
Improving the stability and decreasing the trap state density of mixed-cation perovskite solar cells through compositional engineering†
Kianoosh
Poorkazem
 and
Timothy L.
Kelly
*
and
Timothy L.
Kelly
*
Department of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada. E-mail: tim.kelly@usask.ca
First published on 17th April 2018
Abstract
In order to become a commercially viable technology, perovskite solar cells will require both high efficiencies and long-term stability. Many of the highest efficiency devices reported to date have used complex perovskite formulations synthesized from PbI2, formamidinium iodide, PbBr2, and CH3NH3Br; the addition of a small amount of CsI to this composition can further improve the device efficiency. Unfortunately, previous work has also shown that the methylammonium cation can not only make the perovskite more hygroscopic, it also renders it susceptible to photochemical decomposition; additionally, mixed-halide perovskite compositions have been shown to suffer from a phenomenon known as halide segregation. Here we show that both PbBr2 and CH3NH3Br can be omitted from Cs-containing perovskite devices with little change in device efficiency. We also show that replacing these components with PbI2 and guanidinium iodide leads to a decrease in the trap state density and an increase in the open-circuit voltage. Importantly, the mixed-cation perovskite shows improved moisture resistance and photochemical stability in both thin films and devices.
Introduction
Perovskite solar cells have emerged as one of the most promising new solar technologies in decades. These devices are made from low-cost materials, and their low temperature fabrication processes allow them to be used in flexible1–3 and lightweight4 applications. Additionally, the power conversion efficiency (PCE) of these devices is on-par with market-leading multi-crystalline silicon technology. The high efficiency of these devices is driven by the relatively low bandgap of methylammonium (MA) lead iodide (1.53 eV) and a low loss-in potential (the difference between the bandgap energy and the device open-circuit voltage).5,6 With single-junction perovskite solar cells now exceeding 20% PCE, further efficiency increases are expected to come from further reductions in the loss-in potential,7–9 which can be done by decreasing the density of trap states that act as charge recombination centers in a device.10–13Compositional engineering of the perovskite is one strategy to improve device efficiency. Although MAPbI3 was the first compound to be used in a perovskite solar cell, in the ABX3 perovskite structure, the A-, B-, and X-site ions can all be replaced by suitable alternatives. Since the bandgap of the perovskite-structured formamidinium (FA) lead iodide (α-FAPbI3) is narrower (1.48 eV)14 than that of MAPbI3 (1.53 eV),15 it is possible to entirely replace MA with FA and achieve higher photocurrents. However, at room temperature, pure FAPbI3 is easily converted from the α to a δ phase, which has a wider bandgap and a one-dimensional, non-perovskite structure.16,17 Therefore, one of the important goals of compositional engineering is the stabilization of the α-FAPbI3 phase. Initially, FAPbI3 was mixed with 15 mol% of either MAPbI3,18 MAPbBr3,14 or CsPbI3.19 Later, Jacobsson et al. comprehensively studied the effect of FA(1−x)MAxPbI(3−y)Bry perovskite composition on device efficiency, and reported that the highest efficiency could be achieved when the FA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MA and I:Br ratios were 2:1 and 5:1, respectively.20 To date, the majority of devices with >20% PCE have used this perovskite composition (or slight modifications thereof).8,20–28
:MA and I:Br ratios were 2:1 and 5:1, respectively.20 To date, the majority of devices with >20% PCE have used this perovskite composition (or slight modifications thereof).8,20–28
Other examples of compositional engineering have looked at the use of different A-site cations entirely. A few reports have examined the use of guanidinium (GA) cations, owing to their similarity in structure to FA. A computational study calculated a low formation energy for the replacement of FA with GA in FAPbI3,29 and an experimental study demonstrated a decrease in the number of recombination centers when a small amount of MA was replaced with GA in MAPbI3.30 However, the compatibility of GA with various state-of-the-art perovskite compositions, and its effect on device efficiency and lifetime, remains unknown.
Although improving device efficiency is important, the instability of perovskite devices is the most crucial issue to be addressed. Stability improvements have been made by modifying interfacial layers,8,31–36 changing the perovskite deposition method,23,37–39 or by altering the perovskite composition.7,25,40–43 Among these, perovskite compositional engineering is the most general strategy, since it can be applied to a wide variety of different device configurations.
In one notable example of compositional engineering, Saliba et al. showed that adding CsI to the FA(1−x)MAxPbI(3−y)Bry perovskite improved device efficiency, stability, and reproducibility.22 One reason for this is the small size of Cs+, which decreases the large Goldschmidt tolerance factor44 of FAPbI3, making the cubic structure more favorable.19 Additionally, the α phases of FAPbI3 and CsPbI3 have more similarities in terms of their atomistic structure and unit cell volume, which is not the case for δ phases. This makes the mixing of the α phases (and not the δ phases) energetically favorable,45 and helps remove δ-phase impurities from the perovskite film. Later, a third reason was shown to be a more compact perovskite layer, resulting from the stronger interaction of the A-site cations with the PbI3− framework.46 A device that used only PbI2, FAI, and CsI precursors was shown to be both efficient and relatively stable.47 This calls into question the necessity of including PbBr2 and MABr in the perovskite precursor solution.
Such a complex perovskite formulation (which includes three different A-site cations and two different halide anions) is not necessarily ideal. One drawback is the halide segregation phenomenon observed in mixed I/Br perovskites, where distinct I-rich and Br-rich phases are formed under illumination.48,49 This affects the long-term stability of even encapsulated solar cells. Although this segregation is shown to be suppressed in FA(1−x)CsxPbI(3−y)Bry perovskites when x = 0.1–0.3, the results are dependent on both the perovskite fabrication method and the homogeneity of the Cs distribution.50 Eliminating the possibility of halide segregation, while retaining excellent device efficiency, would be highly desirable. Another problem with this complex composition is the instability of MA-containing perovskites.51 In a humid environment, MAPbI3 absorbs water, crystallizes into distinct hydrate phases, and eventually decomposes irreversibly.52–54 The MA-containing perovskites are also prone to oxidative photobleaching. Photoexcitation of the perovskite produces conduction band electrons, which can reduce O2 to superoxide, which in turn deprotonates the MA cation.55 In our previous work, we observed improved moisture resistance and photostability for FA(1−x)MAxPbI3 perovskites with higher percentages of FA.51 The results suggest that the elimination of MA may substantially improve perovskite stability.
Here, we report on a mixed-cation perovskite that improves perovskite stability, but at the same time does not reduce PCE. We show that the methylammonium and bromide components of a Cs-containing perovskite are unnecessary; although the elimination of bromide reduces the bandgap (decreasing Voc), this is compensated for by an increase in Jsc, and no overall change is observed in the PCE. Furthermore, by adding GAI and PbI2 to the perovskite precursor solution, we decreased the density of trap states in the perovskite layer and minimized the device loss-in potential. Ultimately, the elimination of unstable perovskite components leads to an improvement in the moisture resistance and photostability of these perovskite devices.
