
Lignosulfonate biomass derived N and S co-doped porous carbon for efficient oxygen reduction reaction†
Mingli
Zhang‡
 a,
Yanliang
Song‡
b,
Hengcong
Tao
a,
Chao
Yan
c,
Justus
Masa
de,
Yongchao
Liu
c,
Xiaoyou
Shi
b,
Shizhen
Liu
a,
Xu
Zhang
*b and
Zhenyu
Sun
*a
a,
Yanliang
Song‡
b,
Hengcong
Tao
a,
Chao
Yan
c,
Justus
Masa
de,
Yongchao
Liu
c,
Xiaoyou
Shi
b,
Shizhen
Liu
a,
Xu
Zhang
*b and
Zhenyu
Sun
*a
aState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: sunzy@mail.buct.edu.cn; Tel: +86 13301308339
bBeijing Key Lab of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: zhangxu@mail.buct.edu.cn; Tel: +86 13301308339
cSchool of Material Science & Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, China
dAnalytische Chemie-Elektroanalytik & Sensorik, Ruhr-University Bochum, 44780 Bochum, Germany
eDepartment of Chemistry, Kyambogo University, P. O. Box 1, Kyambogo (Kampala), Uganda
First published on 26th June 2018
Abstract
Finding sustainable, active, inexpensive, and stable electrocatalysts to replace Pt-based materials for the oxygen reduction reaction (ORR) remains a significant challenge. Here we report the use of functionalized lignin biomass as both carbon and dopant source for the direct synthesis of N and S dual-doped carbon sheet networks with abundant mesopores and high surface area. The resulting N and S-doped porous carbon exhibits good activity, long-term durability, and high selectivity for metal-free oxygen reduction electrocatalysis, approaching the performance of 20 wt% Pt/C, the state of the art ORR catalyst. Our method using natural biological resources thus offers a promising way to produce very promising noble metal-free ORR catalysts with potential application in alkaline fuel cells and metal–air batteries.
Introduction
The oxygen reduction reaction (ORR) represents a key cathode reaction in fuel cells and metal–air batteries.1 Unfortunately, the kinetics of the ORR is intrinsically slow, requiring multi-electron and multi-proton transfer. Efficient electrocatalysts are thus needed to reduce reaction overpotential and accelerate reaction kinetics. To date, Pt remains the most active ORR catalyst, affording four-electron (4e−) reduction of O2 to water with minimal or no hydrogen peroxide formation. However, the high cost of Pt, its scarcity and insufficient catalytic stability under reaction conditions make it unsuitable for large-scale implementation. Therefore, finding cost-effective and earth-abundant nonprecious metals or metal-free catalysts has been a focus of interest for decades.Carbon-based materials have shown potential as non-metallic2–8 or non-precious metal9–12 ORR catalysts owing to their good corrosion resistance, inexpensiveness, high electrical conductivity, and tunable surface properties. The advantages of low cost and abundance in nature of bio-renewable materials make them particularly attractive as precursors for the synthesis of carbon-based electrocatalysts. Lignocellulosic biomass is one such material that could provide a ubiquitous inexhaustible raw material of low-cost ORR catalysts. More than 140 billion metric tons of this biomass are generated annually as waste from agricultural and forestry products.13 15–30% of lignocellulosic materials are comprised of lignin in addition to cellulose (30–50%) and hemicellulose (20–35%).14,15 However, direct conversion of lignin into valuable chemicals or fuels is difficult due to its complex and heterogeneous structure. Most of the lignin generated in the pulp and paper industry is burned to provide heat. To make full use of this renewable biomass, transformation of lignin into metal-free electrocatalysts has recently spurred great interest.16 In 2014, Fong and co-workers first used lignin as a carbon source to produce carbon fibres which were further decorated with Ag nanoparticles (NPs) for ORR electrocatalysis.17 Graglia et al. synthesized nitrogen-doped porous carbon for ORR by using lignin as a precursor.18 Despite recent advances in this regard, multiple heteroatom doped carbon materials derived from (functionalized) lignin have been rarely reported, thus their synthesis and application in ORR warrants further exploration.
Here we report the scalable synthesis of N and S co-doped mesoporous carbon by simply annealing a cheap biomass precursor, ammoniated lignosulfonate, in nitrogen. The resulting materials showed outstanding catalytic activity toward ORR in alkaline media, favoring a 4e− process, with the performance approaching that of 20 wt% Pt/C. Control of the thermal treatment temperature permits the graphitization degree to be readily tuned, as well as the N and S doping levels, and thus the electrocatalytic properties of the resulting carbon.
Experimental
2.1 Materials
All the chemicals used in this work were of analytical grade and used as supplied. Sodium lignosulfonate was purchased from Yuanye Biotechnology Co. Ltd. (Shanghai, China) and used without further treatment.2.2 Ammoxidation of lignosulphonate
Ammoxidation was carried out by dissolving sodium lignosulfonate in deionized water, mixed with ammonia solution (25%) and hydrogen peroxide (30%) at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture was sealed in a batch autoclave reactor and heated at 120 °C for 90 min. After cooling to room temperature, the ammoniated sodium lignosulfonate was subjected to freeze-drying.
:1. The mixture was sealed in a batch autoclave reactor and heated at 120 °C for 90 min. After cooling to room temperature, the ammoniated sodium lignosulfonate was subjected to freeze-drying.
