An efficient atom-economical chemoselective CO2 cycloaddition using lanthanum oxide/tetrabutyl ammonium bromide†
Received
26th February 2018
, Accepted 5th April 2018
First published on 6th April 2018
Abstract
The cycloaddition reaction between carbon dioxide (CO2) and epoxides is an attractive example for the synthesis of value-added chemicals exploiting CO2 greenhouse emissions gas. In this study, we reported on propylene carbonate (PC) production mediated by a lanthanum oxide (La–O)/tetrabutyl ammonium bromide (TBAB) binary mixture. The effect of different reaction parameters including catalyst/co-catalyst loading, the amount of substrate, CO2 pressure, temperature, and reaction time was investigated. The binary catalytic system showed high catalytic conversion and selectivity. Catalyst reusability experiments indicated that the binary system was recyclable over five times without losing its activity/selectivity. In addition, a plausible reaction mechanism based on spectroscopic and kinetic measurements was presented, which was supported by density functional theory (DFT) calculations.
1. Introduction
Carbon dioxide (CO2) is considered as a cheap, abundant, non-toxic, non-flammable, and chemically stable C1 chemical feedstock.1,2 It has a great potential to act as a green solvent.3 However, its increased atmospheric accumulation is the main cause to the overall global warming phenomenon,4,5 which is considered nowadays as an unsolved problem. In order to resolve this issue, it is essential to reduce the growing amounts of CO2 resulting from the anthropogenic effect.6 The mitigation policy of CO2 built-up in the atmosphere lies in three main subcategories, viz., carbon capture and sequestration (CCS), carbon capture and recycling (CCR), and carbon capture and utilisation (CCU).7–10
The catalytic conversion of CO2 into useful chemicals is of potential importance to tackle environmental problems and support sustainable development.11 Cyclic carbonates12–14 are one of the interesting commercially available products that can be synthesized from CO2, due to their 100% atom economical synthesis. One of these interesting carbonates is propylene carbonate (PC), a safer alternative to hazardous dipolar aprotic solvents like hexamethylphosphoramide (HMPA), together with its implementation as an accelerator for curing silicate-systems used as foundry sand binders.12 PC has been the main focus of several researchers due to being a green carbonylating agent,15 ease of preparation with almost quantitative yields,16 and its implementation as an organic solvent in electrolytes for high-energy density batteries, and electrolytic capacitors.17 Therefore, it is an important aspect to catalyse the production of the titular molecules under mild conditions with low catalyst loading and high efficiency/selectivity.18
Cyclic carbonates have been synthesized through the use of different homogeneous or heterogeneous catalysts that promote the cycloaddition of CO2 with epoxides, e.g. salen–metal compounds,19 metalloporphyrins,20 metal–organic frameworks (MOFs),21–23 zeolites,24 mesoporous silica,25,26 polyoxometalates (POMs),27 smectite,16,28 ionic liquids,29–33 and most importantly metallic oxides due to their commercial availability.34 Heterogeneous catalysis, among other processes, offers many advantages compared to homogeneous catalysis processes, viz., extra catalyst stability together with the ease of recovery/reusability, minimal cost for product separation and solvent-free processes.35,36 In this respect, metallic oxides have been studied extensively as heterogeneous catalysts for the cycloaddition reaction of CO2 and epoxides due to their acid/base character. Yano et al.37 first reported on the use of a commercial magnesium oxide in N,N-dimethylformamide (DMF) as a solvent with low conversions; however, no data were given regarding the selectivity. Moreover, Arai and coworkers38 utilised different metal oxides (M = Mg, Ca, Al, Zn, Zr, La, and Ce) at high CO2 pressures, up to 80 bars in the same polar solvent. Among these catalysts, La–O was superior in terms of yields. Although MgO, CeO2, and ZnO showed higher selectivity with moderate yields and conversions, the previous figures of merit (conversion, selectivity, and yield were 72.6%, 74.5%, 54.1%, respectively) ranked La–O as the best candidate. A solvent-free, commercial La–O catalyst was superior to its zirconia doped oxide heterogeneous counterparts. It showed a propylene oxide (PO) conversion of ca. 86% and a PC yield and selectivity of 55% and 64%, respectively.39 This makes La–O an interesting case study for further investigations.
Tetrabutylammonium bromide (TBAB) played an important role in the production of cyclic carbonates from epoxides and CO2.40–43 Its role is represented in the nucleophilic attack of the bromide anion on the electrophilic carbon of PO and the formation of a bromo-alkoxide intermediate, which attacks CO2 to form an anionic carbonate that rearranges into PC and subsequently regenerates the TBAB once again for another reaction cycle. Caló et al.44 attributed the reactivity of TBAB to the bulkiness of the tetrahedral cation, where the separation of ions becomes more obvious as less electrostatic attraction is feasible. As a consequence, more bromide will be available for the nucleophilic attack.
To our knowledge, the only metallic oxide used with TBAB as a catalyst combination for the production of PC was introduced by Eddaoudi and coworkers,45 where they used a MOF based Y2O3/TBAB binary mixture. It was reported that the use of Y2O3 resulted in much lower yields compared to the MOF based catalyst. Herein, the synergistic effect between the best performing metal oxide reported in the literature for catalysing the production of PC in the presence of TBAB as a co-catalyst was investigated. In comparison with similar systems reported in the affiliated literature, our catalyst combination, La–O/TBAB, showed high selectivity (>99%) and lower catalyst loading and pressure applied, together with an additional emphasis on the in situ formation of lanthanum carbonate (La2CO3)46 as the active surface rather than La–O, which was common for the cycloaddition reaction of CO2 and PO. In this work, the influence of different experimental parameters and conditions on the cycloaddition reaction has been investigated, namely the effect of catalyst loading, co-catalyst concentration, [PO], CO2 pressure, temperature, and reaction time. Furthermore, the binary catalyst was recycled five times without losing its activity. Density functional theory (DFT) calculations were also used to unveil the mechanism of the cycloaddition reaction.