Results and discussion
Perovskite film deposition and characterization
In order to fabricate perovskites with different compositions, we spin-coated films using three different precursor solutions (Fig. 1). The first perovskite was prepared using three different cations (MA, FA, Cs) in the A-site, and two different halide anions (I, Br); this composition has been used previously to produce some of the highest-efficiency devices to date.22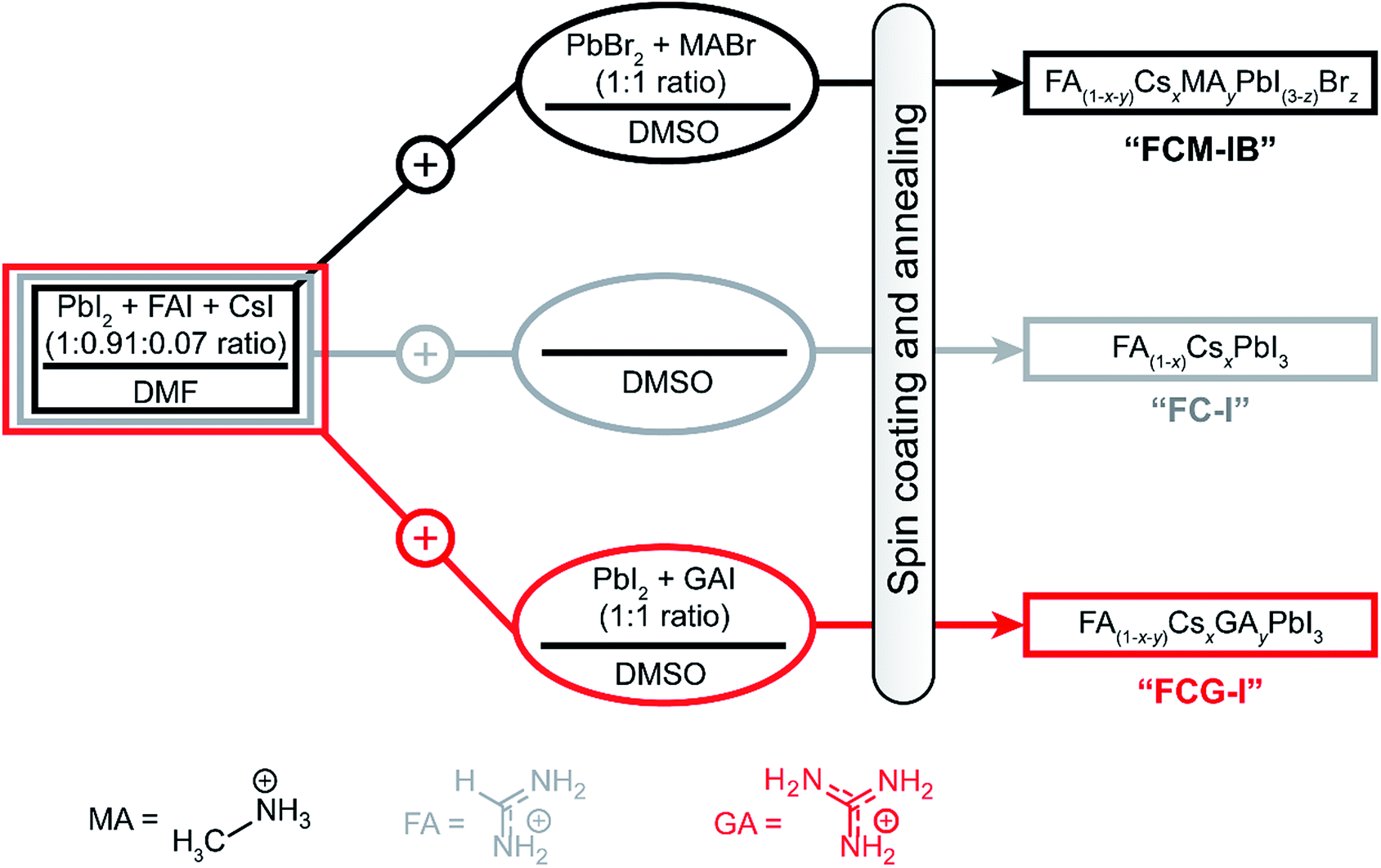 | ||
| Fig. 1 Sample preparation process for the perovskite films used in this study. | ||
The precursor solution was made by mixing a N,N-dimethylformamide (DMF) solution of PbI2, FAI, and CsI with a dimethyl sulfoxide (DMSO) solution of PbBr2 and MABr (refer to ESI for details†). The perovskite fabricated from this solution is denoted FCM-IB, based on the first letters of the constituent A-site and X-site ions. In order to determine whether PbBr2 and MABr are needed to produce high-efficiency devices, the second set of samples eliminated these components from the precursor solution. In this case, pure DMSO was added to the same stock DMF solution as before. The resulting FA(1−x)CsxPbI3 samples are denoted FC-I. Finally, to explore the effect of GA ions, the stock DMF solution was mixed with a DMSO solution of GAI and PbI2. The perovskite produced from this solution is denoted FCG-I. The MA and bromide ions were both eliminated from the FC-I and FCG-I samples.
After spin-coating and annealing, the perovskite films were analyzed by powder X-ray diffraction (pXRD). The pXRD patterns revealed a small amount of residual lead halide in the FCM-IB sample (indicated by a peak at 14.5°); however, this was absent in both FC-I and FCG-I (Fig. 2a), highlighting the better phase purity achieved when PbBr2 and MABr are eliminated from the precursor solution. The absorption and emission spectra also clearly show the effect of this elimination (Fig. 2b). Since pure MAPbBr3 has a higher bandgap (2.24 eV)15 than pure α-FAPbI3 (1.48 eV),14 eliminating MA and bromide from the film led to a red-shift in both the absorption and the emission spectra. The diffraction peaks for FCG-I are also more intense than those of FC-I, suggesting improved crystallinity of the perovskite phase; however, the FCG-I peaks did not shift in position relative to FC-I, and no new peaks were observed in the pXRD pattern. If GA was successfully incorporated into a perovskite lattice, either smaller-angle peaks should appear in the pXRD pattern (for the appearance of a new phase),30 or the peaks should shift to smaller angles (resulting from an enlargement of the existing perovskite unit cell). Since neither of these were observed, we conclude that GA does not incorporate into the perovskite structure. Absorption and emission spectra support this conclusion. Both FCG-I and FC-I were determined to have the same optical bandgap (1.53 eV, Fig. S1, ESI†); however, pure GAPbI3 has a larger bandgap (1.55 eV)30 than α-FAPbI3, and the inclusion of GA would be expected to blue-shift the bandgap.