2.3 Preparation of doped porous carbon
Carbonization was performed by thermal treatment of the raw materials at reaction temperature for 60 min in a quartz-tube furnace with a heating rate of 9 °C min−1 under N2 at a flow rate of 20 sccm. After cooling to room temperature, the carbonized samples were washed with excess deionized water to remove Na+ and then freeze dried.2.4 Characterization
X-ray diffraction (XRD) was performed on a D/MAX-RC diffractometer operated at 30 kV and 100 mA with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a K-Alpha spectrometer (Thermo Fisher Scientific Inc., Switzerland) equipped with a monochromatic Al Kα source operated at 150 W. All spectra were calibrated based on the C 1s binding energy at 284.8 eV. Scanning electron microscopy (SEM) was conducted using a field emission microscope (FEI Quanta 600 FEG) operated at 20 kV and equipped with an energy-dispersive X-ray spectrometer (EDX). Transmission electron microscopy (TEM) was performed using a JEOL 2200 microscope with an accelerating voltage of 80 kV. TEM samples were prepared by depositing a droplet of suspension onto a Cu grid coated with a lacey carbon film. Nitrogen adsorption/desorption measurements were made on a MicroActive ASAP2460 apparatus to determine the Brunauer–Emmett–Teller (BET) surface area. The samples were degassed in vacuum at 200 °C for 8 h prior to N2 adsorption at −195 °C. Raman spectra of samples deposited on 300 nm thick SiO2/Si substrates were collected using a Renishaw in via Raman microscope with a He/Ne Laser excitation at 532 nm.2.5 Electrochemical measurement
Cyclic voltammetry (CV) and linear-scan voltammetry (LSV) were carried out in a three-electrode system using Ag/AgCl as a reference electrode, Pt wire as a counter electrode, and glassy carbon as a working electrode on a CHI 760E electrochemical workstation (Shanghai Chenhua Co., China). Rotating disk electrode (RDE) experiments were run on an AFMSRCE RDE control system (Pine Inc., USA). The details of preparation of catalyst inks are described in the ESI.† In all the electrochemical measurements, a 0.1 M aqueous solution of KOH saturated with O2 was used as the electrolyte. The onset potential of ORR in this work is determined by taking the potential at the intersection point of the tangent at maximum slope. The number of electrons, n, transferred per oxygen molecule during the ORR was estimated by RDE voltammetry using the Koutecky–Levich (K–L) equation [eqn (1)]: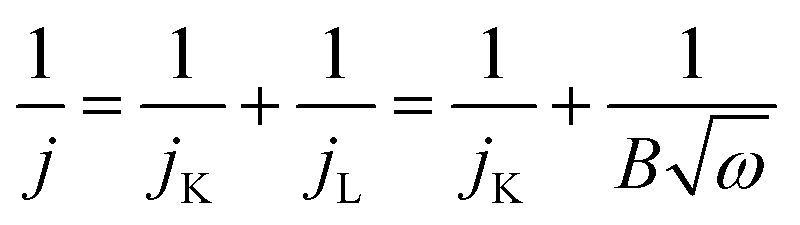 | (1) |
485C mol−1); A, the geometric area of the electrode (0.19625 cm2); ν, the kinematic viscosity of the electrolyte (0.01 cm2 s−1), and C* the solubility of oxygen in the electrolyte, which for the case of KOH (0.1 M) was taken to be 1.2 × 10−6 mol cm−3.
Results and discussion
N–S co-doped porous carbon samples were obtained by thermal treatment of ammoniated lignosulfonate at 600, 700, 800 and 900 °C, designated here after as N–S/C_600, N–S/C_700, N–S/C_800 and N–S/C_900, respectively. The schematic representation of this process is presented in Fig. 1. The morphology and microstructure of the obtained materials were characterized by using a number of techniques. X-ray diffraction (XRD) patterns of original lignosulfonate, ammoniated lignosulfonate, and annealed samples are given in Fig. 2a. The rather broad peak at ca. 23° indicates poor crystallinity of N–S/C_600, which