2. Results and discussion
2.1. Catalyst characterization
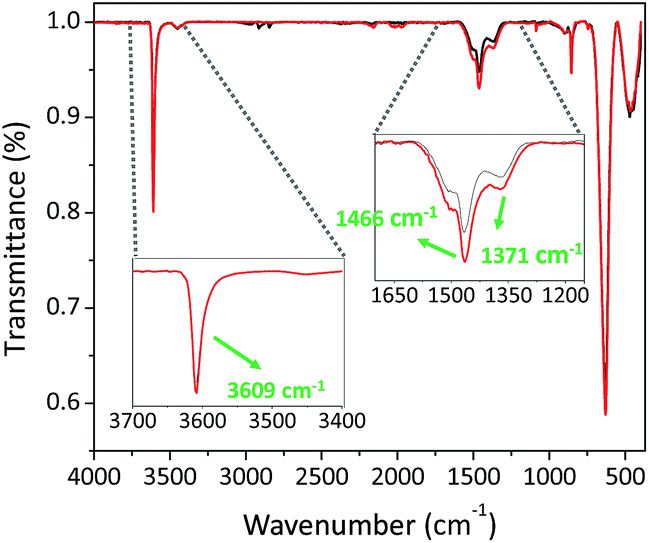
La–O is well known to react with both CO2 and H2O (key components of the atmosphere). Therefore, we analysed the material before submitting the catalyst for testing. A simple Brunauer–Emmett–Teller (BET) analysis revealed that the material is non-porous with a surface area of 7.9 m2 g−1. The material was further subjected to extra analysis using attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, which revealed a carbonated surface as shown by the prominent peaks at 1466 and 1371 cm−1, which can be ascribed to the symmetric vibrations of the monodentate carbonate group (Fig. 1).47
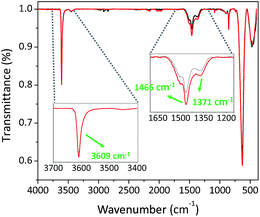 |
| | Fig. 1 ATR-FTIR spectra of La–O before (black) and after (red) the cycloaddition reaction. | |
A further peak was observed at 3609 cm−1, which can be ascribed to OH groups bound to the catalyst, viz., the surface was comprised of both LaCO3OH and La–O. In addition, the absence of peaks within the region of 1620–1780 cm−1 indicated that there was no bulk carbonate.47 Upon performing the cycloaddition reactions, a correspondent spectrum between the sample as received and the reused (washed and dried) La-based catalyst was observed with a higher intensity within the region of 1350–1620 cm−1, namely the peak centred at 1466 cm−1 was red-shifted to 1464 cm−1. This was anticipated by a further modification of the surface under the reaction conditions and fortified by the kinetic measurements with a second-order reaction with respect to CO2 (vide infra). Notably, the surface is modified through La–O rather than La–OH because of having the same peak centred at 3609 cm−1.
Thermogravimetric analysis (TGA) of the material under nitrogen showed a mass loss of about 2% when the sample was heated up to 300 °C which might be attributed to water elimination. Three major steps were recorded upon further heating at ∼332, 487 and 662 °C, with an overall weight loss of about 10%. The first prominent mass loss was consistent with that trace obtained from La(OH)3 as reported by Füglein and Walter.48 The other two weight losses in the TGA pattern corresponded to dioxycarbonate (La2O2CO3) and the oxide species (La–O), respectively.49,50 It was noteworthy that the release of CO2 between 600 and 800 °C (Fig. S1, ESI†) was verified using a coupled TG-FTIR.
2.2. Optimisation of reaction conditions
In the current work, the cycloaddition process using the lanthanum-based catalyst (in the absence of the co-catalyst) showed an insignificant conversion (6.6%), poor selectivity (50%) and low percent yield (<1%) under the investigated arbitrary conditions (Table 1, run 1). The use of the co-catalyst exhibited a mild conversion with respect to PO and gave a yield of about 13.6% PC (Table 1, run 2), and further peaks were observed via1H NMR for tributylamine (TBA) till a cat/co-cat ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (wt/wt) as shown in Fig. S2, ESI.† When a ratio of 1:4 was used, the peaks of TBAB were prominent with no degradation losses. This was a clear indication that TBAB was not decomposed by the catalyst as verified using 1H NMR in the absence of PO (Fig. S3, ESI†). This was not the case when PO was present, in accordance with previous observations by North and coworkers,51–53 where TBA was formed and detected via gas chromatography-mass spectrometry (GC-MS). In agreement with other studies reported in the literature,45 TBAB alone under relevant conditions (120 °C, 20 bar CO2, 6 h, and 7 mL PO) gave a yield of 16%. Individually, no reaction took place in the absence of PO or CO2, and no oligomers, glycols or cyclic ethers were observed as verified by 1H NMR (Table 1, runs 3 & 4). The catalyst combination La–O/TBAB showed a clear synergistic effect with a yield of 45% (152 °C, 10 bar CO2, 2 h, and 3 mL PO), which was consistent with the finding of North and colleagues,51 as well as Eddaoudi and co-workers.45 Regarding the reaction medium, Bhanage et al.38 reported on the use of toluene together with a solvent-free cycloaddition of epoxides with CO2 using different heterogeneous metal oxide-based catalysts. They showed that once toluene was utilised, it gave much lower conversions but higher selectivity when ethylene carbonate was formed (conversion was improved from 41% to 82%, and selectivity was decreased from 78% to 64%, for toluene, and solvent-free trials, respectively), and no justification was given for such behaviour. The former observations were taken into consideration in our studies by using a lower boiling point solvent, namely dichloromethane (DCM), for the ease of product separation purposes.
:2 (wt/wt) as shown in Fig. S2, ESI.† When a ratio of 1:4 was used, the peaks of TBAB were prominent with no degradation losses. This was a clear indication that TBAB was not decomposed by the catalyst as verified using 1H NMR in the absence of PO (Fig. S3, ESI†). This was not the case when PO was present, in accordance with previous observations by North and coworkers,51–53 where TBA was formed and detected via gas chromatography-mass spectrometry (GC-MS). In agreement with other studies reported in the literature,45 TBAB alone under relevant conditions (120 °C, 20 bar CO2, 6 h, and 7 mL PO) gave a yield of 16%. Individually, no reaction took place in the absence of PO or CO2, and no oligomers, glycols or cyclic ethers were observed as verified by 1H NMR (Table 1, runs 3 & 4). The catalyst combination La–O/TBAB showed a clear synergistic effect with a yield of 45% (152 °C, 10 bar CO2, 2 h, and 3 mL PO), which was consistent with the finding of North and colleagues,51 as well as Eddaoudi and co-workers.45 Regarding the reaction medium, Bhanage et al.38 reported on the use of toluene together with a solvent-free cycloaddition of epoxides with CO2 using different heterogeneous metal oxide-based catalysts. They showed that once toluene was utilised, it gave much lower conversions but higher selectivity when ethylene carbonate was formed (conversion was improved from 41% to 82%, and selectivity was decreased from 78% to 64%, for toluene, and solvent-free trials, respectively), and no justification was given for such behaviour. The former observations were taken into consideration in our studies by using a lower boiling point solvent, namely dichloromethane (DCM), for the ease of product separation purposes.
Table 1 Control experiments to study the impact of cat (run 1), co-cat (run 2), CO2 (run 3) and PO (run 4) on the cycloaddition reaction at 152 °C for 2 ha
| Run |
Cat (g) |
Co-cat (g) |
PO (mL) |
CO2 (bar) |
% yield |
|
N. R.: no reaction.
|
| 1 |
0.2 |
0 |
3 |
10 |
<1 |
| 2 |
0 |
0.2 |
3 |
10 |
13.6 |
| 3 |
0.2 |
0.2 |
0 |
10 |
N. R. |
| 4 |
0.2 |
0.2 |
3 |
0 |
N. R. |
a. The effect of catalyst loading.