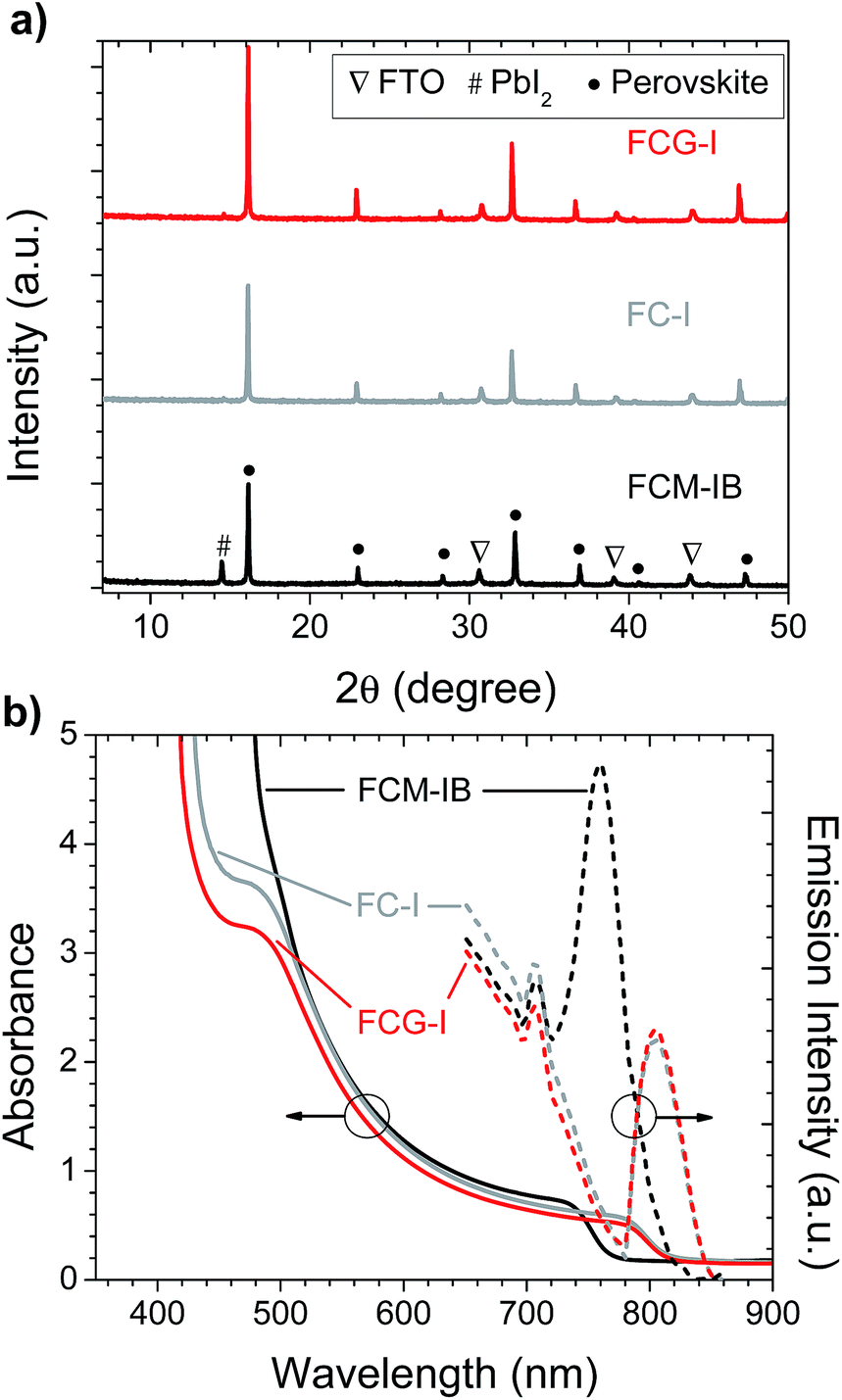 | ||
| Fig. 2 (a) pXRD patterns, and (b) absorption and photoluminescence spectra, of perovskite films prepared on glass/FTO/compact-TiO2 substrates. | ||
Device fabrication and characterization
Having characterized the perovskite films, we set about testing them in perovskite solar cells. We initially used a planar-heterojunction device architecture: FTO/c-TiO2/perovskite/spiro-OMeTAD/Ag (where FTO is fluorine-doped tin oxide and spiro-OMeTAD is 2,2′,7,7′-tetrakis-(N,N′-di-p-methoxyphenylamino)-9,9′-spirobifluorene). Because of the low intrinsic hole mobility of spiro-OMeTAD,56–60 we first attempted to optimize the lithium bis(trifluoromethane)sulfonimide (Li-TFSI) doping level (Table S1, Fig. S2, ESI†). This resulted in efficient devices, but during the subsequent optimization of the GAI concentration in FCG-I cells (Table S2, Fig. S3, ESI†), we observed that excess Li-TFSI led to more rapid perovskite decomposition (Fig. S4, ESI†). This is consistent with previous studies showing that hygroscopic Li-TFSI can facilitate the absorption of moisture,61,62 and that the migration of Li+ into the perovskite layer can further undermine its stability.63 Instead, we used a double hole-transport layer consisting of a thin layer of WO3 deposited on top of a more lightly-doped spiro-OMeTAD layer.64–66 This produced devices with performances similar to those that employed much higher Li-TFSI doping levels (Table S3, Fig. S5, ESI†).Having settled on the use of the spiro-OMeTAD/WO3 hole-transport layer, we fabricated and tested perovskite solar cells with each of the three perovskite films. The electrical parameters of the devices are tabulated in Table 1, and the J–V curves, IPCE spectra, and loss-in potentials are shown in Fig. 3. Comparing the data for the FCM-IB and FC-I devices, the elimination of bromide results in a lower average Voc, but a higher average Jsc and FF. The changes in Voc and Jsc are consistent with the Shockley–Queisser calculations,67 where a narrower bandgap semiconductor (i.e., FC-I) absorbs more light and produces a higher current density, but at the expense of voltage. This results in a net-zero change in performance, and the PCEs of both sets of devices were almost identical. In the case of the GA-doped sample (FCG-I), the addition of GAI and PbI2 to the precursor solution appears to have an overall positive effect on device performance. The current density and fill factor do not materially change; however, the open-circuit voltage increases by ca. 23 mV. As a result, the champion device in this study was prepared using FCG-I, and had a PCE of 17.7%.
| Perovskite | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | |
|---|---|---|---|---|---|
| FCM-IB | Average | 1.08 ± 0.01 | 20.6 ± 0.5 | 0.71 ± 0.01 | 15.7 ± 0.5 |
| Best | 1.091 | 21.7 | 0.70 | 16.7 | |
| FC-I | Average | 0.990 ± 0.007 | 21.6 ± 0.7 | 0.74 ± 0.01 | 15.8 ± 0.5 |
| Best | 1.001 | 22.0 | 0.76 | 16.7 | |
| FCG-I | Average | 1.013 ± 0.006 | 21.7 ± 0.7 | 0.74 ± 0.02 | 16.3 ± 0.5 |
| Best | 1.025 | 23.4 | 0.74 | 17.7 | |
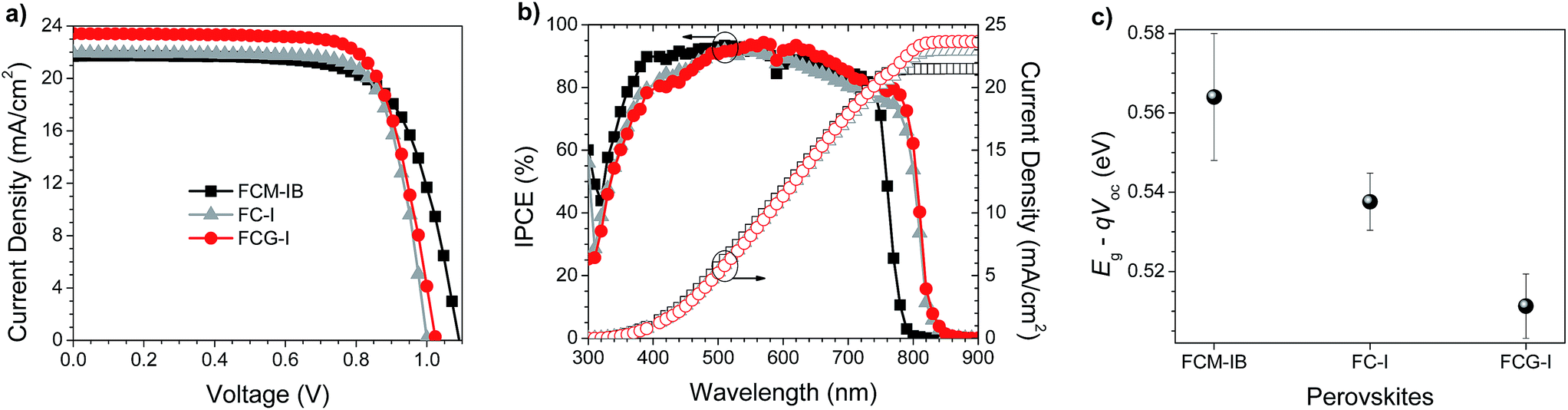 | ||
| Fig. 3 (a) J–V curves and (b) IPCE spectra (solid symbols) and integrated current density (open symbols), of champion devices. (c) Loss-in potential of perovskite solar cells; values (and their associated uncertainties) are calculated from the average Eg and Voc values. | ||
The incident photon-to-current efficiency (IPCE) spectra of champion devices were then measured (Fig. 3b). Eliminating MA and bromide from the composition red-shifted the IPCE spectra, consistent with the behavior observed in the optical absorption spectra (Fig. 2b). The effect on short-circuit current densities is immediately apparent; both FC-I and FCG-I devices had noticeably higher Jsc values. All of the Jsc values calculated from the IPCE spectra (measured under the pseudo-steady-state conditions of the IPCE experiment) agree to within 5% of the Jsc values obtained from the J–V curves.