however became sharper and was shifted to lower diffraction angles with increase of the annealing temperature most probably due to enhancement in crystallinity. A weak diffraction peak at ca. 43° corresponding to the (100) planes of a graphitic structure was also observed for the samples that were annealed at temperatures above 600 °C. Fourier-transform infrared spectroscopy (FTIR) confirmed the formation of nitrogen and sulfur functionalities. The FTIR spectra (ESI, Fig. S1a†) showed characteristic absorption bands of C–N (1196 cm−1), C–S (760 cm−1), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1420 cm−1 and 1597 cm−1) in the N–S/C_700 sample.18,19
C (1420 cm−1 and 1597 cm−1) in the N–S/C_700 sample.18,19
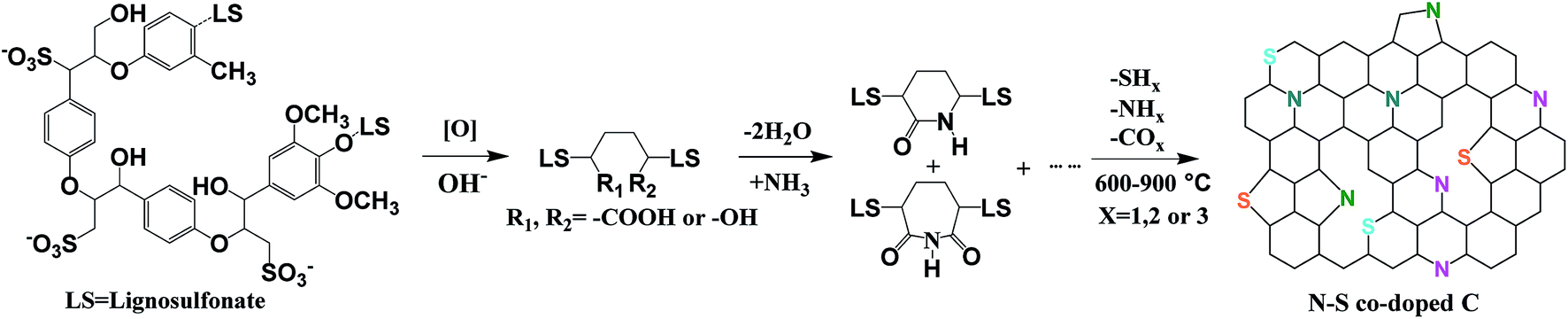 | ||
| Fig. 1 Illustration of the preparation processes of N and S co-doped porous carbon. | ||
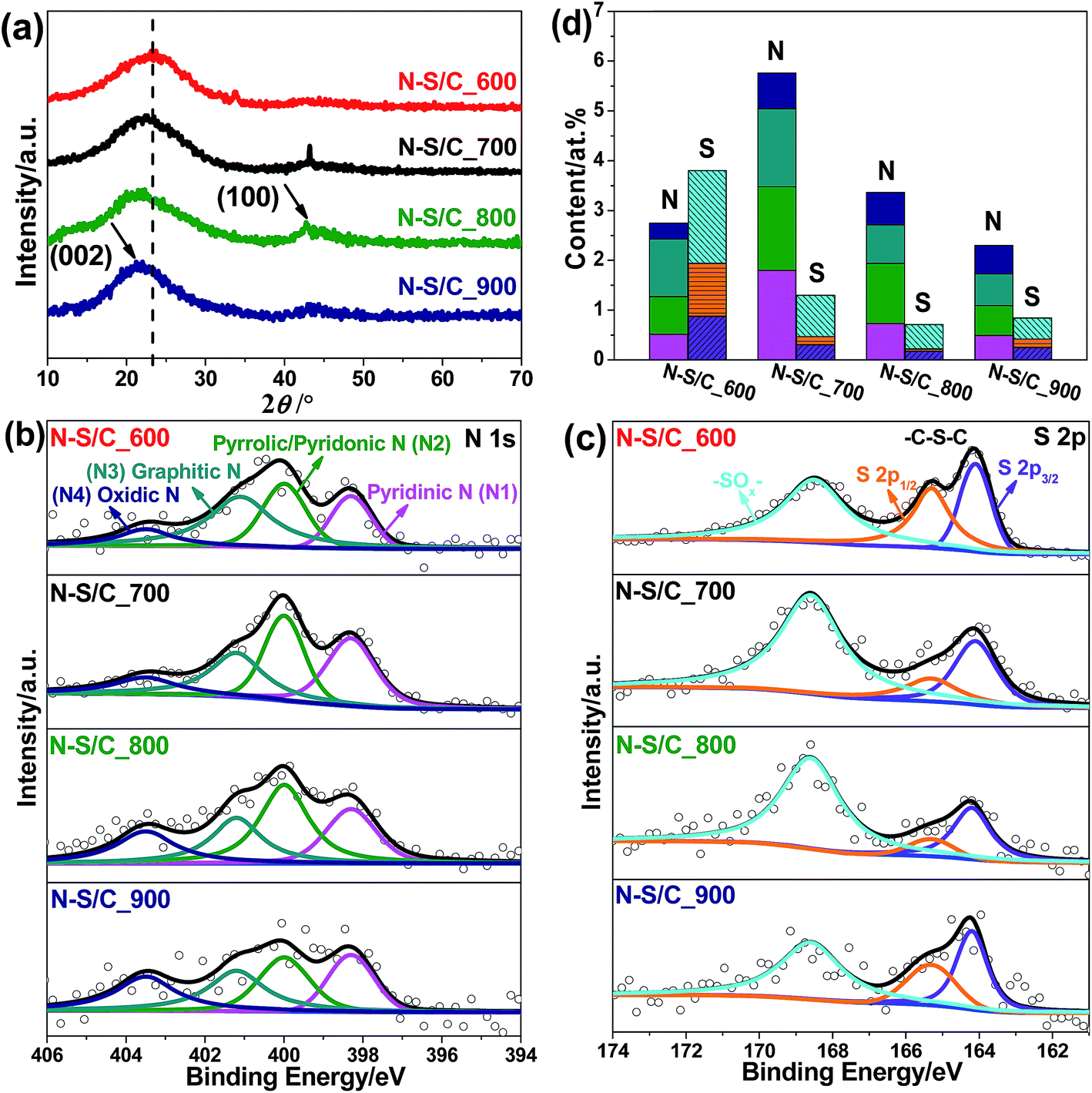 | ||
| Fig. 2 (a) XRD patterns of annealed samples. (b) High resolution N 1s and (c) S 2p XPS spectra, and (d) atomic contents of N species including pyridinic N (N1), pyrrolic/pyridonic N (N2), graphitic N (N3), and oxidic N (N4), and S species including –C–S–C– and oxidized –C–SOx–C– of annealed samples. | ||
X-ray photoelectron spectroscopy (XPS) was further applied to investigate the surface elemental composition and chemical state of the samples. The wide-survey XPS spectra displayed the existence of N and S besides C after annealing (Fig. S2†). The N 1s XPS spectra were deconvoluted into three prominent peaks which can be attributed to pyridinic N (N1) at ∼398.3 eV, pyrrolic/pyridonic N (N2) at ∼399.99 eV, and graphitic (quaternary) N (N3) at ∼401.2 eV, as well as a weak peak from oxidic N (N4) at ∼403.5 eV (Fig. 2b).20 The relative contribution of the various N configurations was determined from the deconvoluted N 1s spectra and the results were shown in Fig. 2b. The absolute contents of all the four N configurations were found to increase with annealing temperature (T) from 2.75% at 600 °C to 5.76% at 700 °C but decreased with T above 700 °C. The fraction of pyridinic N in the four N configurations increased with T and reached a maximum of ca. 36% at 700 °C while the fraction of oxidic N increased monotonically with T. An increase in the percentage of pyrrolic/pyridonic N was observed when the temperature was increased up to 800 °C before receding afterwards, whereas the percentage of graphitic N exhibited a different trend. This may suggest that pyrrolic/pyridonic N species were converted to graphitic N and oxidic N at T > 800 °C. Deconvolution of the S 2p XPS spectra indicated that S mainly existed in two chemical states as –C–S–C– (thiophene S) with two peaks corresponding to 2p1/2 at ∼165.3 eV and 2p3/2 at ∼164.1 eV as a result of spin–orbit coupling, and oxidized –C–SOx–C– (x = 2, 3 or 4) with binding energy at ∼168.6 eV (Fig. 2c).21,22 No other sulfur species were identified in the XPS spectra. An obvious decrease of both thiophene and oxidized S species was observed with the increase of T from the sum of 3.8% at 600 °C to 1.3% at 700 °C. Above 700 °C, the content of thiophene S remained unchanged, whereas the content of oxidized S further decreased, indicating a low stability of such species at high T. The overall content of S in the as-prepared carbon structures at 700–900 °C is in the range of ca. 0.8–1.3%, much lower than the N content (2.3–5.76%) (Fig. 2d).