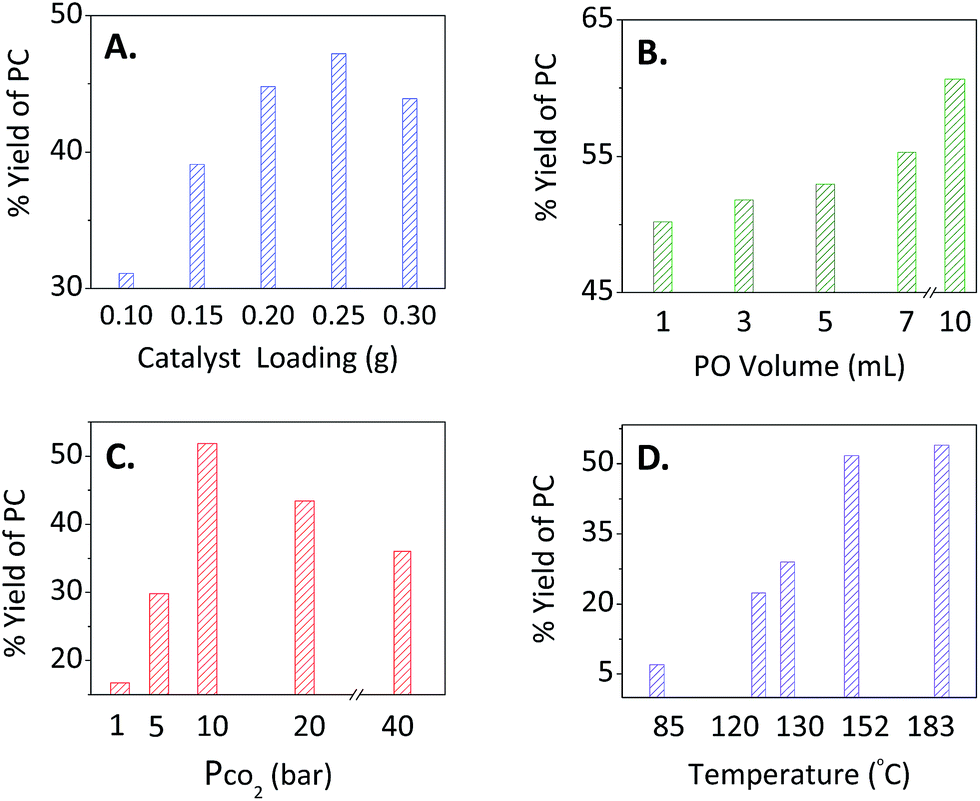
For optimisation purposes, the impact of catalyst loading on the percentage yield of PC was tested by varying its amount keeping the other variables fixed (Fig. 2A). As expected, increasing the catalyst loading increased the yield presumably due to the increase of active sites. At a catalyst loading of ≥0.2 g, La–O showed a maximum PC conversion with a selectivity of >99% as evidenced by 1H NMR. Therefore, this catalyst loading was chosen as an optimal loading for further experiments.
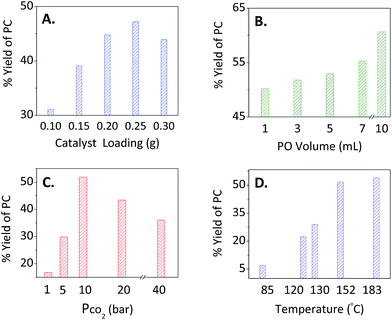 |
| | Fig. 2 Optimisation of the reaction conditions upon varying (A) catalyst loading, (B) substrate (PO) volume, (C) the pressure of CO2 and (D) temperature for the overall % yield of PC while fixing the other parameters. For (A), the PO volume = 3 mL, PCO2 = 10 bar, Temp. = 152 °C, mass of TBAB = 0.2 g and a reaction time of 2 h. For (B) and (C), the mass of La–O was 0.2 g, while keeping the other variables the same as those mentioned in (A). In (D), the optimised reaction conditions in terms of epoxide volume (3 mL), CO2 pressure (10 bar), masses of cat and co-cat (0.2 g:0.2 g La–O/TBAB) and a reaction time of 2 h were adopted. | |
b. The effect of [PO].
PO was selected as a model substrate to optimise the cycloaddition reaction conditions. Slight changes were observed at different PO concentrations (Fig. 2B, 1–7 mL, yielding 50–55%). Upon increasing the amount up to three-fold (10 mL of PO), a yield of 61% was obtained. The reason behind the poor performance might be attributed to the fixed number of the lanthanum-Lewis acid active sites found in the catalyst.
c. The effect of CO2 pressure.
The effect of pressure was also studied over a range of 1–40 bar of CO2 (Fig. 2C). Applying 1 bar gave the lowest yield (17%) among different trials. Increasing the pressure from 5 to 10 bars yielded 30 to 52%, respectively. When higher pressures were applied (20 and 40 bars), lower yields of 43 and 36% were obtained, respectively. This might be explained by the competition between CO2 and PO to react with the lanthanum-Lewis acid active sites. This makes 10 bar of CO2 the optimal parameter to be used for the binary catalytic system of La–O/TBAB.
d. The effect of temperature.
The effect of temperature was a crucial factor to deal with upon studying this reaction at different temperatures. The stability of the TBAB co-catalyst increased as shown by the TGA. It revealed that co-catalyst decomposition temperature raised from 112 to 126 °C upon mixing with a basic catalyst (Fig. S4, ESI†). Moreover, the optimal selectivity and yields for the PC production were considered. Starting at 85 °C, modest yields were obtained (ca. 7%). Increasing the temperature improved the reaction yield, where a maximum value of 54% was reached at 183 °C; however, a very dark brown colour was observed indicating the decomposition of TBAB. At 152 °C, the reaction maintained the activity and selectivity of the La–O/TBAB binary system.
e. The effect of time.
