. Bellare, R. J. Albalak, W. Hu and G. Reiter, Science, 2009, 323, 760–764 CrossRef PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414(6861), 345–352 CrossRef PubMed.
- V. G. Anju, R. Manjunatha, P. M. Austeria and S. Sampath, J. Mater. Chem. A, 2016, 4(14), 5258–5264 RSC.
- K. Shimizu, L. Sepunaru and R. G. Compton, Chem. Sci., 2016, 7(5), 3364–3369 RSC.
- L. M. Roen, C. H. Paik and T. D. Jarvi, Electrochem. Solid-State Lett., 2004, 7(1), A19 CrossRef.
- D. A. Stevens and J. R. Dahn, Carbon, 2005, 43(1), 179–188 CrossRef.
- Z. Siroma, K. Ishii, K. Yasuda, Y. Miyazaki, M. Inaba and A. Tasaka, Electrochem. Commun., 2005, 7(11), 1153–1156 CrossRef.
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef PubMed.
- N. Achar, G. M. Fohlen, J. A. Parker and J. Keshavayya, Polyhedron, 1987, 6, 1463–1467 CrossRef.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00221e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Zahid Manzoor
Bhat
a,
Shahid Pottachola
Shafi
a,
Harish Makri
Nimbegondi Kotresh
*c,
Alagar Raja
Kottaichamy
a,
Kallam Ramareddy
Venugopala Reddy
*d and
Musthafa Ottakam
Thotiyl
a,
Zahid Manzoor
Bhat
a,
Shahid Pottachola
Shafi
a,
Harish Makri
Nimbegondi Kotresh
*c,
Alagar Raja
Kottaichamy
a,
Kallam Ramareddy
Venugopala Reddy
*d and
Musthafa Ottakam
Thotiyl