We used Raman spectroscopy to study the degree of defects in the obtained carbon structures. The Raman spectra of the N–S/C samples exhibited two remarkable peaks at about 1336 and 1590 cm−1, corresponding to the D and G band, respectively (Fig. 3a). The D-band corresponds to the breathing mode of aromatic rings while the G-band is related to the tangential vibration mode of C sp2 atoms.23 The peak intensity ratio of the D band to the G band (ID/IG) was used as a metric for the defect degree. We found that ID/IG increased with annealing temperature probably due to the corresponding increase in N doping level, up to 700 °C beyond which it tended to decrease, due to the removal of some sp3 defects and enhanced graphitic degree at higher temperatures (Fig. 3b). The Raman maps of ID/IG obtained from 144 different regions for all the N–S/C samples display quasi uniform distribution of defects, as can be observed in Fig. 3c–f.
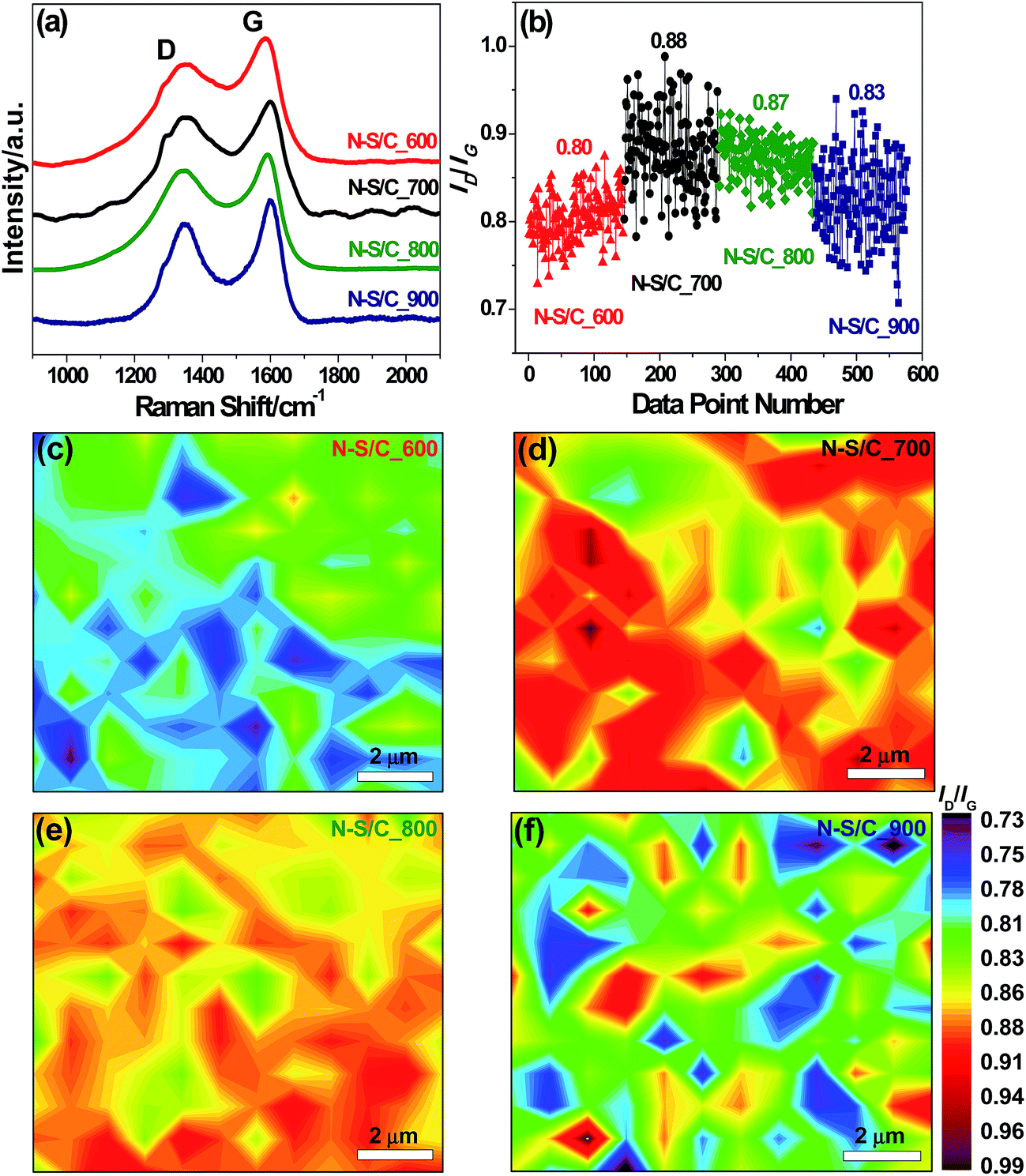 | ||
| Fig. 3 (a) Raman spectra of annealed samples. The spectra were normalized to the D-mode peak intensity. (b) The D to G peak intensity ratios of annealed samples. Raman mapping images of (c) N–S/C_600, (d) N–S/C_700, (e) N–S/C_800, and (f) N–S/C_900. The color gradient bar to the right of the map indicates the D to G peak intensity ratio. | ||
Typical scanning electron microscope (SEM) images of N–S/C_700 clearly displayed the formation of honeycomb-like flake networks with macropores of several hundred nanometers in diameter (Fig. 4a and b). EDX elemental mapping recorded during the SEM observations revealed distribution of both N and S, in addition to C and O, within the flakes (Fig. 4c–f), confirming the successful N and S co-doping of the carbon. Transmission electron microscopy (TEM) images (Fig. 4g and h) further disclosed that the porous networks were composed of thin flakes with slightly wrinkled and folded feature resembling graphene20 and other two-dimensional materials.24 Similar morphologies were also observed in other annealed samples (not shown here). We believe that such porous and crumpled structure is beneficial for enhancing catalytic activities by providing a high fraction of exposed edge sites, a high density of active sites and a large surface-to-mass ratio.