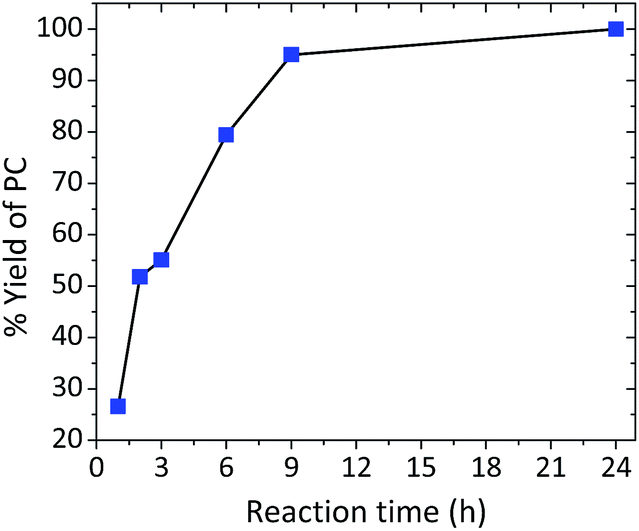
The effect of time was also another crucial factor to be investigated. A semi-logarithmic relationship was observed with a reaction time ranging from 1 to 24 h with an achieved selectivity of over 99%. Reaction time ranging from 1 to 6 h resulted in a yield from 27 to 79%. Almost quantitative yields were obtained after 9 h, and a full conversion was obtained after 24 h (Fig. 3). Turnover numbers (TONs) and turnover frequencies (TOFs) of the investigated binary catalytic system are shown in Table S1, ESI.†
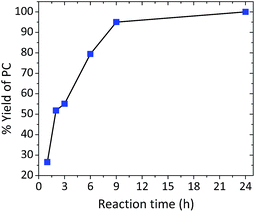 |
| | Fig. 3 Reaction profile of PO (3 mL) and CO2 (10 bar) using 1:1 by weight La–O/TBAB at 152 °C as a function of time. | |
2.3. Cycloaddition of different substrates
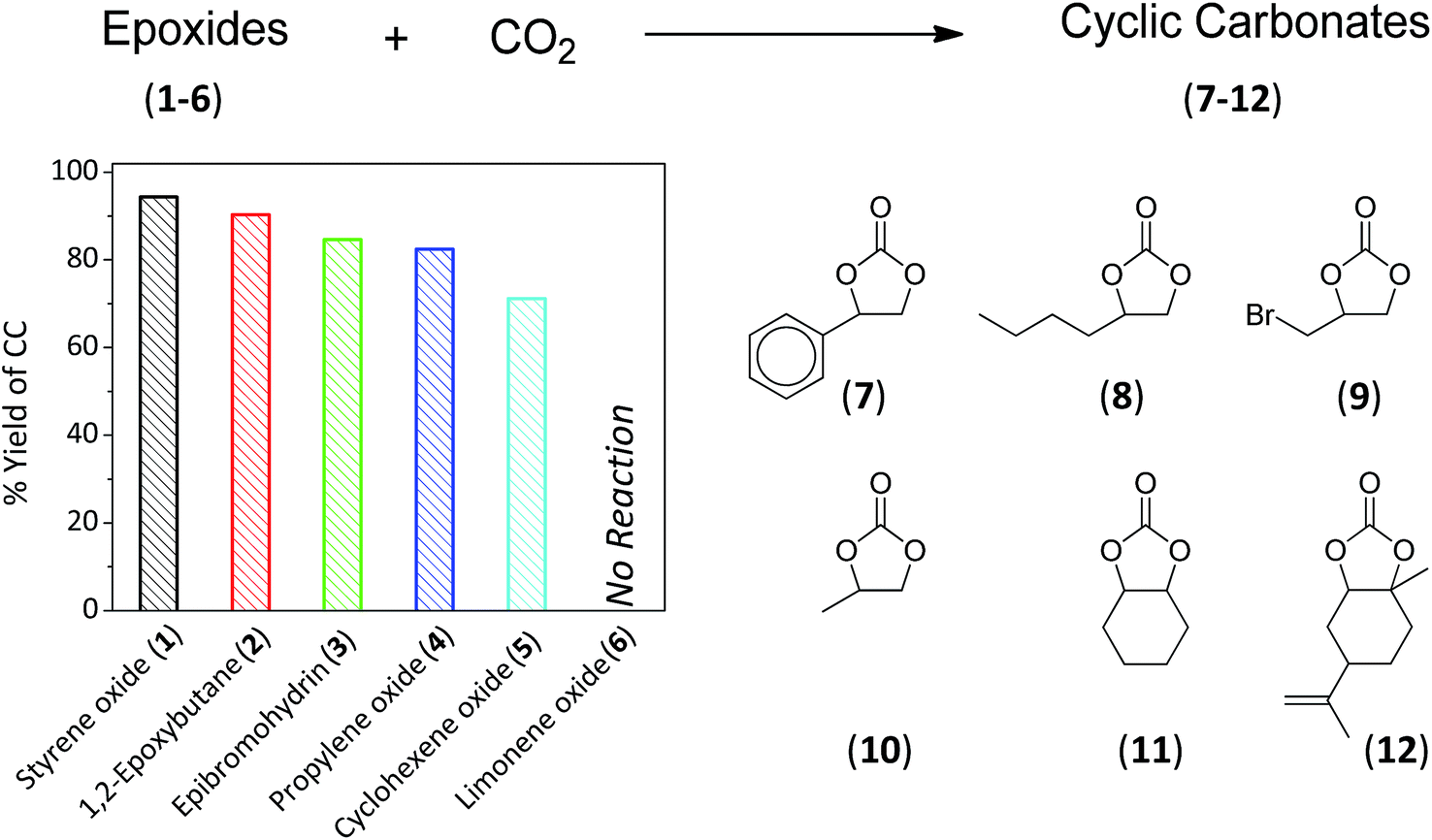
Different substrates were reported ranging from aromatic, cycloaliphatic to sustainable epoxides (1–6, Fig. 4). Under the optimised conditions, cyclic carbonate esters (7–12) were produced in a descending order as follows: styrene oxide > 1,2-epoxybutane > epibromohydrin > PO > cyclohexene oxide with yields of 94, 90, 85, 83 and 71%, respectively. It was anticipated that increasing the bulkiness of the substrate will prohibit the access to the active sites, thus resulting in a lower reaction yield. This was confirmed while no reaction was observed for limonene oxide (6).
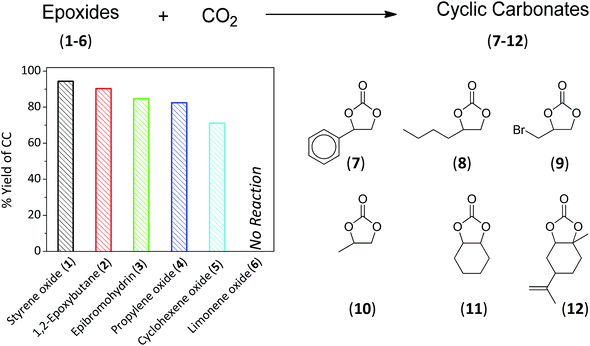 |
| | Fig. 4 Percentage yield of cyclic carbonate esters (7–12) using different epoxides (1–6) under the optimized reaction conditions. Epoxide (3 mL) and CO2 (10 bar) using 0.2 g:0.2 g La–O/TBAB at 152 °C and a reaction time of 6 h. | |
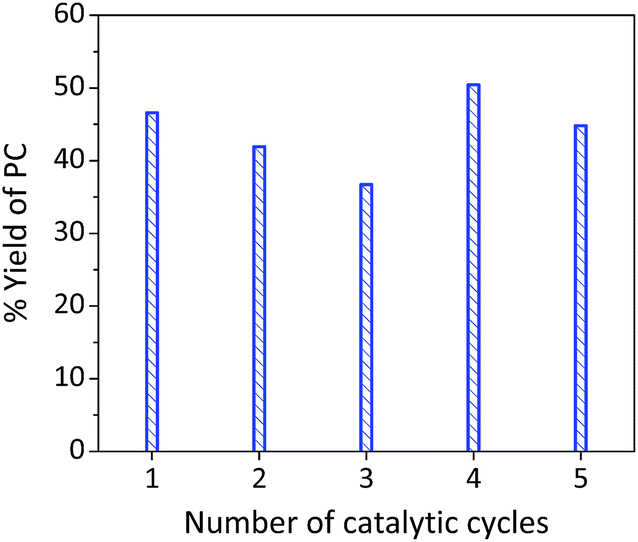
2.4. Catalyst reusability
The system showed a reasonable stability and reusability (Fig. 5) after five cycles without losing its activity and selectivity. The performance was mainly attributed to the synergistic effect rather than individual contributions of the binary system components. The increased reaction yield after the third catalytic cycle might be attributed to measurement uncertainty and/or carbonation of the La–O catalyst, which created new active sites for the cycloaddition reaction (vide infra).