To better understand the reason for the higher performance of the FCG-I devices, the average loss-in potential for each device set was calculated (Fig. 3c). The loss-in potential was calculated as the difference between the optical bandgap (Fig. S1, ESI†) and the open-circuit voltage (qVoc). Although the FCM-IB devices had the highest Voc, they also had the highest loss-in potential. The loss-in potential was slightly lower for FC-I devices, and lower again for FCG-I. This suggests that the elimination of MA and bromide may result in a reduction in the number of trap states, and that the addition of GAI may help further passivate traps (Fig. 4).
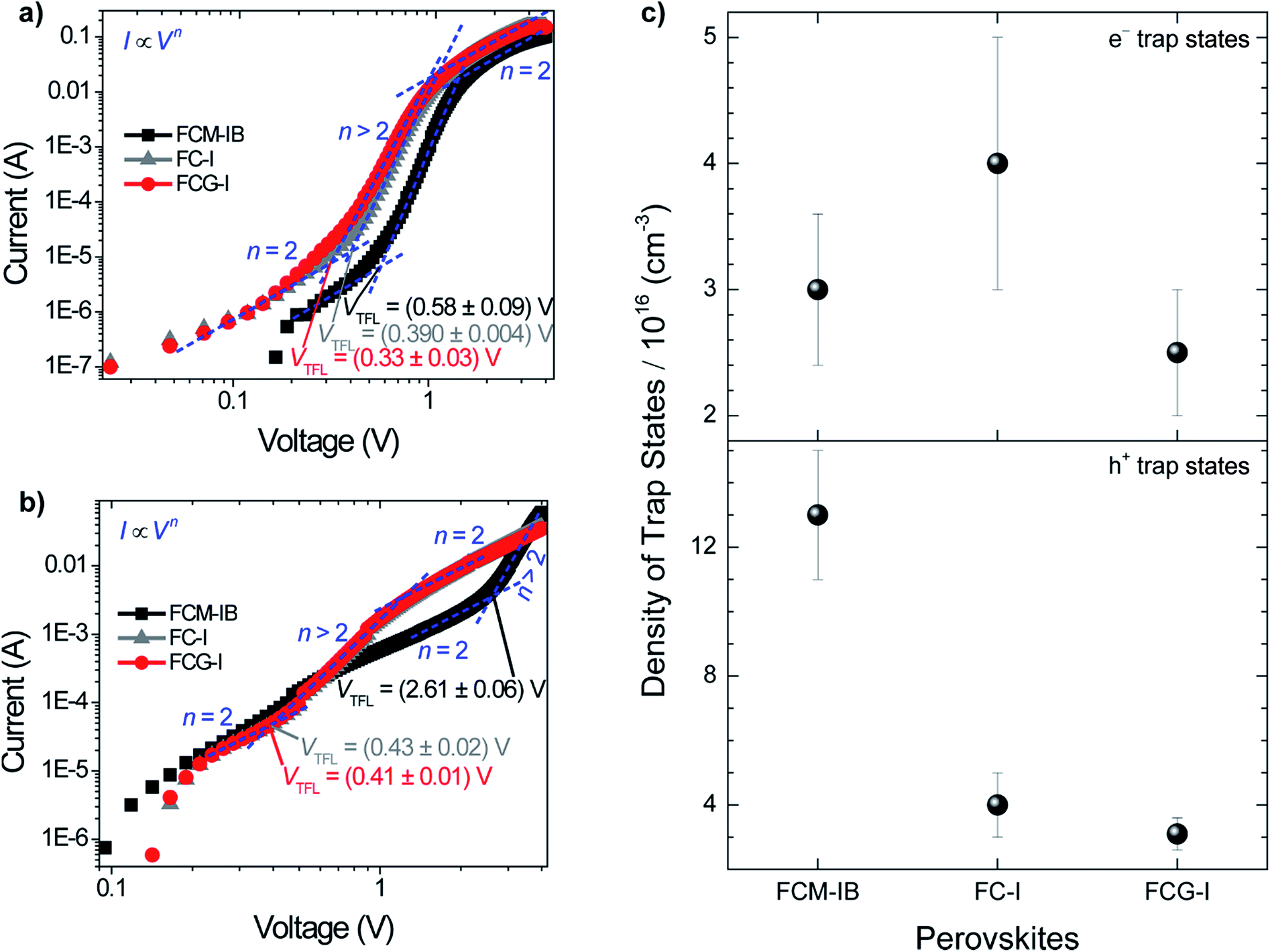 | ||
| Fig. 4 Representative dark current measurements for: (a) e−-only (FTO/c-TiO2/perovskite/PC61BM/Au) and (b) h+-only (FTO/PEDOT:PSS/perovskite/spiro-OMeTAD/WO3/Au) devices. The reported uncertainties are the standard deviations of measurements on 5–11 devices of each type. (c) The density of trap states for electrons and holes in different perovskite layers. Errors are propagated from the standard deviations of film thicknesses and VTFL values. | ||
To evaluate the trap state density in our devices, we fabricated electron-only and hole-only devices by sandwiching each of the three perovskites between two sets of electron transport layers or two sets of hole transport layers, respectively.11,68 This produces the conditions for space-charge limited current (SCLC).69 An I–V curve was acquired in the dark and plotted on a double logarithmic graph.69 Under ideal SCLC conditions, four distinct regions are observed. They are defined by n, the slope of the best-fit line in each region: ohmic (n = 1), trap-limited SCLC (n = 2), trap-filled limit (n = ∞), and trap-free SCLC (n = 2). A value of n = ∞ for the trap-filled limit region occurs only when the energy levels of the trap states are identical. However, a value of n > 2 is representative of this region in real cases, where a distribution of energy levels leads to a gradual increase in current with increasing voltage. The end of the trap-limited SCLC region is when the trap states become filled with charge carriers. The voltage at this point, known as the trap-filled limit voltage (VTFL), is used to calculate the density of trap states based on the following equation:
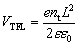 | (1) |
The decreased density of trap states in FCG-I could be due to either the passivation of defects or the suppression of iodide migration. A previous study on MAPbI3 devices suggested that two underlying phenomena could be responsible for the decreased density of trap states when MA was replaced by GA.30 One was the passivation of undercoordinated iodide ions at grain boundaries by the Lewis acidic GA ion; the other was the suppression of deep iodide vacancies within the perovskite structure. Other recent work showed the ability of KI to suppress iodide migration in a perovskite with similar composition to the FCM-IB formulation in this work.72 Similar to GAI, KI cannot incorporate into the perovskite structure and stay in grain boundaries. Therefore, it is fair to assume that GAI could suppress iodide migration, which led to the observed reduction in trap state density. This is consistent with the increase in film crystallinity observed for the FCG-I films, as compared to the FC-I controls (Fig. 2a).
Perovskite film stability
In order to determine the relative stability of the various perovskite compositions to moisture, light, and oxygen, the pXRD patterns of the perovskite films were monitored while the films were irradiated at 1 sun intensity in air at 85% relative humidity (RH, Fig. 5). This was accomplished by diluting a flow of saturated water vapor to 85% RH with dry air, and directing the flow into a transparent sample chamber; samples were irradiated from below using a white LED array.51 We focused our stability studies on a high-performance perovskite composition from the literature (FCM-IB) and our highest-performing perovskite composition (FCG-I). For FCM-IB, the intensity of the perovskite diffraction peaks decreased rapidly; after 25 h of exposure, the most intense peak at ∼16° had almost entirely disappeared (Fig. 5). Previous reports51 have shown that MAPbI3 decomposes into crystalline PbI2. In contrast, the amount of crystalline PbI2 in FCM-IB actually decreased during exposure, as evidenced by a reduced intensity of the diffraction peak at ∼14.5°. Similarly, FCG-I, which initially showed no crystalline PbI2 in the pXRD pattern (Fig. 2a), did not produce any crystalline PbI2 after 30 h of exposure. The lack of PbI2 was also evidenced by the lack of its distinct yellow color during the decomposition process. Other non-perovskite crystalline phases were also missing from the pXRD pattern (e.g., δ-FAPbI3 at ∼13.6° (ref. 14 and 51) or δ-CsPbI3 at ∼13.1° (ref. 50)). These results might imply a gradual amorphization of the layers, or an entirely different decomposition mechanism for the FAPbI3-based perovskites (as compared to MAPbI3). Comparing the overall rate of decomposition, the FCG-I samples decomposed more slowly than FCM-IB; the intensity of the perovskite diffraction peaks in the FCG-I sample dropped to approximately half of their initial intensity after 30 h of exposure, whereas the FCM-IB samples had completely decomposed. Similar results were obtained in a duplicate experiment (Fig. S6, ESI†). Control samples kept in the dark in a glovebox (∼1 ppm O2 and H2O) showed virtually no decomposition over the same time period (Fig. S7, ESI†), highlighting the moisture and photochemically-driven nature of the decomposition process.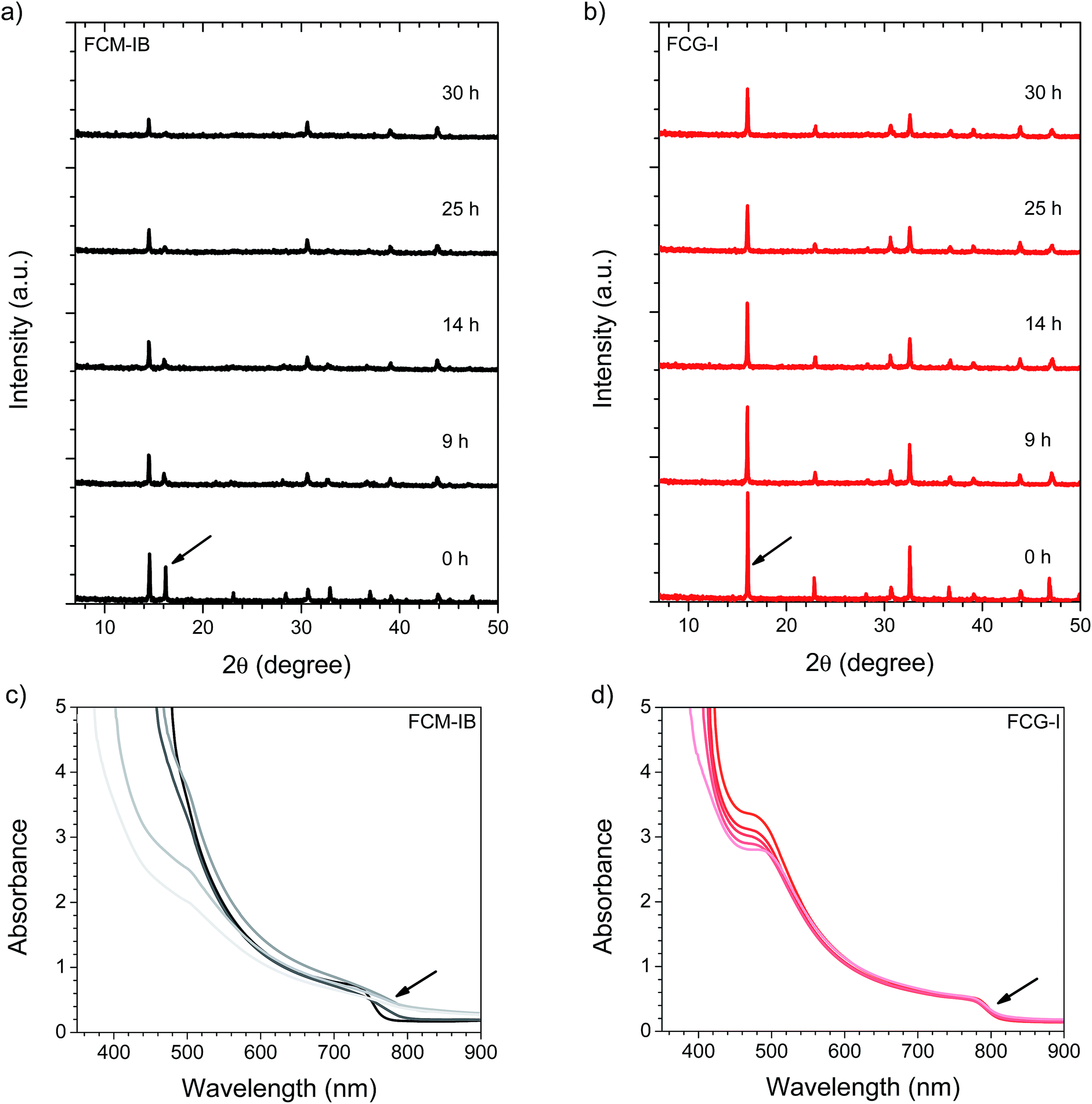 | ||
| Fig. 5 (a and b) pXRD patterns and (c and d) absorbance spectra of the perovskite films throughout the 30 h exposure to 85% RH and 1 sun irradiation in air (the UV/vis time series goes from dark to light). Arrows highlight: (a and b) the most intense perovskite peak in the pXRD patterns, and (c and d) the band edge region of the UV/vis spectra. | ||
Absorption spectroscopy and scanning electron microscopy (SEM) were used to evaluate the optical and morphological changes that occurred during our stability study. In the UV/vis spectrum, the band-edge features in FCM-IB started to flatten after only the first few hours of exposure (Fig. 5c); this was accompanied by a pronounced loss of absorbance in the blue region of the spectrum (400–500 nm). One key observation is that the band edge of this sample red-shifts after exposure to our environmental chamber. This is consistent with our previous work on (MA)1−x(FA)xPbI3 perovskites, where selective decomposition of MA-rich domains led to a red-shift in the apparent bandgap.51 In contrast, the absorption spectra of FCG-I showed smaller changes in the blue region and almost no changes near the band edge over the course of the study (Fig. 5d). Again, a duplicate experiment (Fig. S8, ESI†) showed very similar results, and control samples stored in the dark in the glovebox showed negligible decomposition (Fig. S9, ESI†). To see the effect of these conditions on the film morphology, we captured top-view SEM images before and after 29 h of exposure (Fig. S10, ESI†). Consistent with the pXRD and UV/vis results, the FCM-IB film had noticeably decomposed, taking on a more porous, sponge-like appearance. In contrast, the FCG-I film retained more of its original structure, with the only noticeable change being the appearance of tiny pinholes at the grain boundaries.
Device stability
Finally, we evaluated the stability of complete devices under the same set of environmental conditions (85% RH, 1 sun illumination in air). Gold electrodes were used instead of silver, since silver electrodes have already been demonstrated to accelerate the device degradation process.51,73 After 40 min of exposure to the relatively harsh conditions used for our stability test, none of the devices was able to tolerate the applied bias needed to acquire a J–V curve. This may be due to more rapid ion migration within the perovskite layer after it has absorbed moisture, and will be the subject of further study. Nonetheless, the IPCE of the devices could still be measured, since it is measured under short-circuit conditions. We measured the IPCE spectrum of 4–5 devices for each type of perovskite and averaged the spectra, a process repeated for each exposure time (Fig. 6). In both the FCM-IB and FCG-I devices, the IPCE spectra decreased evenly across the spectrum as a function of aging time, and there was also relatively little change at the red-edge of the spectra. In contrast, however, the FCM-IB devices displayed a more rapid loss in IPCE than the FCG-I cells. After 180 min of exposure, the highest points in their IPCE spectra were ∼40% and ∼60%, respectively. The Jsc values associated with each of the IPCE spectra were then calculated and plotted against exposure time (Fig. 6c). At early exposure times, the FCG-I devices retained almost all of their initial photocurrent, whereas there was a significant loss of performance in the FCM-IB cells. After 40 min of aging, however, the rate of decomposition was similar for both sets of cells.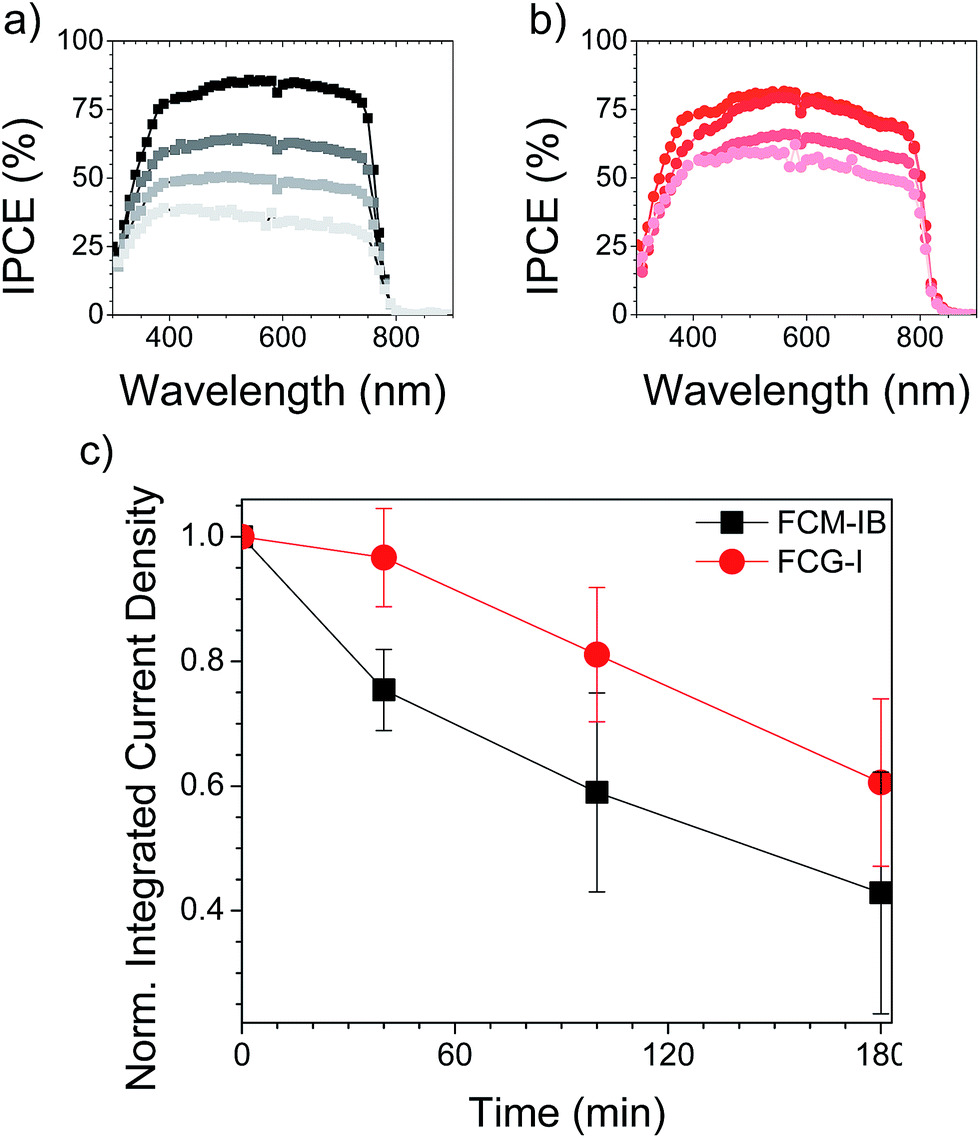 | ||
| Fig. 6 Average IPCE spectra (measured on 4–5 devices each) of (a) FCM-IB and (b) FCG-I devices as a function of exposure time to 85% RH and 1 sun irradiation in air. The time series goes from dark to light. (c) Integrated current densities calculated from the IPCE spectra in (a) and (b). The error bars represent plus-or-minus one standard deviation from the mean. | ||
To see how the device stack was affected by the test, we captured cross-sectional SEM images after 6 h of exposure (Fig. 7). The SEM images of the fresh devices were quite similar to each other; however, the perovskite/spiro-OMeTAD interface in the FCM-IB device was completely destroyed after exposure. There was also a marked increase in the total thickness of the perovskite layer, which may be due to swelling of the film after it absorbs moisture during the test. This process appears to have just started in the FCG-I sample; there is a roughening of the perovskite/spiro-OMeTAD interface, but the effect is much less pronounced than in the FCM-IB device. Overall, these results are consistent with our analysis of the perovskite thin films, which showed improved stability of the FCG-I layer.
 | ||
| Fig. 7 Cross-sectional SEM images of glass/FTO/c-TiO2/perovskite/spiro-OMeTAD/WO3/Au devices before and after 6 h of exposure to 85% RH and 1 sun irradiation in air. All scale bars are 1 μm. | ||
Conclusions
In this work, we have demonstrated that the MA and bromide ions are not essential components in high-performing perovskite solar cells. Devices made using FC-I had performances equal to those of their FCM-IB counterparts. We additionally showed that GA ions can be used to passivate trap states in these formamidinium/cesium mixed-cation perovskites, resulting in a decrease in the loss-in potential and an improvement in PCE. Furthermore, these perovskite compositions appear to be more stable than the FCM-IB systems widely used in the literature, and completely eliminate the possibility of halide segregation. Ultimately, this is a step toward a perovskite solar cell that is both highly efficient and more stable.Experimental section
Device fabrication
FTO-coated (2.5 cm × 2.5 cm) glass slides were cleaned by sequential sonication in 2% aqueous Extran 300 detergent, deionized water, and 2-propanol (20 min each), and drying in an oven at 120 °C for at least 2 h. A 0.1 M solution of titanium diisopropoxide bis(acetylacetonate) in 1-butanol was then spin-coated three times at 4000 rpm for 10 s. In between each coating step, the samples were annealed at 125 °C for 5–10 min and allowed to cool for 1 min.47 After sintering at 450 °C for 0.5 h and cooling to room temperature, a 0.1 M solution of Li-TFSI in acetonitrile was spin-coated at 3000 rpm for 10 s, followed by annealing at 450 °C for 0.5 h.22,74 The samples were transferred while hot (T > 150 °C) to a nitrogen-filled glovebox (H2O and O2 levels ∼1 ppm). To deposit the perovskite layer, a stock solution of 1.375 M PbI2, 1.250 M FAI, and 0.0987 M CsI in anhydrous DMF was first prepared. Anhydrous DMSO solutions of 0.792 M MABr and 0.792 M PbBr2 (FCM-IB), pure DMSO (FC–I), and 0.0833 M GAI and 0.0833 M PbI2 (FCG-I) were prepared separately. The DMF and DMSO solutions were combined in a 76/24 (v/v) ratio, and filtered through 0.45 μm polytetrafluoroethylene syringe filters. The filtered solution was dispensed on the TiO2-coated substrates and spin-coated at 1000 rpm for 10 s followed by 6000 rpm for 20 s. In the last 5 s of the spin-coating, 500 μL of a 50/50 (v/v) mixture of chlorobenzene and chloroform was dispensed as an anti-solvent. Immediately after spinning, the slides were annealed at 140 °C for about 0.5 h, and slowly cooled to room temperature. The samples were removed from the glovebox. 18 μL of 4-tert-butyl pyridine and 18 μL of a 2.0 M Li-TFSI solution in acetonitrile were added to 1000 μL of a 70.0 mM spiro-OMeTAD solution in chlorobenzene, and the resulting solution was syringe filtered through a 0.45 μm polytetrafluoroethylene filter. After filtering, this solution was spin-coated onto the perovskite layer at 500 rpm for 3 s, followed by 4000 rpm for 20 s. The films were returned to the glovebox, and allowed to dry in an open Petri dish overnight. A second WO3 hole-transport layer (∼45 nm thick) was deposited by thermal evaporation (∼0.7 Å s−1) at a base pressure of 1 × 10−6 mbar. Finally, Au or Ag top electrodes were again deposited by thermal evaporation at a base pressure of 1 × 10−6 mbar.Characterization
pXRD patterns were measured on a PANalytical Empyrean diffractometer with a Co X-ray source. Absorption and fluorescence spectra were acquired on a Cary 6000i UV-vis-NIR spectrophotometer and a PTI fluorometer (excitation wavelength of 460 nm), respectively. SEM images were taken by a Hitachi SU8010 microscope. J–V curves were acquired using a Keithley 2400 source-measure unit. Devices were measured in the glovebox using a 450 W Class AAA solar simulator equipped with a AM1.5G filter (Sol3A, Oriel Instruments); a standard silicon reference cell (91150V, Oriel Instruments) was used to set the light intensity at 1 sun. A non-reflective black-anodized aluminum aperture mask was used to define a 0.101 cm2 cell area. IPCE measurements were performed on the highest efficiency devices under ambient conditions using a QE-PV-SI system (Oriel Instruments) consisting of a 300 W Xe arc lamp, monochromator, chopper, lock-in amplifier and certified silicon reference cell. Measurements were made using a 30 Hz chop frequency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (NSERC) and the University of Saskatchewan are acknowledged for financial support. T. L. K. is a Canada Research Chair in Photovoltaics. The research was undertaken, in part, thanks to funding from the Canada Research Chair program. The Western College of Veterinary Medicine Imaging Centre is acknowledged for SEM imaging. Mr K. M. Fransishyn is acknowledged for experimental assistance.References
- K. Poorkazem, D. Liu and T. L. Kelly, J. Mater. Chem. A, 2015, 3, 9241–9248 CAS.
- D. Liu and T. L. Kelly, Nat. Photonics, 2014, 8, 133–138 CrossRef CAS.
- M. Ye, X. Hong, F. Zhang and X. Liu, J. Mater. Chem. A, 2016, 4, 6755–6771 CAS.
- M. Kaltenbrunner, G. Adam, E. D. Glowacki, M. Drack, R. Schwodiauer, L. Leonat, D. H. Apaydin, H. Groiss, M. C. Scharber, M. S. White, N. S. Sariciftci and S. Bauer, Nat. Mater., 2015, 14, 1032–1039 CrossRef CAS PubMed.
- P. K. Nayak and D. Cahen, Adv. Mater., 2014, 26, 1622–1628 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, A. J. Barker, G. Grancini, W. Zhang, J. M. Ball, A. R. S. Kandada, H. J. Snaith and A. Petrozza, Energy Environ. Sci., 2016, 9, 3472–3481 CAS.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green, J. T.-W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot, R. May, A. Palmstrom, D. J. Slotcavage, R. A. Belisle, J. B. Patel, E. S. Parrott, R. J. Sutton, W. Ma, F. Moghadam, B. Conings, A. Babayigit, H.-G. Boyen, S. Bent, F. Giustino, L. M. Herz, M. B. Johnston, M. D. McGehee and H. J. Snaith, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- E. H. Anaraki, A. Kermanpur, L. Steier, K. Domanski, T. Matsui, W. Tress, M. Saliba, A. Abate, M. Gratzel, A. Hagfeldt and J.-P. Correa-Baena, Energy Environ. Sci., 2016, 9, 3128–3134 CAS.
- M. T. Klug, A. Osherov, A. A. Haghighirad, S. D. Stranks, P. R. Brown, S. Bai, J. T. W. Wang, X. Dang, V. Bulovic, H. J. Snaith and A. M. Belcher, Energy Environ. Sci., 2017, 10, 236–246 CAS.
- M. Hadadian, J.-P. Correa-Baena, E. K. Goharshadi, A. Ummadisingu, J.-Y. Seo, J. Luo, S. Gholipour, S. M. Zakeeruddin, M. Saliba, A. Abate, M. Grätzel and A. Hagfeldt, Adv. Mater., 2016, 28, 8681–8686 CrossRef CAS PubMed.
- D. Yang, X. Zhou, R. Yang, Z. Yang, W. Yu, X. Wang, C. Li, S. Liu and R. P. H. Chang, Energy Environ. Sci., 2016, 9, 3071–3078 CAS.
- S. M. Jain, Z. Qiu, L. Haggman, M. Mirmohades, M. B. Johansson, T. Edvinsson and G. Boschloo, Energy Environ. Sci., 2016, 9, 3770–3782 CAS.
- J. T.-W. Wang, Z. Wang, S. Pathak, W. Zhang, D. W. deQuilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. Mohd Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan, F. Yu, X. Zhang, C. Zhao and S. Liu, Adv. Mater., 2015, 27, 5176–5183 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- A. Binek, F. C. Hanusch, P. Docampo and T. Bein, J. Phys. Chem. Lett., 2015, 6, 1249–1253 CrossRef CAS PubMed.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS.
- T. Jesper Jacobsson, J.-P. Correa-Baena, M. Pazoki, M. Saliba, K. Schenk, M. Gratzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1706–1724 CAS.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 Search PubMed.
- J.-P. Correa-Baena, M. Anaya, G. Lozano, W. Tress, K. Domanski, M. Saliba, T. Matsui, T. J. Jacobsson, M. E. Calvo, A. Abate, M. Grätzel, H. Míguez and A. Hagfeldt, Adv. Mater., 2016, 28, 5031–5037 CrossRef CAS PubMed.
- G. Giorgi, J.-I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. C, 2015, 119, 4694–4701 CAS.
- N. D. Marco, H. Zhou, Q. Chen, P. Sun, Z. Liu, L. Meng, E.-P. Yao, Y. Liu, A. Schiffer and Y. Yang, Nano Lett., 2016, 16, 1009–1016 CrossRef CAS PubMed.
- J. Yang, K. M. Fransishyn and T. L. Kelly, Chem. Mater., 2016, 28, 7344–7352 CrossRef CAS.
- F. Bella, G. Griffini, J.-P. Correa-Baena, G. Saracco, M. Grätzel, A. Hagfeldt, S. Turri and C. Gerbaldi, Science, 2016, 354, 203–206 CrossRef CAS PubMed.
- Y. Yue, N. Salim, Y. Wu, X. Yang, A. Islam, W. Chen, J. Liu, E. Bi, F. Xie, M. Cai and L. Han, Adv. Mater., 2016, 28, 10738–10743 CrossRef CAS PubMed.
- S. Gholipour, J.-P. Correa-Baena, K. Domanski, T. Matsui, L. Steier, F. Giordano, F. Tajabadi, W. Tress, M. Saliba, A. Abate, A. Morteza Ali, N. Taghavinia, M. Grätzel and A. Hagfeldt, Adv. Energy Mater., 2016, 6, 1601116 CrossRef.
- H.-C. Kwon, A. Kim, H. Lee, D. Lee, S. Jeong and J. Moon, Adv. Energy Mater., 2016, 6, 1601055 CrossRef.
- D. Koushik, W. J. H. Verhees, Y. Kuang, S. Veenstra, D. Zhang, M. A. Verheijen, M. Creatore and R. E. I. Schropp, Energy Environ. Sci., 2017, 10, 91–100 CAS.
- M. Long, T. Zhang, Y. Chai, C.-F. Ng, T. C. W. Mak, J. Xu and K. Yan, Nat. Commun., 2016, 7, 13503 CrossRef CAS PubMed.
- C. Qin, T. Matsushima, T. Fujihara and C. Adachi, Adv. Mater., 2017, 29, 1603808 CrossRef PubMed.
- Y. Sun, Y. Wu, X. Fang, L. Xu, Z. Ma, Y. Lu, W.-H. Zhang, Q. Yu, N. Yuan and J. Ding, J. Mater. Chem. A, 2017, 5, 1374–1379 CAS.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- Z. Yang, A. Rajagopal, C.-C. Chueh, S. B. Jo, B. Liu, T. Zhao and A. K. Y. Jen, Adv. Mater., 2016, 28, 8990–8997 CrossRef CAS PubMed.
- F. Wang, W. Geng, Y. Zhou, H.-H. Fang, C.-J. Tong, M. A. Loi, L.-M. Liu and N. Zhao, Adv. Mater., 2016, 28, 9986–9992 CrossRef CAS PubMed.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Gratzel, S. M. Zakeeruddin, U. Rothlisberger and M. Gratzel, Energy Environ. Sci., 2016, 9, 656–662 CAS.
- T. Matsui, J.-Y. Seo, M. Saliba, S. M. Zakeeruddin and M. Grätzel, Adv. Mater., 2017, 29, 1606258 CrossRef PubMed.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- D. J. Slotcavage, H. I. Karunadasa and M. D. McGehee, ACS Energy Lett., 2016, 1, 1199–1205 CrossRef CAS.
- W. Rehman, D. McMeekin, J. Patel, R. Milot, M. B. Johnston, H. Snaith and L. M. Herz, Energy Environ. Sci., 2017, 10, 361–369 CAS.
- K. Poorkazem and T. L. Kelly, ACS Appl. Energy Mater., 2018, 1, 181–190 CrossRef CAS.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- Z. Song, A. Abate, S. C. Watthage, G. K. Liyanage, A. B. Phillips, U. Steiner, M. Graetzel and M. J. Heben, Adv. Energy Mater., 2016, 6, 1600846 CrossRef.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 CAS.
- D. Shi, X. Qin, Y. Li, Y. He, C. Zhong, J. Pan, H. Dong, W. Xu, T. Li, W. Hu, J.-L. Brédas and O. M. Bakr, Sci. Adv., 2016, 2, e1501491 Search PubMed.
- V. W. Bergmann, S. A. L. Weber, F. Javier Ramos, M. K. Nazeeruddin, M. Grätzel, D. Li, A. L. Domanski, I. Lieberwirth, S. Ahmad and R. Berger, Nat. Commun., 2014, 5, 5001 CrossRef CAS PubMed.
- Z. Li, J. Tinkham, P. Schulz, M. Yang, D. H. Kim, J. Berry, A. Sellinger and K. Zhu, Adv. Energy Mater., 2017, 7, 1601451 CrossRef.
- P. Wang, J. Zhang, Z. Zeng, R. Chen, X. Huang, L. Wang, J. Xu, Z. Hu and Y. Zhu, J. Mater. Chem. C, 2016, 4, 9003–9008 RSC.
- T. Miletić, E. Pavoni, V. Trifiletti, A. Rizzo, A. Listorti, S. Colella, N. Armaroli and D. Bonifazi, ACS Appl. Mater. Interfaces, 2016, 8, 27966–27973 Search PubMed.
- T. Leijtens, T. Giovenzana, S. N. Habisreutinger, J. S. Tinkham, N. K. Noel, B. A. Kamino, G. Sadoughi, A. Sellinger and H. J. Snaith, ACS Appl. Mater. Interfaces, 2016, 8, 5981–5989 CAS.
- A. Pellaroque, N. K. Noel, S. N. Habisreutinger, Y. Zhang, S. Barlow, S. R. Marder and H. J. Snaith, ACS Energy Lett., 2017, 2, 2044–2050 CrossRef CAS.
- Z. Li, C. Xiao, Y. Yang, S. P. Harvey, D. H. Kim, J. A. Christians, M. Yang, P. Schulz, S. U. Nanayakkara, C.-S. Jiang, J. M. Luther, J. J. Berry, M. C. Beard, M. M. Al-Jassim and K. Zhu, Energy Environ. Sci., 2017, 10, 1234–1242 CAS.
- R. Hock, T. Mayer and W. Jaegermann, J. Phys. Chem. C, 2012, 116, 18146–18154 CAS.
- Y. Liu, Q. Chen, H.-S. Duan, H. Zhou, Y. Yang, H. Chen, S. Luo, T.-B. Song, L. Dou and Z. Hong, J. Mater. Chem. A, 2015, 3, 11940–11947 CAS.
- E. Della Gaspera, Y. Peng, Q. Hou, L. Spiccia, U. Bach, J. J. Jasieniak and Y.-B. Cheng, Nano Energy, 2015, 13, 249–257 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- V. Adinolfi, M. Yuan, R. Comin, E. S. Thibau, D. Shi, M. I. Saidaminov, P. Kanjanaboos, D. Kopilovic, S. Hoogland, Z.-H. Lu, O. M. Bakr and E. H. Sargent, Adv. Mater., 2016, 28, 3406–3410 CrossRef CAS PubMed.
- G. Mazzanti and M. Marzinotto, Extruded Cables for High-Voltage Direct-Current Transmission: Advances in Research and Development, JohnWiley & Sons, Inc., The Institute of Electrical and Electronics Engineers, Inc., 2013 Search PubMed.
- Q. Han, S.-H. Bae, P. Sun, Y.-T. Hsieh, Y. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi and G. Li, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS PubMed.
- Y. Huang, L. Li, Z. Liu, H. Jiao, Y. He, X. Wang, R. Zhu, D. Wang, J. Sun, Q. Chen and H. Zhou, J. Mater. Chem. A, 2017, 5, 8537–8544 CAS.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497 CrossRef CAS PubMed.
- Y. Kato, L. K. Ono, M. V. Lee, S. Wang, S. R. Raga and Y. Qi, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- F. Giordano, A. Abate, J. P. Correa Baena, M. Saliba, T. Matsui, S. H. Im, S. M. Zakeeruddin, M. K. Nazeeruddin, A. Hagfeldt and M. Graetzel, Nat. Commun., 2016, 7, 10379 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and syntheses, Tauc plots, results of device optimization, and results of stability tests. See DOI: 10.1039/c8se00127h |
| This journal is © The Royal Society of Chemistry 2018 |