 | ||
| Fig. 4 (a) Low-magnification and (b) enlarged SEM images of N–S/C_700. EDX maps of (c) C, (d) O, (e) N, and (f) S collected from the area shown in (a). (g) Low-magnification and (h) enlarged TEM images of N–S/C_700. | ||
The nitrogen adsorption/desorption isotherms of the N–S/C samples exhibited a type IV isotherm according to the Brunauer, Deming, Deming and Teller (BDDT) classification (Fig. S3a and b†).25 The N2 adsorption gradually increased in the 0.2–0.4 relative pressure (P/P0) range, indicative of a mesoporous character. Meanwhile, relatively smaller adsorption occurred in the 0–0.05 P/P0 range originating from some microporosity. A slow increase in adsorption was observed in the mid P/P0 range, ascribed to multilayer adsorption. The N–S/C samples displayed a type H3 loop which does not exhibit any limiting adsorption at P/P0 > 0.8, being associated with a capillary condensation mechanism in the slit-shaped mesopores.25 The Brunauer–Emmett–Teller (BET) surface area and single-point total pore volume of N–S/C_700 were calculated to be approximately 1165.4 m2 g−1 and 0.71 cm3 g−1, respectively (Table S1†), both of which are similar to those of other annealed samples but dramatically higher than the values of the starting lignosulfonate and ammoniated lignosulfonate. The average Barrett–Joyner–Halenda (BJH) pore diameter estimated from the desorption branch of the isotherm was about 3.9 nm (Fig. S3c and d and Table S1†). The high BET surface areas and pores of the N–S/C samples likely correlate with the formation and release of COx, SHx, and NHx species resulting from the decomposition and carbonization of ammoniated lignosulfonate upon annealing. Such structural features are expected to be beneficial for ORR electrocatalysis.26
We studied the electrocatalytic properties of the N–S/C samples for ORR by cyclic voltammetry (CV) and rotating disc electrode (RDE) voltammetry in an alkaline electrolyte (0.1 M KOH). No obvious electrocatalytic response occurred under N2, whereas a pronounced cathodic current with a peak appearing at ∼0.72 V (vs. RHE) was observed upon introducing oxygen (Fig. 5a). Fig. 5b shows the linear scan voltammogram (LSV) curves of original lignosulfonate, ammoniated lignosulfonate, and the N–S/C samples obtained at various annealing temperatures under O2-saturated conditions. Notably, all the N–S/C catalysts displayed more positive ORR onset potentials (0.77–0.80 V vs. RHE) compared to both original lignosulfonate (0.66 V vs. RHE) and ammoniated lignosulfonate (0.63 V vs. RHE) (Fig. 5c), suggesting faster reaction kinetics. The onset potential of the ORR in the N–S/C_700 sample was approximately 0.80 V (vs. RHE), comparable to the values of 0.74 V (vs. RHE) reported for electrochemically synthesized N-doped graphene,27 0.70 V (vs. RHE) for N/S co-doped carbon nanofiber networks,28 0.81 V (vs. RHE) for N/S co-doped graphene,29 and other carbon materials30 (Table S2†). The current densities at 0.7, 0.6, 0.5 and 0.2 V being approximately 2.22, 2.50, 2.63 and 2.86 mA cm−2, respectively, outperform those (≈0.2, 0.5, 0.9, and 1.3 mA cm−2 at 0.7, 0.6, 0.5, and 0.2 V, respectively) reported for N-doped graphene.27 The N–S dual-doped C exhibited better performances than S single-doped C (Fig. S4†). Interestingly, the N–S/C_700 sample outperformed 5 wt% Pt/C in terms of both onset potential (0.78 V vs. RHE) and current density. The limiting current density of N–S/C_700 approaches that of the 20 wt% Pt/C despite a smaller onset potential [0.80 V (vs. RHE) of N–S/C_700 relative to 0.95 V (vs. RHE) of 20% Pt/C obtained in this work] (Fig. 5b).