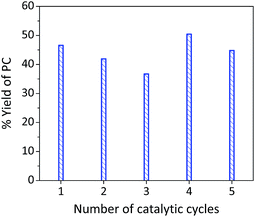 |
| | Fig. 5 Catalyst reusability of the binary system La–O/TBAB (under the optimised reaction conditions). Drying of the catalyst was carried out in a vacuum oven. Epoxide (3 mL) and CO2 (10 bar) using 1:1 by weight La–O/TBAB at 152 °C and a reaction time of 6 h. | |
2.5. Reaction kinetics
The integrated rate law of the cycloaddition reaction was obtained by varying the concentration/pressure of one reactant as a function of time while keeping excess quantities of the other reactant together with the cat/co-cat binary system at a constant temperature. The results of the kinetic study obey the following rate equation:
It was found that the reaction followed a pseudo-first-order kinetics with respect to PO and a second-order kinetics with respect to CO2 as confirmed by the straight lines obtained by plotting ln[PO] and the reciprocal of CO2 pressure against time (Fig. S5, ESI†). The first-order dependence of the substrate was consistent when a bimetallic aluminium–salen complex combined with TBAB was exploited for the cycloaddition of CO2 to epoxides.52,54 The chemisorption of CO2 by the strong Brönsted basic sites (O2−) within La–O might be the main reason for the second-order kinetics of CO246,47,55 in which one mole reacts with the surface and the other mole reacts to form PC accordingly.
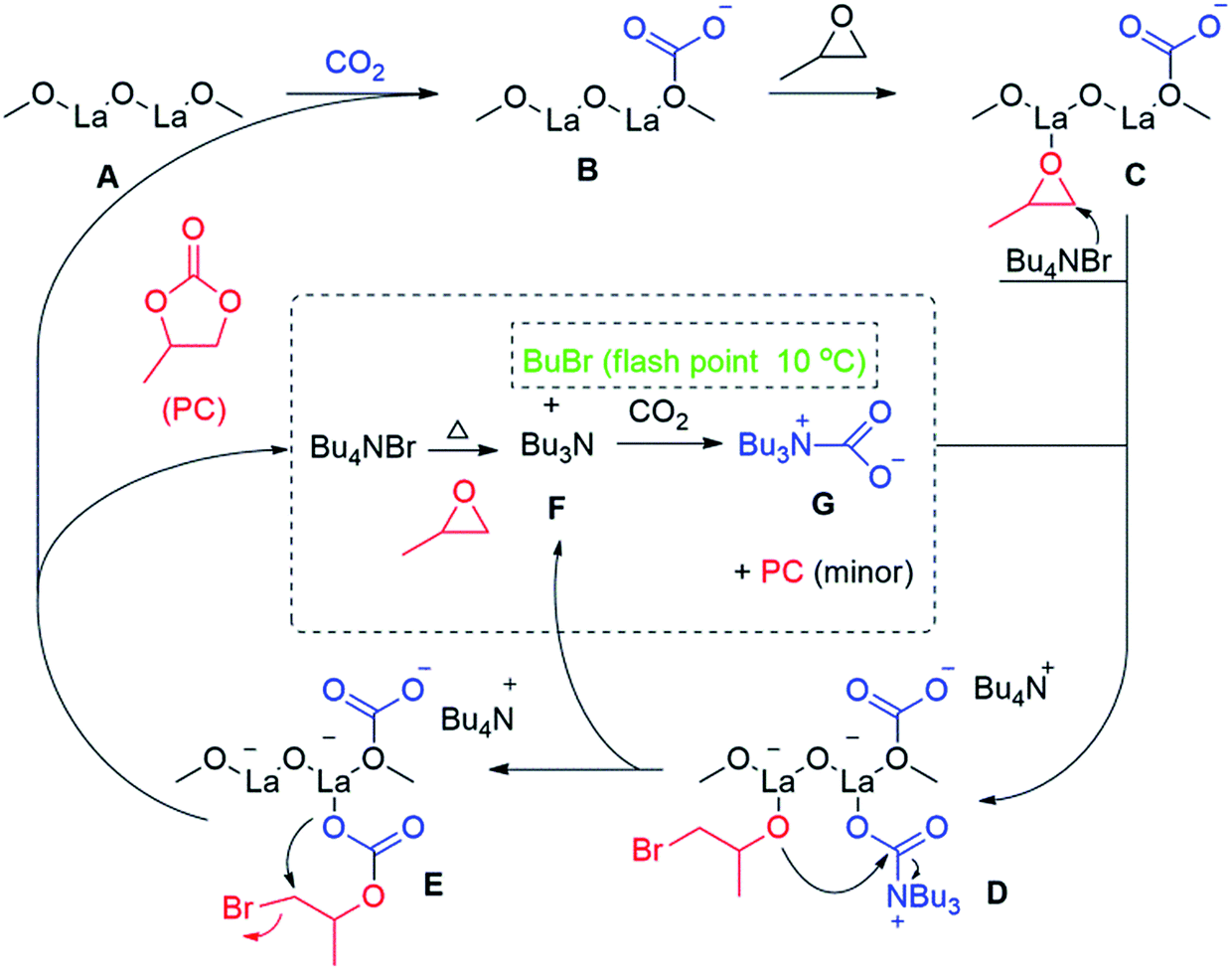
2.6. Reaction mechanism
Based on the demonstrated spectroscopic and kinetic data, Scheme 1 shows a plausible catalytic cycle of the investigated cycloaddition reaction between CO2 and PC exploiting the La–O/TBAB binary system. Upon feeding the autoclave with CO2, the surface of the basic La–O catalyst (A, Scheme 1) reacts with CO2 to form the carbonated species (B, Scheme 1) as verified by ATR-FTIR spectroscopy (Fig. 1). Meanwhile, an epoxide molecule coordinated to Lewis acidic sites of lanthanum (C, Scheme 1) which tend to orient the molecule to be ring-opened by a TBAB bromide ion to give an alkoxide ion (D, Scheme 1). Instantaneously, as shown in the 1H NMR (Fig. S2, ESI†), TBAB was decomposed in the presence of PO to give tributylamine (TBA, F, Scheme 1) and a presumable formation of butyl bromide which was not shown earlier. We suggested that the butyl bromide decomposed under the investigated conditions due to its low flash point (10 °C), which was the presumable reason behind the brownish colour of the solution upon the reaction completion. Unfortunately, we could not verify this assumption via a suitable spectroscopic technique. The formed tertiary amine reacts with the CO2 to form a carbamate activated species52 (G, Scheme 1) which tends to coordinate to the active species (D, Scheme 1), that rearranges upon an intramolecular nucleophilic attack of the coordinated alkoxide ion to carbamate ion. Afterwards, a TBA molecule was eliminated as a leaving group, which can be reintroduced once again within the catalytic cycle. Upon the formation of the coordinated ionic carbonate (E), another intermolecular rearrangement formed the organic carbonate, PC, as a target molecule to regenerate the stabilized carbonated species (B) to start the catalytic cycle once again.
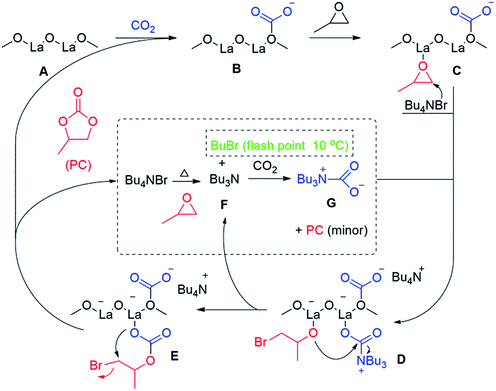 |
| | Scheme 1 A plausible reaction mechanism for the formation of PC catalyzed by the La–O/TBAB binary system as suggested by ATR-FTIR spectroscopy, kinetic data and DFT calculations. | |
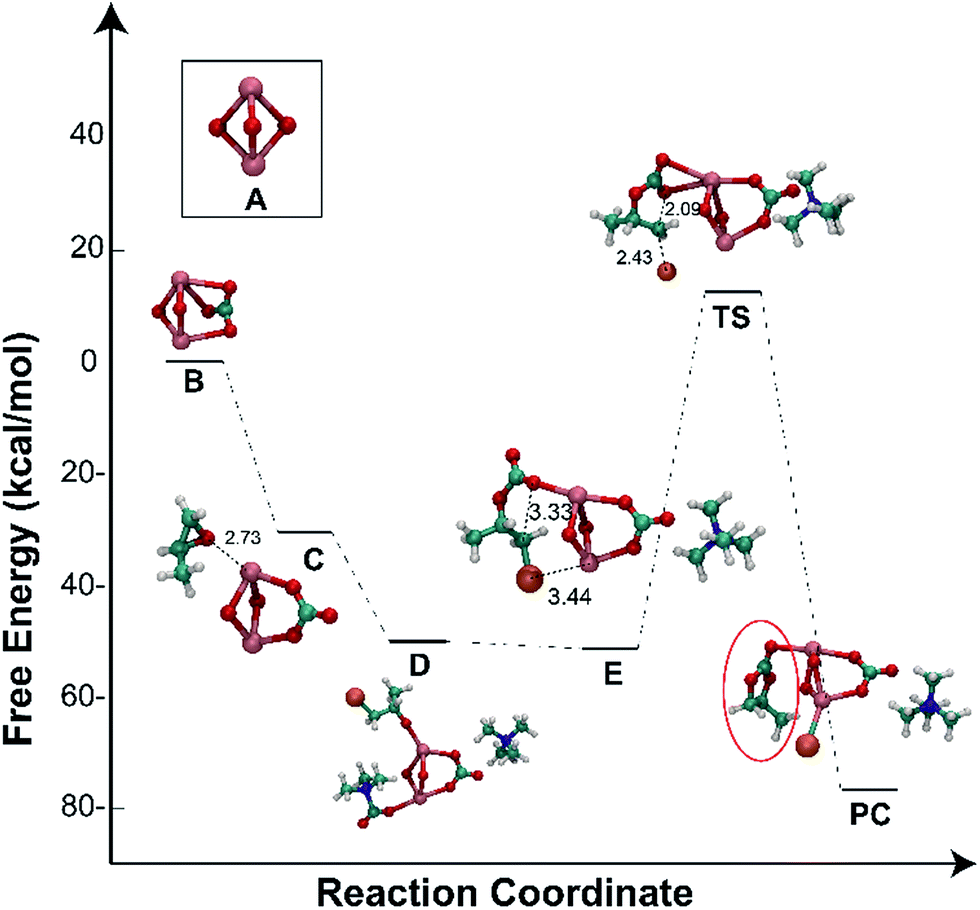
2.7. Density functional theory (DFT) calculations
The reaction mechanism was further verified by using quantum-chemical calculations, employing the DFT method (B3LYP/aug-cc-PVDZ). For the sake of simplicity and less expensive calculations, we chose to use a simplified cluster of lanthanum oxide, namely La–O,56 in order to understand the energetic transformations during the cycloaddition process. All calculations were performed in the gas-phase. The energy diagram and the DFT-optimised structures for the catalysed reaction are shown in Fig. 6. Firstly, CO2 was sorbed by La–O to form the carbonate species (B, Fig. 6) which was stabilised through multiple dipole–dipole interactions. Later, the epoxide molecule coordinated to the lanthanum centre (C, Fig. 6), which led to an overall stabilising in the energy of −31 kcal mol−1. The ring-opening reaction of the epoxide by tetramethylammonium bromide (TMAB) (for simplicity, it was chosen instead of TBAB) was found to introduce additional stabilisation through ion–ion interactions, in which the ammonium cation is in close proximity to the anionic carbonate anionic, whereas the bromide anion was accessible to attack the epoxide (D, Fig. 6). The next step involved the intermolecular rearrangement and elimination of trimethylamine (E, Fig. 6). The ring-closure step involved an intramolecular nucleophilic attack of the O atom towards the carbon bearing the bromide (E, Fig. 6), excluding the latter as a good leaving group to produce PC, while the bromide anion bound to the lanthanum. The transition state for this step is associated with an energy barrier (65 kcal mol−1). The ring-closing step was further studied as a function of the C⋯O bond distance (Fig. S6, ESI†). The most stable structure is obtained once the PC is formed with a bond distance of ∼1.4 Å, while shorter and longer distances were less stable. An energy barrier was obtained at 2.2 Å, which resembled the transition state.
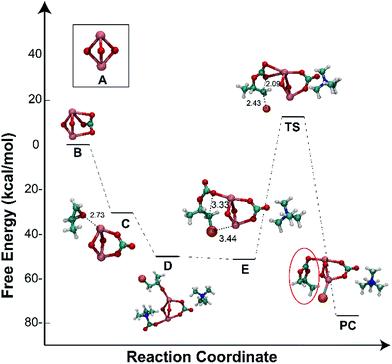 |
| | Fig. 6 Free energy profile of the minimum energy path for the formation of PC catalyzed by the La–O/TMAB binary system (it was chosen instead of TBAB for simplicity) calculated at the B3LYP/aug-cc-PVDZ level of theory. | |
2.8. A comparative study
Compared with other catalysts, the binary catalytic system showed a reasonable activity with extra selectivity towards PC formation. Our binary catalyst of La–O/TBAB (run 1, Table 2) showed a lower catalyst loading, better selectivity and yields, and lower periods of cycloaddition together with a lower pressure of CO2 applied without the formation of oligomeric materials as shown by Bhanage et al. (run 2, Table 2). Moreover, the same research group utilised different metal oxides (M = Mg, Ca, Al, Zn, Zr, La, and Ce) at higher pressures (PCO2 = 80 bar) in DMF as a reaction medium. Among these catalysts, La–O was superior in terms of yield and selectivity. Adeleye et al.39 reported on using commercial La–O with a yield and selectivity of 55% and 64%, respectively. On the other hand, the use of main group oxides such as magnesium oxide (MgO) gave 41% PC in the presence of DMF as a solvent as reported by Yano et al.37 Kaneda and colleagues57 reported on the use of a mixed Mg–Al oxide and yielded 88% PC and a PO conversion of 96%. Moreover, the activity of the Mg–Al mixed oxide was greatly larger than that of a physical mixture of MgO and Al2O3. Eddaoudi et al.45 prepared an yttrium-based catalyst as a heterogeneous catalyst which showed a good performance once combined with TBAB. Upon the use of Y2O3 as a control sample, it gave much lower yields than the corresponding yttrium-based MOF, viz., gea-MOF-1, when accompanied by tetrabutylammonium bromide (TBAB) as a cocatalyst, and the authors attributed the lower reactivity of the lanthanide-based oxide to the higher accessibility of the Lewis acid Y-sites, which is a considered as a pre-requisite for the epoxide activation step.
Table 2 Different heterogeneous catalysts reported in the literature together with our La–O/TBAB binary catalyst
| Run |
Catalyst |
Yield (%) |
Selectivity (%) |
Reference |
|
Reaction conditions: PO used: 3 mL; CO2: 10 bar; catalyst: 0.2 g (DCM: 5 mL); temperature: 152 °C; time: 9 h.
Reaction conditions: PO used: 57 mmol; CO2: 8 MPa (80 bar); catalyst: 1 g (DMF: 4 mL); temperature: 150 °C; time: 15 h. Co-oligomer of PO and PC was formed.
Reaction conditions: supercritical conditions, catalyst loading, 10% (w/w); reaction temperature: 170 °C; CO2: 70 bar; reaction time: 20 h; stirring speed: 350 rpm.
Reaction conditions: MgO (0.4 g, calcined for 3 h), CO2 pressure 20 kg cm2 (19.6 bar); PO used (10 mmol); DMF (3 mL); 135 °C; 12 h.
Reaction conditions: Mg–Al mixed oxide ((Mg/Al) = 5, calcined at 400 °C) 0.5 g; epoxide 4 mmol; DMF: 3 mL; CO2: 5 atm; 100 °C; and 24 h.
Conversion of PO was 96%.
Reaction conditions: PO used (100 mmol); Y2O3 (0.15 mmol); TBAB (0.15 mmol); 120 °C; 20 bar; 6 h.
Reaction conditions: PO used (100 mmol); gea-MOF-1 (60 mg, corresponding to 0.15 mmol yttrium), TBAB (0.15 mmol); 120 °C; 20 bar; 6 h. n.a.: not available.
|
| 1 |
La–O/TBABa |
95 |
>99 |
Current work |
| 2 |
La–Ob |
54.1 |
74.5 |
38
|
| 3 |
La–Oc |
55 |
64 |
39
|
| 4 |
MgOd |
41 |
n.a. |
37
|
| 5 |
Mg–Al mixed oxidee |
88 |
n.a.f |
57
|
| 6 |
Y2O3/TBABg |
29 |
n.a. |
45
|
| 7 |
gea-MOF-1, TBABh |
88 |
n.a. |
45
|
3. Conclusions
The utilisation of a highly selective catalyst (La–O/TBAB) for the cycloaddition reaction was reported. Upon addition of TBAB as a co-catalyst, the conversion was enhanced dramatically due to the presumable nucleophilic attack of the bromide ion while epoxides are bound to the La-active sites. All substrates were active towards CO2 cycloaddition except sterically bulk substrates due to the presumable inaccessibility of the epoxide to La atoms. The reaction mechanism was found in accordance with both DFT calculations and kinetics. The La–O/TBAB binary catalyst showed modest reusability performance over five cycles.
4. Experimental
4.1. Chemicals
Unless otherwise stated, all chemicals were used without further purification. La–O was purchased from Sigma-Aldrich and used as received without any activation. The morphology and structural properties of lanthanum oxide were reported previously by Adeleye et al.39 CO2 (99.95%, Food Grade) was purchased from Advanced Technical Gases Co. (Amman, Jordan). Propylene oxide, limonene oxide, styrene oxide, epibromohydrin, 1,2-epoxybutane, and CDCl3 were purchased from Sigma-Aldrich, and dichloromethane and tetrabutylammonium bromide were purchased from TEDIA and Fluka, respectively.
4.2. Instruments
Solution 1H nuclear magnetic resonance (NMR) spectra were collected at room temperature using an AVANCE-III 400 MHz (1H: 400 MHz), FT-NMR NanoBay spectrometer (Bruker, Switzerland) in CDCl3. Ex situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra were recorded on a Bruker Vertex 70-FT-IR spectrometer at room temperature coupled with a Vertex Pt-ATR accessory. Thermogravimetric analysis (TGA) of the TBAB and La–O/TBAB (1:4) was performed using a NETZSCH STA 409 PC LUXX instrument (heating rate 10 K min−1). TGA of the La–O material was carried out using a NETZSCH TG 209F1 Libra with a heating rate of 10 K min−1 from 22 to 900 °C using Al2O3 crucibles. Gas chromatography (GC) was performed using helium as a carrier gas with a Hewlett Packard (HP 6890) using a nonpolar capillary column (length: 15 m, diameter: 0.53 μm, and film thickness: 1.5 μm). The GC oven was heated from 40 °C till 140 °C, with a heating rate of 10 K min−1. An FID detector temperature of 260 °C was used. Brunauer–Emmett–Teller (BET) was analysed using a Quantachrome Autosorb Automated Gas Sorption System, Quantachrome Corporation, the material was activated at 60 °C (in vacuo, 3 h), and the software used was Autosorb for Windows®, version 1.20.
4.3. Cycloaddition reaction
The cycloaddition reaction was carried out in a 300 mL stainless steel autoclave equipped with a magnetic stirring bar in a thermostatted oil bath maintained at 152 °C, as optimised reaction conditions. Upon loading the catalyst, the cocatalyst and epoxide together with DCM were added. The reactor was left to pressurize to an appropriate amount of CO2 and was heated to the desired temperature. After fulfilling the reaction time, the reactor was cooled, and the gases were vented off, followed by evaporating the DCM. The reaction mixture was filtered through a filter paper to remove the catalyst. Yields and selectivity were determined using a representative sample using either 1H NMR or GC. For reproducibility purposes, each trial for the cycloaddition reaction was repeated two times and the average value was given in the corresponding tables.
4.4. Kinetic measurements
A simplified kinetic study was carried out in a 100 mL stainless autoclave that was heated in a thermo-coupled heating mantle. The reaction order of PO and CO2 was evaluated using 1H NMR and GC while keeping the La–O/TBAB constant according to the optimised conditions.
4.5. Density functional theory (DFT) calculations
All calculations were performed using the Gaussian 09 program.58 The B3LYP function was chosen to optimise the geometries and calculate the energies of the reactants, intermediates, transition states, and products. The aug-cc-PVDZ basis set was applied to the hydrogen (H), carbon (C), oxygen (O), nitrogen (N), and bromine (Br) atoms, while the LANL2DZ ECP59 basis set was used to describe the lanthanum (La) atom. Harmonic vibrational frequency calculations were also performed to verify the local minima points of the potential energy surface with all the positive frequencies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. F. E. acknowledges the Deanship of Scientific Research at the Hashemite University for the financial support. The effort of Brett Urash (University of California Santa Barbara, UCSB) in the discussion and selection of the lanthanum oxide cluster for DFT calculations is highly appreciated.
References
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 CAS.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 CAS.
-
A. Galia and G. Filardo, inCarbon Dioxide as Chemical Feedstock, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 15–31 Search PubMed.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- P. T. Brown and K. Caldeira, Nature, 2017, 552, 45 CrossRef CAS PubMed.
- G. P. Peters, R. M. Andrew, T. Boden, J. G. Canadell, P. Ciais, C. Le Quere, G. Marland, M. R. Raupach and C. Wilson, Nat. Clim. Change, 2013, 3, 4–6 CrossRef.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernandez, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- A. K. Qaroush, H. S. Alshamaly, S. S. Alazzeh, R. H. Abeskhron, K. I. Assaf and A. F. Eftaiha, Chem. Sci., 2018, 9, 1088–1100 RSC.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- A. K. Qaroush, A. S. Al-Hamayda, Y. K. Khashman, S. I. Vagin, C. Troll and B. Rieger, Catal. Sci. Technol., 2013, 3, 2221–2226 CAS.
- Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518–523 RSC.
-
D. Mohler and J. R. Ritter, Electrolytic capacitor and electrolyte therefor, US Pat., US3065389A, 1962.
- J. Qin, P. Wang, Q. Li, Y. Zhang, D. Yuan and Y. Yao, Chem. Commun., 2014, 50, 10952–10955 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- D. Bai, S. Duan, L. Hai and H. Jing, ChemCatChem, 2012, 4, 1752–1758 CrossRef CAS.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- J. Liang, Y.-Q. Xie, X.-S. Wang, Q. Wang, T.-T. Liu, Y.-B. Huang and R. Cao, Chem. Commun., 2018, 54, 342–345 RSC.
- J. Zhu, P. M. Usov, W. Xu, P. J. Celis-Salazar, S. Lin, M. C. Kessinger, C. Landaverde-Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, J. Am. Chem. Soc., 2018, 140, 993–1003 CrossRef CAS PubMed.
- M. Tu and R. J. Davis, J. Catal., 2001, 199, 85–91 CrossRef CAS.
- M. Liu, K. Gao, L. Liang, J. Sun, L. Sheng and M. Arai, Catal. Sci. Technol., 2016, 6, 6406–6416 CAS.
- M. Liu, X. Lu, L. Shi, F. Wang and J. Sun, ChemSusChem, 2017, 10, 1110–1119 CrossRef CAS PubMed.
- H. Yasuda, L.-N. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119–122 CrossRef CAS.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- Y. Xiong, H. Wang, R. Wang, Y. Yan, B. Zheng and Y. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC.
- Y. Xiong, J. Liu, Y. Wang, H. Wang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS PubMed.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- M. Liu, J. Lan, L. Liang, J. Sun and M. Arai, J. Catal., 2017, 347, 138–147 CrossRef CAS.
-
S. Fujita, M. Arai and B. M. Bhanage, in Transformation and Utilization of Carbon Dioxide, ed. B. M. Bhanage and M. Arai, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 39–53 Search PubMed.
-
O. Deutschmann, H. Knözinger, K. Kochloefl and T. Turek, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2009 Search PubMed.
- V. B. Saptal and B. M. Bhanage, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 1–10 CrossRef.
- T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129–1130 RSC.
- B. M. Bhanage, S. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS.
- A. I. Adeleye, D. Patel, D. Niyogi and B. Saha, Ind. Eng. Chem. Res., 2014, 53, 18647–18657 CrossRef CAS.
- W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- J. Sun, S. Fujita, B. M. Bhanage and M. Arai, Catal. Today, 2004, 93–95, 383–388 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- S. Senthilkumar, M. S. Maru, R. S. Somani, H. C. Bajaj and S. Neogi, Dalton Trans., 2018, 47, 418–428 RSC.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- V. Guillerm, L. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, L. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CrossRef PubMed.
- O. V. Manoilova, S. G. Podkolzin, B. Tope, J. Lercher, E. E. Stangland, J.-M. Goupil and B. M. Weckhuysen, J. Phys. Chem. B, 2004, 108, 15770–15781 CrossRef CAS.
- B. Klingenberg and M. A. Vannice, Chem. Mater., 1996, 8, 2755–2768 CrossRef CAS.
- E. Füglein and D. Walter, J. Therm. Anal. Calorim., 2012, 110, 199–202 CrossRef.
- M. Hoang, A. E. Hughes, J. F. Mathews and K. C. Pratt, J. Catal., 1997, 171, 313–319 CrossRef CAS.
- P. Jeevanandam, Y. Koltypin, O. Palchik and A. Gedanken, J. Mater. Chem., 2001, 11, 869–873 RSC.
- M. North, P. Villuendas and C. Young, Tetrahedron Lett., 2012, 53, 2736–2740 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828–6843 CrossRef CAS PubMed.
- C. Chu, Y. Zhao, S. Li and Y. Sun, Phys. Chem. Chem. Phys., 2016, 18, 16509–16517 RSC.
- C. Chu, Y. Zhao, S. Li and Y. Sun, J. Phys. Chem. C, 2016, 120, 2737–2746 CAS.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- P. Jeffrey Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00092a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Fatima A.
Alsoubani
b,
Ala'a M.
Al-Khateeb
b,
Enas
Nabih
b,
Esraa
Al-Ramahi
b,
Mohammad F.
Khanfar
c,
Khaleel I.
Assaf
*a,
Fatima A.
Alsoubani
b,
Ala'a M.
Al-Khateeb
b,
Enas
Nabih
b,
Esraa
Al-Ramahi
b,
Mohammad F.
Khanfar
c,
Khaleel I.
Assaf