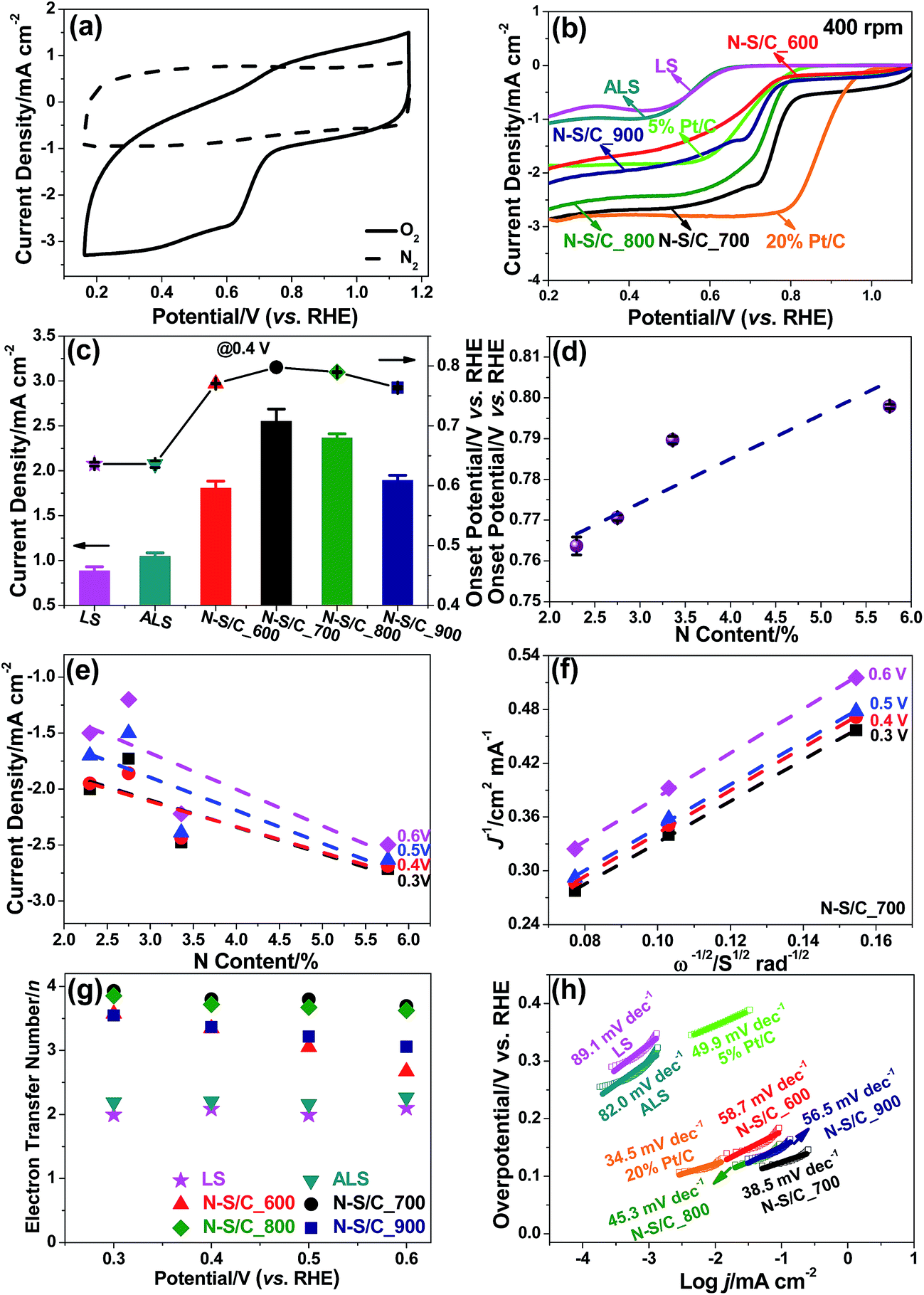 | ||
| Fig. 5 (a) CVs of N–S/C_700 in N2-(dashed line) and O2-saturated (solid line) aqueous 0.1 M KOH at a scan rate of 100 mV s−1. (b) LSVs of original lignosulfonate (LS), ammoniated lignosulfonate (ALS), N–S/C_600, N–S/C_700, N–S/C_800, N–S/C_900, 5 wt% Pt/C, and 20 wt% Pt/C obtained at 400 rpm and 5 mV s−1 in oxygen-saturated 0.1 M KOH. (c) Comparison of onset potential and current density at 0.4 V (vs. reversible hydrogen electrode, RHE) for LS, ALS, N–S/C_600, N–S/C_700, N–S/C_800, and N–S/C_900. Correlation of (d) onset potential and (e) current densities at 0.6, 0.5, 0.4, 0.3 V (vs. RHE) with N contents. (f) K–L plot derived from voltammograms at different rotation speeds. (g) Number of electrons transferred in the ORR, n. (h) Tafel plots, with the respective slopes indicated, for LS, ALS, N–S/C_600, N–S/C_700, N–S/C_800, N–S/C_900, 5 wt% Pt/C, and 20 wt% Pt/C extracted from the LSV results. | ||
We attempted to correlate the onset potential and current density at different potentials for each ORR with overall N content (Fig. 5d and e), graphitic N (Fig. S5a and b†), and pyridinic N concentrations (Fig. S5c and d†). N–S/C_700 with the highest N content, graphitic N and pyridinic N concentrations exhibited the best ORR activity. Although N–S/C_600 contains more total N dopant compared to N–S/C_900, it showed inferior current density. This may be due to the enhanced graphitization degree at higher temperatures, promoting electron transfer that is beneficial for high ORR performance.
The Koutecky–Levich (K–L) lines exhibit a linear relationship between 1/i and 1/w1/2 (Fig. 5f and Fig. S6a–e†). The average number of electrons transferred during oxygen reduction obtained by K–L analysis remained nearly constant in the potential range of 0.3–0.6 V (vs. RHE) for N–S/C_700 and N–S/C_800, corresponding to ∼3.8 for the former and ∼3.7 for the latter (Fig. 5g). Both are higher than those of S- (3.2 at 0.477 V vs. RHE)22 and N-doped graphene (3.2 at 0.75 V vs. RHE),3 and other carbon materials.29,30 This suggests that the ORR catalyzed by N–S/C_700 is primarily dominated by a one-step 4e− process. An increase of the number of transferred electrons with overpotential was observed for N–S/C_600, as illustrated by the increase in the slope of the K–L plots from 0.3 V to 0.6 V (Fig. S6c†), corresponding to a change in n from ca. 2.7 electrons at 0.7 V to ca. 3.6 electrons at 0.3 V (Fig. 5g). This implies that the sequential two step, two-electron pathway with the formation of OOH− as an intermediate predominates oxygen reduction on the N–S/C_600 modified electrode as is commonly observed for metal-free catalysts.31,32 All the N–S/C samples showed remarkably larger values of electrons transferred than original lignosulfonate and ammoniated lignosulfonate (Fig. 5g). The superior ORR activity of N–S/C_700 is also reflected in the much smaller Tafel slope of 38.5 mV dec−1 at low overpotentials than those of N–S/C_600 (58.7 mV dec−1) N–S/C_800 (45.3 mV dec−1) and N–S/C_900 (56.5 mV dec−1) in 0.1 M KOH (Fig. 5h). In addition, the N–S/C_700 displayed a significantly higher double layer capacitor than ammoniated lignosulfonate and other annealed samples (Fig. S7†).
We also explored the ORR electrocatalytic properties of the four N and S co-doped porous carbons in O2-saturated H2SO4 solution (0.5 M). As seen in Fig. S8a,† N–S/C_700 shows a more positive onset potential and higher oxygen reduction current densities than N–S/C_800, N–S/C_900, and N–S/C_600, in line with their catalytic results in alkaline media. Nevertheless, an increase of the number of transferred electrons was observed for the N–S/C_700 sample with ca. 2.8 electrons at 0.1 V to ca. 3.9 electrons at −0.2 V (Fig. S8b†), suggesting a sequential two step two-electron reaction pathway.
We further examined the electrochemical stability and tolerance towards methanol crossover of the N–S/C catalysts. Chronoamperometric measurements showed that the N–S/C_700 catalyst remains stable for ORR for at least 12 h (Fig. 6a). This is in stark contrast to a dramatic loss in activity of up to 50% for 20% Pt/C after a short period time of 4 h. Meanwhile, the N–S/C_700 catalyst displayed excellent immunity towards methanol crossover without evident degradation in oxygen reduction current density upon the addition of 3 M methanol (Fig. 6b). However, the 20% Pt/C electrode showed a dramatic drop in current density when adding methanol as a result of oxidation of methanol on the Pt catalyst.
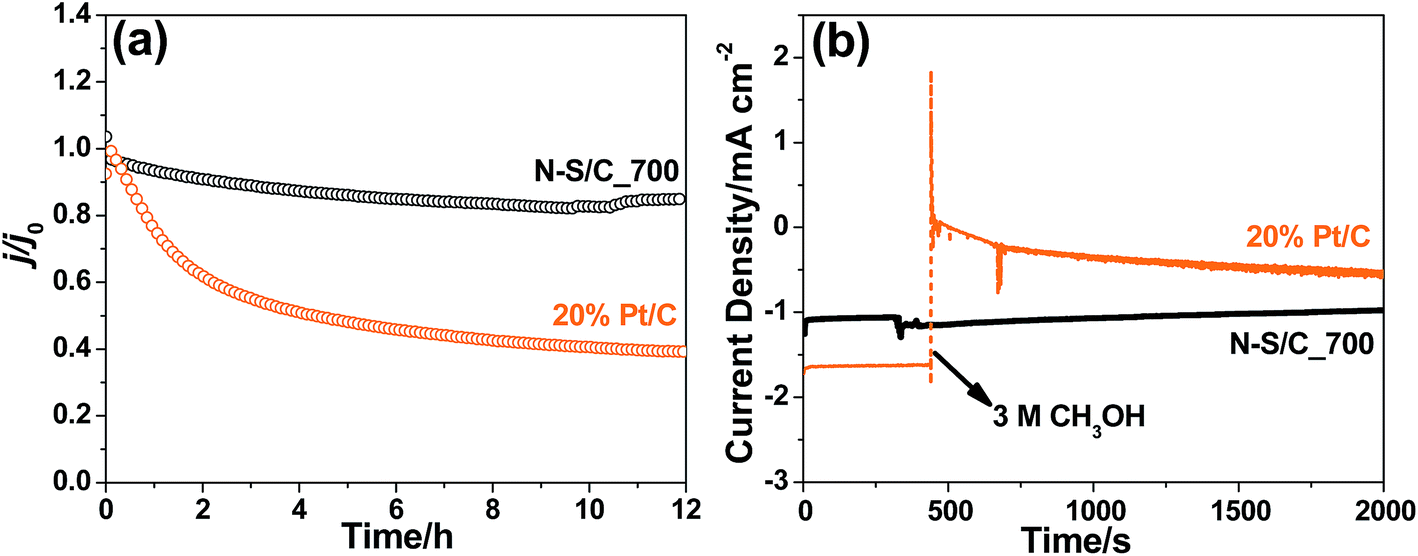 | ||
| Fig. 6 (a) Long-term chronoamperometric stability plots of N–S/C_700 and 20% Pt/C at 0.46 V (vs. RHE) obtained at a rotation speed of 100 rpm in oxygen-saturated 0.1 M KOH. (b) Chronoamperometric responses at 0.46 V (vs. RHE) in O2-saturated 0.1 M KOH on N–S/C_700 and 20% Pt/C electrodes (100 rpm) after adding CH3OH (3 M) at 300 s. | ||
Conclusions
In summary, we have demonstrated that annealing ammoniated lignosulfonate, a bio-renewable material, enabled the synthesis of N and S co-doped porous carbon. SEM, TEM, and N2 adsorption/desorption measurements revealed the formation of honeycomb-like flake networks with large surface areas. XPS confirmed the successful N and S co-doping in the resultant samples. The doping levels and dopant configurations as well as graphitization degree were readily tuned by controlling the thermal treatment temperature. The N and S dual-doped carbon materials exhibited good electrocatalytic activity for the ORR, approaching the benchmark Pt/C (20%) catalyst but showed significantly better durability, which makes the catalysts very promising cathode ORR catalysts in fuel cells. Our results provide a sustainable pathway for exploring heteroatom-doped porous carbons applicable to energy conversion and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Laboratory of Organic–Inorganic Composites (No. oic-201503005); the National Key Research and Development Program of China (2017YFB0306800); the 111 project (B13005); the Fundamental Research Funds for the Central Universities (No. buctrc201525); Beijing National Laboratory for Molecular Sciences (BNLMS20160133); State Key Laboratory of Separation Membranes and Membrane Processes (Tianjin Polytechnic University, No. M2-201704). Justus Masa acknowledges support from The World Academy of Sciences (TWAS), Grant number: 16-072 RG/CHE/AF/AC_I – FR3240293327.References
- H. C. Tao, et al., Two-dimensional nanosheets for electrocatalysis in energy generation and conversion, J. Mater. Chem. A, 2017, 5, 7257–7284 RSC.
- L. T. Qu, et al., Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells, ACS Nano, 2010, 4, 1321–1326 CrossRef PubMed.
- Z. Y. Sun, et al., High-quality functionalized few-layer graphene: facile fabrication and doping with nitrogen as a metal-free catalyst for the oxygen reduction reaction, J. Mater. Chem. A, 2015, 3, 15444–15450 RSC.
- Z. Y. Sun, et al., Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials, Chem, 2017, 3, 560–587 Search PubMed.
- T. Ma, et al., Heterogeneous electrochemical CO2 reduction using nonmetallic carbon-based catalysts: current status and future challenges, Nanotechnology, 2017, 28, 472001 CrossRef PubMed.
- Z. Y. Sui, et al., Nitrogen-doped and nanostructured carbons with high surface area for enhanced oxygen reduction reaction, Carbon, 2018, 126, 111–118 CrossRef.
- Y. Lv, et al., Nitrogen and fluorine-codoped forous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction in fuel cells, ACS Appl. Mater. Interfaces, 2017, 9, 32859–32867 CrossRef PubMed.
- Z. Xiang, et al., Nitrogen-doped holey graphitic carbon from 2D covalent organic polymers for oxygen reduction, Adv. Mater., 2014, 26, 3315–3320 CrossRef PubMed.
- L. Yang, et al., Recent progress in MOF-derived, heteroatom-doped porous carbons as highly efficient electrocatalysts for oxygen reduction reaction in fuel cells, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- L. Yang, et al., Biomass-derived FeNi alloy and nitrogen-codoped porous carbons as highly efficient oxygen reduction and evolution bifunctional electrocatalysts for rechargeable Zn-air battery, Energy Storage Mater., 2018, 12, 277–283 CrossRef.
- L. Yang, et al., Co, N-codoped nanotube/graphene 1D/2D heterostructure for efficient oxygen reduction and hydrogen evolution reactions, J. Mater. Chem. A, 2018, 6, 3926–3932 RSC.
- H. Yu, et al., Cu, N-codoped hierarchical porous carbons as electrocatalysts for oxygen reduction reaction, ACS Appl. Mater. Interfaces, 2016, 8, 21431–21439 CrossRef PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Lignin up for break-down, Nat. Chem., 2014, 6, 1035–1036 CrossRef PubMed.
- B. M. Upton and A. M. Kasko, Strategies for the conversion of lignin to high-value polymeric materials: review and perspective, Chem. Rev., 2016, 116, 2275–2306 CrossRef PubMed.
- Z. R. Zhang, et al., Catalytic transformation of lignocellulose into chemicals and fuel products in ionic liquids, Chem. Rev., 2017, 117, 6834–6880 CrossRef PubMed.
- M. Borghei, et al., Advanced biomass-derived electrocatalysts for the oxygen reduction reaction, Adv. Mater., 2017, 1703691 Search PubMed.
- C. L. Lai, et al., Lignin-derived electrospun carbon nanofiber mats with supercritically deposited Ag nanoparticles for oxygen reduction reaction in alkaline fuel cells, Electrochim. Acta, 2014, 130, 431–438 CrossRef.
- M. Graglia, et al., Nitro lignin-derived nitrogen-doped carbon as an efficient and sustainable electrocatalyst for oxygen reduction, ACS Nano, 2016, 10, 4364–4371 CrossRef PubMed.
- Y. Su, et al., Sulfur-enriched conjugated polymer nanosheet derived sulfur and nitrogen co-doped porous carbon nanosheets as electrocatalysts for oxygen, Adv. Funct. Mater., 2016, 26, 5893–5902 CrossRef.
- H. C. Tao, et al., N-Doping of graphene oxide at low temperature for the oxygen reduction reaction, Chem. Commun., 2017, 53, 873–876 RSC.
- X. Y. Yu, et al., Ultrathin MoS2 nanosheets supported on N-doped carbon nanoboxes with enhanced lithium storage and electrocatalytic properties, Angew. Chem., Int. Ed., 2015, 54, 7395–7398 CrossRef PubMed.
- S. B. Yang, et al., Efficient synthesis of heteroatom (N or S)-doped graphene based on ultrathin graphene oxide-porous silica sheets for oxygen reduction reactions, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef.
- M. Borghei, et al., Porous N, P-doped carbon from coconut shells with high electrocatalytic activity for oxygen reduction: Alternative to Pt–C for alkaline fuel cells, Appl. Catal., B, 2017, 204, 394–402 CrossRef.
- Y. Zhang, et al., Graphene/porous beta TiO2 nanocomposites prepared through a simple hydrothermal method, Current Graphene Science, 2017, 1, 64–70 CrossRef.
- K. S. W. Sing, et al., Reporting physisorption data for gas/solid systems, Pure Appl. Chem., 1985, 57, 603–619 Search PubMed.
- C. R. Raj, et al., Emerging new generation electrocatalysts for the oxygen reduction reaction, J. Mater. Chem. A, 2016, 4, 11156–11178 RSC.
- Y. Jiao, et al., Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: A roadmap to achieve the best performance, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef PubMed.
- T. Liu, et al., CoO nanoparticles embedded in three-dimensional nitrogen/sulfur co-doped carbon nanofiber networks as a bifunctional catalyst for oxygen reduction/evolution reactions, Carbon, 2016, 106, 84–92 CrossRef.
- W. Ai, et al., Nitrogen and sulfur codoped graphene: multifunctional electrode materials for high-performance li-ion batteries and oxygen reduction reaction, Adv. Mater., 2014, 26, 6186–6192 CrossRef PubMed.
- T. Akhter, et al., Self-assembled N/S codoped flexible graphene paper for high performance energy storage and oxygen reduction reaction, ACS Appl. Mater. Interfaces, 2016, 8, 2078–2087 CrossRef PubMed.
- J. Masa, et al., Trace metal residues promote the activity of supposedly metal-free nitrogen-modified carbon catalysts for the oxygen reduction reaction, Electrochem. Commun., 2013, 34, 113–116 CrossRef.
- J. Masa, et al., Metal-free catalysts for oxygen reduction in alkaline electrolytes: Influence of the presence of Co, Fe, Mn and Ni inclusions, Electrochim. Acta, 2014, 128, 271–278 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00231b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |