
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Passivation against oxygen and light induced degradation by the PCBM electron transport layer in planar perovskite solar cells†
Chieh-Ting
Lin
 ab,
Sebastian
Pont
a,
Jinhyun
Kim
a,
Tian
Du
ab,
Shengda
Xu
b,
Xiaoe
Li
a,
Daniel
Bryant
ac,
Martyn A.
Mclachlan
b and
James R.
Durrant
*ac
ab,
Sebastian
Pont
a,
Jinhyun
Kim
a,
Tian
Du
ab,
Shengda
Xu
b,
Xiaoe
Li
a,
Daniel
Bryant
ac,
Martyn A.
Mclachlan
b and
James R.
Durrant
*ac
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk
bDepartment of Materials and Centre for Plastic Electronics, Imperial College London, Exhibition Road, London SW7 2AZ, UK
cSPECIFIC IKC, College of Engineering, Swansea University, Bay Campus, Fabian Way, Swansea SA1 8EN, UK
First published on 21st June 2018
Abstract
Herein, we investigate the causes of a 20 fold improved stability of inverted, planar structure devices (ITO/PTAA/CH3NH3PbI3/PCBM/BCP/Cu) compared to conventional structure devices (FTO/compact-TiO2/meso-TiO2/CH3NH3PbI3/spiro-OMeTAD/Au) under oxygen and light stress. The PCBM layer is shown to function as an oxygen diffusion barrier and passivation layer against superoxide mediated degradation. The passivation properties of the PCBM layer are shown to depend on the electron affinity of the fullerene acceptor, attributed to the low LUMO level of PCBM energetically inhibiting superoxide generation. We also find that planar structure devices show slower lateral oxygen diffusion rates than mesoporous scaffold devices, with these slower diffusion rates (days per 100 μm) also being a key factor in enhancing stability. Faster degradation is observed under voltage cycling, attributed to oxygen diffusion kinetics being ion motion dependent. We conclude by discussing the implications of these results for the design of perovskite solar cells with improved resistance to oxygen and light induced degradation.
Inorganic–organic hybrid perovskite materials such as CH3NH3PbI3 are attractive materials for photovoltaic applications due to their high device performance, tunable band gap, high carrier mobility, ease of processing and low cost.1–3 Perovskite photovoltaic device efficiencies have increased from 10% to over 20% in the past three years.1,4 However, achieving adequate device stability remains a key priority for realising this technology on a large scale.5 The causes of potential instability include the intrinsic properties of the perovskite material, which is sensitive to oxygen/light, moisture, and temperature, the stability of electron and hole transporting layers (ETL and HTL respectively), and the diffusion of electrode metal ions.6–11 In particular several reports have shown that oxygen and light exposure can cause a rapid loss of power conversion efficiency (PCE) in conventionally structured perovskite solar cells (structure: FTO/compact-TiO2/meso-TiO2/CH3NH3PbI3/spiro-OMeTAD/Au), indicating that this can be the dominant degradation mechanism limiting the operational lifetime of devices exposed to air ingress.12–14 This oxygen/light induced degradation has been attributed to oxygen reduction by photogenerated CH3NH3PbI3, resulting in superoxide formation.7 Consistent with this mechanism, this degradation has been observed to be most severe under open rather than short circuit conditions.9 It has also been shown that this degradation is oxygen level dependent and can be suppressed by surface treatments to the CH3NH3PbI3 photoactive layer and by iodide/bromide exchange.13–16 It is also partially suppressed by the use of a mesoporous TiO2 layer, attributed to rapid electron transfer to the TiO2 reducing the yield of superoxide formation.12,17 Other studies have reported stable operational performance of unencapsulated devices using various strategies, including the use of mixed cation/halide light absorber layers,18,19 but have not addressed directly the origins of improved resistance to light and oxygen induced degradation in these devices.20–22 To date most studies on light/oxygen induced degradation have focused on ‘conventional architecture’ devices employing mesoporous TiO2 electron collection layers. We have previously shown that such mesoporous TiO2 layers can partially suppress oxygen/light induced degradation, attributed to fast electron extraction reducing superoxide formation.12,17 In this work we focus on the stability under light and oxygen stress of inverted architecture planar perovskite solar cells employing the most widely used perovskite light absorber, organic electron transporting layer (ETL) and hole transporting layer (HTL), with the structure ITO/PTAA/CH3NH3PbI3/PCBM/BCP/Cu. There is increasing interest in such inverted device structures with organic transport layers as they facilitate fabrication at lower processing temperatures and typically exhibit low current/voltage hysteresis.4,23,24 Remarkably we find that such devices show greatly suppressed light/oxygen induced degradation relative to conventional devices employing mesoporous TiO2 as the ETL, and investigate the origins of this improved stability.
Conventional structure (FTO/compact-TiO2/meso-TiO2/CH3NH3PbI3/spiro-OMeTAD/Au) and inverted structure (ITO/PTAA/CH3NH3PbI3/PCBM/BCP/Cu) devices were fabricated and then exposed to dry air and simulated AM1.5 illumination under open circuit conditions without encapsulation (see the ESI† for details). We note that the open circuit test represents the most severe stability test, and slower degradation would be expected under short circuit and maximum power point conditions due to electron extraction from the device under these conditions.12,25 The initial device efficiencies for the conventional structure devices averaged 16.7% (champion 17.0%) and for the inverted structure devices averaged 17.0% (champion 17.7%) (see Fig. S1 and Table S1†). External quantum efficiency (EQE) data are provided in Fig. S1d.† Unlike other reports, herein we employed Cu as a stable contact for inverted structure devices rather than Ag or Al, due to their previously reported interfacial instability under oxygen exposure.15,24,26Fig. 1a shows typical time courses of the loss of device power conversion efficiency (PCE) under these stress conditions. The conventional structure device showed a rapid drop in PCE of 50% within 1 hour, consistent with previous reports.12 In contrast, the inverted structure device exhibited remarkably improved stability, taking 20 hours for such a 50% PCE drop, corresponding to a 20-fold improvement in device stability. This improvement was also seen for the inverted devices with the Au contact, highlighting that the contact was not limiting device stability under these conditions (Fig. S2a†). Moreover, control data for the inverted structure devices in N2 showed a 13% PCE drop after 20 hours of irradiation (Fig. S2b†), confirming that the degradation studied herein is oxygen induced.
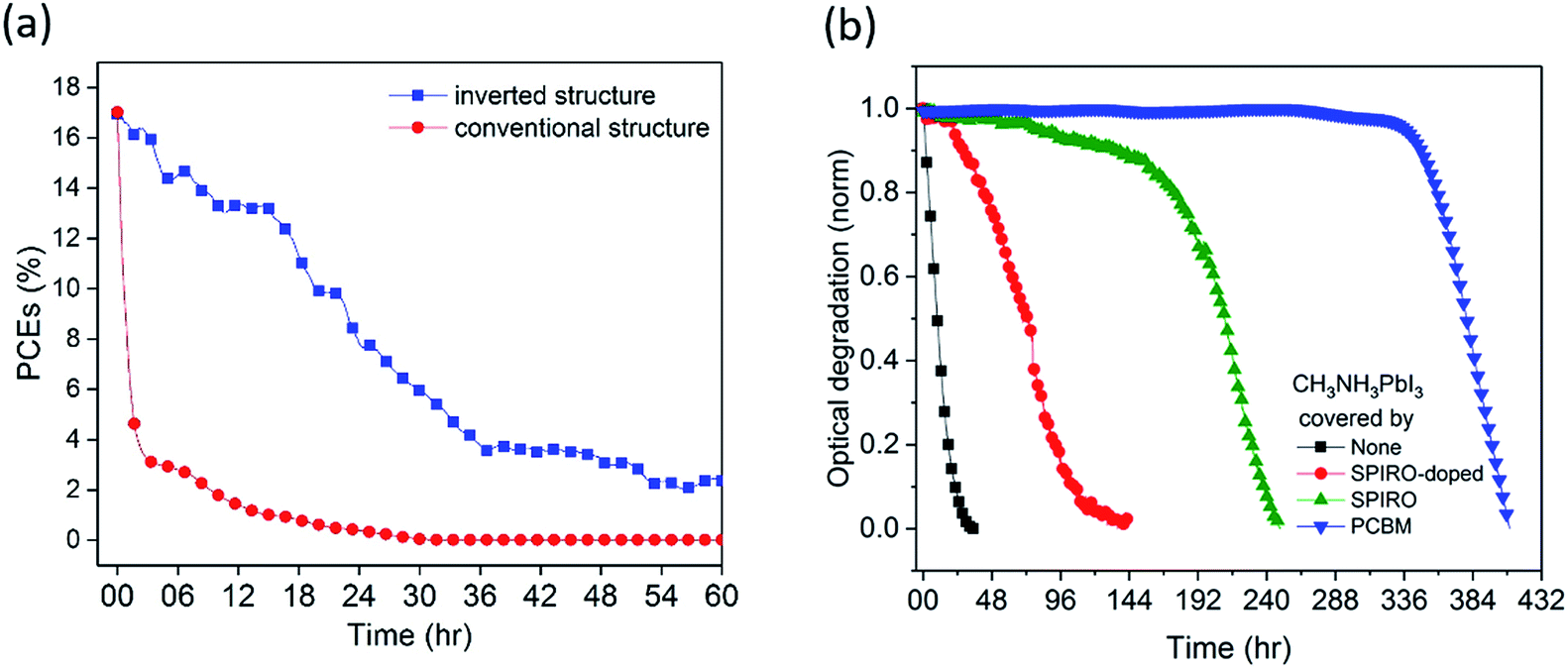 | ||
| Fig. 1 (a) Device stability curve of inverted structure (ITO/PTAA/CH3NH3PbI3/PCBM/BCP/Cu) and conventional structure (FTO/compact-TiO2/meso-TiO2/CH3NH3PbI3/spiro-OMeTAD/Au) devices. (b) Normalized optical degradation of CH3NH3PbI3, CH3NH3PbI3/doped spiro-OMeTAD, CH3NH3PbI3/spiro-OMeTAD, and CH3NH3PbI3/PCBM films. Stability measurements were done in dry air and under continuous illumination from an LED calibrated to give an equivalent Jsc produced with an AM1.5 solar simulator. | ||
Understanding the origin of this improved stability is necessary to further design environmentally stable perovskite device architectures. Haque et al. have previously reported that the photoexcitation of CH3NH3PbI3 in the presence of oxygen can lead to superoxide formation and subsequent degradation of the material, with this degradation occurring particularly at iodide vacancy sites.13 Possible strategies to improve stability under oxygen exposure include (1) use of a capping layer on the CH3NH3PbI3 to act as an oxygen barrier to slow down oxygen diffusion into the device, (2) accelerating electron extraction from the CH3NH3PbI3 to interlayers to reduce the rate of superoxide formation, (3) passivation of CH3NH3PbI3 defect sites associated with superoxide induced degradation, and (4) the use of superoxide scavenger materials, accepting electrons from superoxide. We first of all investigated the role of various transport layers as oxygen diffusion barriers in the device using, as previously reported, photobleaching of the CH3NH3PbI3 optical absorption as a convenient assay of light/oxygen induced photodegradation (see the ESI† for details).14 Photobleaching was monitored using a CCD camera to monitor in situ film coloration, which has been previously shown to be an effective assay of perovskite decomposition, correlating with material XRD and efficiency degradation (see the Experimental methods for details).14 Films were excited from the glass side, to further minimise the optical excitation of the PCBM. We note that the degradation kinetics monitored by this optical assay can be expected to be significantly slower than those determined from device PCE due to PCE being sensitive to even partial CH3NH3PbI3 degradation. Typical data are shown in Fig. 1b, obtained under white light irradiation in dry air for CH3NH3PbI3 films with spiro-OMeTAD or PCBM overlayers of thickness ∼60 nm. Without any overlayer, 5% photobleaching was observed in 1 hour, whereas with the coverage of a spiro-OMeTAD overlayer, this 5% photobleaching took 77 hours. However including the dopants usually added to the spiro-OMeTAD layer to improve its hole conductivity reduced this photobleaching time to only 22 hours. Remarkably, CH3NH3PbI3 covered by an PCBM overlayer with an equivalent thickness took over 300 hours to undergo equivalent photobleaching, indicating that PCBM overlayers cause a significant increase in oxygen/light stability compared to spiro-OMeTAD. In order to rule out that this enhancement was due primarily to the electron extraction properties of PCBM, control data were collected using PCBM as an underlayer beneath the CH3NH3PbI3. In this case, only a very modest 2 fold stability enhancement was observed (see Fig. S3†), indicating that the 300 fold stability enhancement observed for the PCBM overlayer in this assay relative to CH3NH3PbI3 alone results primarily from its passivation/oxygen barrier function rather than from its electron extraction properties.
In order to investigate the effectiveness of the PCBM overlayer as an oxygen diffusion barrier, we performed further CH3NH3PbI3 photobleaching measurements as a function of the thickness of the PCBM overlayer. The thickness of the PCBM overlayer was varied between 30 nm and 90 nm, controlled by the concentration of PCBM in the spin-coating solution. All the resulting CH3NH3PbI3/PCBM films showed similar CH3NH3PbI3 photoluminescence quenching (PLQ ∼ 82%) relative to CH3NH3PbI3 alone, Fig. 2a, indicative of similar efficiencies of electron extraction, with the exception of the thinnest, 30 nm thick, PCBM overlayer. This thinnest overlayer resulted in a lower PLQ efficiency of ∼ 54%, attributed to incomplete film formation, as confirmed by SEM data showing pinholes in this PCBM overlayer, Fig. 2b, consistent with the only modest stabilization observed with this overlayer. Fig. 2c shows the photobleaching characterization of CH3NH3PbI3 stability with PCBM films of different thicknesses, which were exposed to dry air under white light irradiation for 16 days. The 30 nm PCBM overlayer, which achieves only partial film formation, results in a modest enhancement in film stability from 1 hour to 38 hours (quantified as above as the 5% photobleaching time). Strikingly, when the PCBM layer was thick enough to form a pinhole-free capping layer, the CH3NH3PbI3 stability is increased significantly to over 310 hours. This observation indicates that a full coverage without pin-holes plays a key role in suppressing this light and oxygen induced photobleaching. With a further increase in the thickness of PCBM from 50 nm to 90 nm, the degradation time extended from 310 hours to 380 hours. Fig. 2d plots these degradation times versus film thickness. It is apparent that a linear dependence is observed, consistent with the fact that thicker PCBM overlayers provide a physical barrier for oxygen diffusion into the CH3NH3PbI3. It is also apparent that this dependence on film thickness is relatively modest compared to the stability enhancement already observed for the thinnest complete PCBM layer. This suggests that the PCBM overlayer not only functions as an oxygen diffusion barrier but also provides a form of passivation of the CH3NH3PbI3 against oxygen/light induced degradation, as we discuss further below.
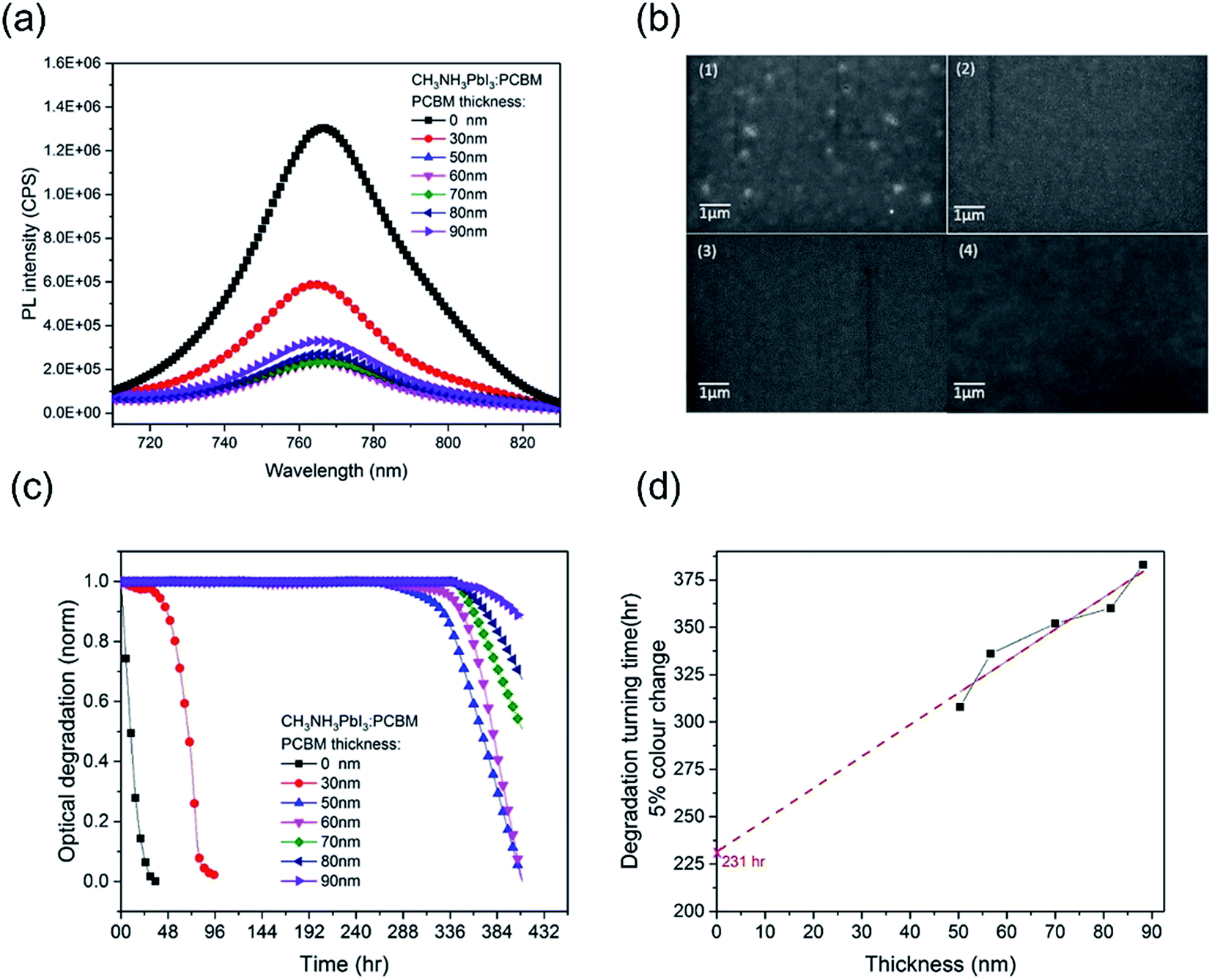 | ||
| Fig. 2 (a) Photoluminescence of the CH3NH3PbI3/PCBM film under 1 sun in N2. The range of thickness is from 31 nm to 88 nm controlled by PCBM solution concentration. The light intensity is 1 sun provided by an LED with a filter. (b) SEM image of CH3NH3PbI3 with (1) 30 nm, (2) 50 nm, (3) 70 nm and (4) 90 nm PCBM layers. (c) Normalized optical degradation of CH3NH3PbI3 with PCBM films of different thicknesses. The thickness of PCBM was from 30 nm to 90 nm controlled by concentration. Dry air conditions were controlled in an environmental chamber with RH 10%. Light was provided by an LED with 1 sun calibrated to give an equivalent Jsc of a sealed CH3NH3PbI3 device under 1 sun measured by using a solar simulator. (d) Fitting plot of thickness versus degradation turning time. | ||
Aristidou et al. investigated in detail the mechanism of oxygen and light induced degradation.7,13 In particular they proposed that this degradation is initiated by oxygen diffusion into the film and incorporation into iodine vacancies within the perovskite lattice. Superoxide, which is then formed in the presence of photoexcited electrons, consequently results in the degradation of the perovskite.13 Brenes et al. also observed shallow traps associated with surface iodide vacancies, which can be passivated by oxygen, and that they dominated the oxygen/light induced degradation.27 As a result, the stability of the perovskite under oxygen/light conditions is related to defects in the perovskite lattice and can be enhanced by passivating these ion vacancies. It has been shown that PCBM can effectively passivate the traps, suppress ion migration in the perovskite layer and minimize device J/V hysteresis.28 We suggest PCBM can play the same role here, passivating surface traps and inhibiting oxygen penetration into the active sites for oxygen/light degradation.
We now turn to the consideration of the potential role of PCBM energetics in enabling this passivation against superoxide mediated degradation. It has been previously shown that the light and oxygen induced degradation of polymer:fullerene films is strongly dependent upon the acceptor LUMO level energy, with the low LUMO energy of PCBM being critical for suppressing superoxide mediated photodegradation.29 Herein, to test whether this dependence extends to the perovskite devices studied herein, fullerene derivatives ICBA, ICTA, and PCBM were deposited on CH3NH3PbI3 and aged under oxygen and light, as shown in Fig. 3a. The LUMO levels of O2, ICTA, ICBA and PCBM have been reported to be −3.75 eV, −3.77 eV, −3.90 eV, and −4.02 eV, respectively (Fig. 3b).29 The CH3NH3PbI3 covered with the highest LUMO level fullerene, ICTA, shows the fastest photobleaching (5% photobleaching after 117 hours), whereas the films covered with ICBA and PCBM took 165 hours and 337 hours to display this 5% photobleaching, respectively. CH3NH3PbI3 PL quenching was employed to assay the efficiency of electron transfer from the perovskite to these ETL layers, with ICTA, ICBA and PCBM ETLs resulting in 74%, 78%, and 82% PL quenching, respectively (Fig. 3c). It is apparent that a deeper fullerene LUMO level increases the efficiency of electron transfer and is thus likely to explain in part the enhanced stability with a deeper LUMO level. However this trend of PL quenching is only modest, much less than the trend in the film photobleaching time. It therefore appears that the enhanced passivation properties of PCBM relative to ICTA and ICBA are primarily due to its LUMO level lying below the O2/O2− reduction couple. As such PCBM anions are unable to reduce oxygen, and rather PCBM is able to quench superoxide by electron transfer from superoxide to the fullerene, thereby potentially further stabilizing the system against superoxide mediated degradation. In contrast, as has been discussed previously in the context of organic solar cell stability,29 anions of the more electropositive fullerenes ICBA and ICTA are both able to reduce oxygen to superoxide as illustrated in Fig. 3b29 and can therefore drive superoxide mediated degradation. We note that TiO2 conduction band electrons have sufficient energy to reduce oxygen to superoxide (a process key to the efficiency of TiO2 photocatalysis), such that this protection mechanism will not operate with TiO2 ETLs.
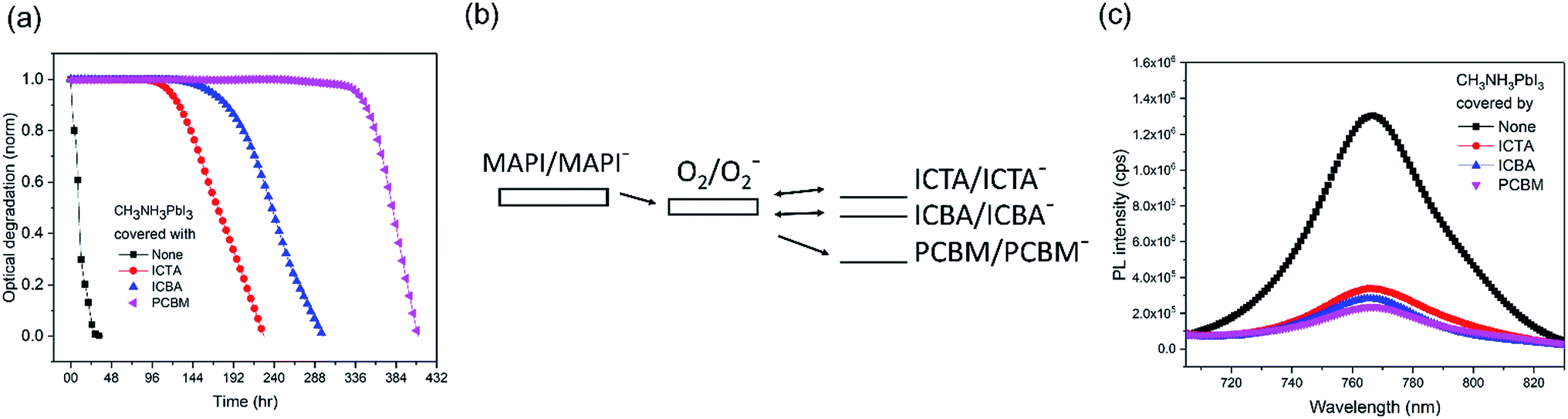 | ||
| Fig. 3 (a) Normalized optical degradation of CH3NH3PbI3 with the coverage of ICTA, ICBA and PCBM films. (b) Suggested mechanism of electron transfer from superoxide to fullerene derivative acceptors. CH3NH3PbI3 is represented as MAPI. (c) Photoluminescence of CH3NH3PbI3 films covered with ICTA, ICBA and PCBM under 1 sun in N2. | ||
We turn now to the consideration of the lateral diffusion kinetics of oxygen into the device. Fig. 4a shows a photo of a light and oxygen exposed inverted device (1.5 × 3 mm) which has lost 50% of its initial efficiency. It is apparent that yellow coloration of the device, assigned to the formation of the PbI2 degradation product, is only observed around the margins of the tested pixel. This suggests that, as expected, the metal top contact functions as an oxygen diffusion barrier, and that oxygen penetrates the photoactive layer of the device through the PCBM uncovered by the electrode around the pixel edges (Fig. 4b). It is also apparent that the yellow margin coloration is only apparent for one pixel of the device, with the other five pixels showing no colour change. The pixel showing yellow coloration was the tested pixel in this study (with the JV performance being scanned every 10 minutes and held at VOC for the rest of the time), whilst the other pixels were held constantly at VOC under the same light/oxygen exposure. This observation suggests that the degradation of inverted perovskite devices under oxygen and light stress is accelerated by repeated JV-scans. Whilst a full analysis of this observation is beyond the scope of this study, we suggest that this is most likely related to the ion migration processes likely to happen under JV scanning. The migration of ions/ion vacancies may increase the possibility of oxygen diffusion into the perovskite, thus accelerating the rate of degradation to PbI2. It has been suggested that PCBM deposition may suppress ion migration in perovskite devices, which may also be a factor in the improved stability of inverted perovskite devices under oxygen/light conditions.30,31
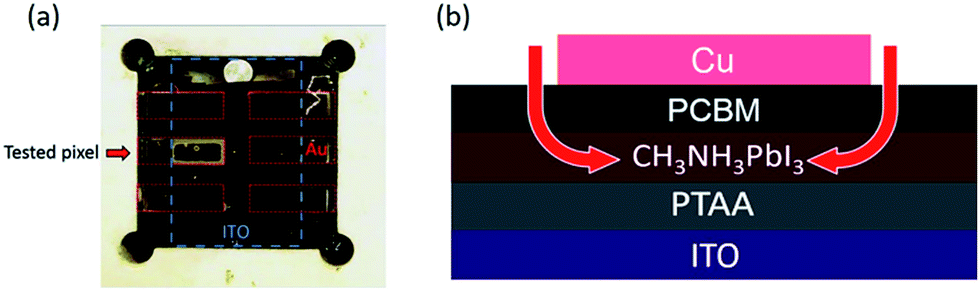 | ||
| Fig. 4 (a) Picture of an aged inverted structure device with a 50% PCE drop. Tested pixel was measured by JV scanning every 10 minutes and held at the open circuit voltage for the rest of the time. Other pixels were held at the open circuit voltage persistently. (b) Illustration of oxygen diffusion in the tested pixel of the aged inverted structure device. | ||
We now turn to the more quantitative consideration of lateral oxygen ingress through the CH3NH3PbI3 layer itself. To test the oxygen diffusion rate laterally though the perovskite film, we fabricated CH3NH3PbI3 films on mesoporous TiO2 and planar substrates and covered them by a PMMA film, a well-established oxygen diffusion barrier.32 A 1 mm wide scratch was then made in this PMMA barrier (Fig. 5a), and the CH3NH3PbI3 film photobleaching was subsequently monitored spatially (with a spatial resolution of ∼30 μm) under dry air/white light stress conditions, as plotted in Fig. 5b and c. Directly under the PMMA scratch, 50% photobleaching of the CH3NH3PbI3 absorption on both the planar and mesoporous substrates was observed in 3 hours (and 5% in 1 hour), consistent with Fig. 1b above and consistent with the rapid perpendicular diffusion of oxygen through the 500 nm thick film.13 For both films, the rate of degradation slowed with an increased lateral distance from the scratch. CH3NH3PbI3 deposited on mesoporous TiO2 showed faster degradation, namely 50% photobleaching was observed in 28 hours at 200 μm from the scratch and in 330 hours at 500 μm. CH3NH3PbI3 on the planar substrates was significantly more resistant to photobleaching, with the 50% photobleaching time increasing to 190 hours at a distance of ∼200 μm from the scratch and material more than 500 μm from the scratch exhibiting negligible (<2%) photobleaching over the 480 hour timescale of this measurement. We note that the slower timescale of these oxygen diffusion kinetics compared to those reported by Haque et al. results from the different length scales of lateral diffusion (hundreds of μm, studied herein) and perpendicular diffusion (∼500 nm (ref. 33)). According to Fig. 5d, CH3NH3PbI3 on mesoporous substrates shows 2–3 fold longer distances from the scratch for 5% photobleaching than that on the planar substrates. The CCD camera images of degraded films over time are shown in Fig. S4.† This variance of the lateral oxygen diffusion rate can most obviously be attributed to the ∼25 fold smaller size of CH3NH3PbI3 crystallites in the mesoporous scaffold compared to that on the planar substrates; this difference in crystallite size was confirmed by cross-sectional SEM images (Fig. S5†). These differences in lateral oxygen diffusion kinetics are likely to be a further reason for the improved stability of the planar devices compared to the mesoporous devices observed in Fig. 1. Such slow lateral oxygen diffusion kinetics have also been observed in organic photovoltaic devices and suggested to be critical to device stability.34 They also suggest that incorporating oxygen getter materials, which is already well established in the OLED field, may be an effective strategy for achieving large area and long term stability perovskite modules.
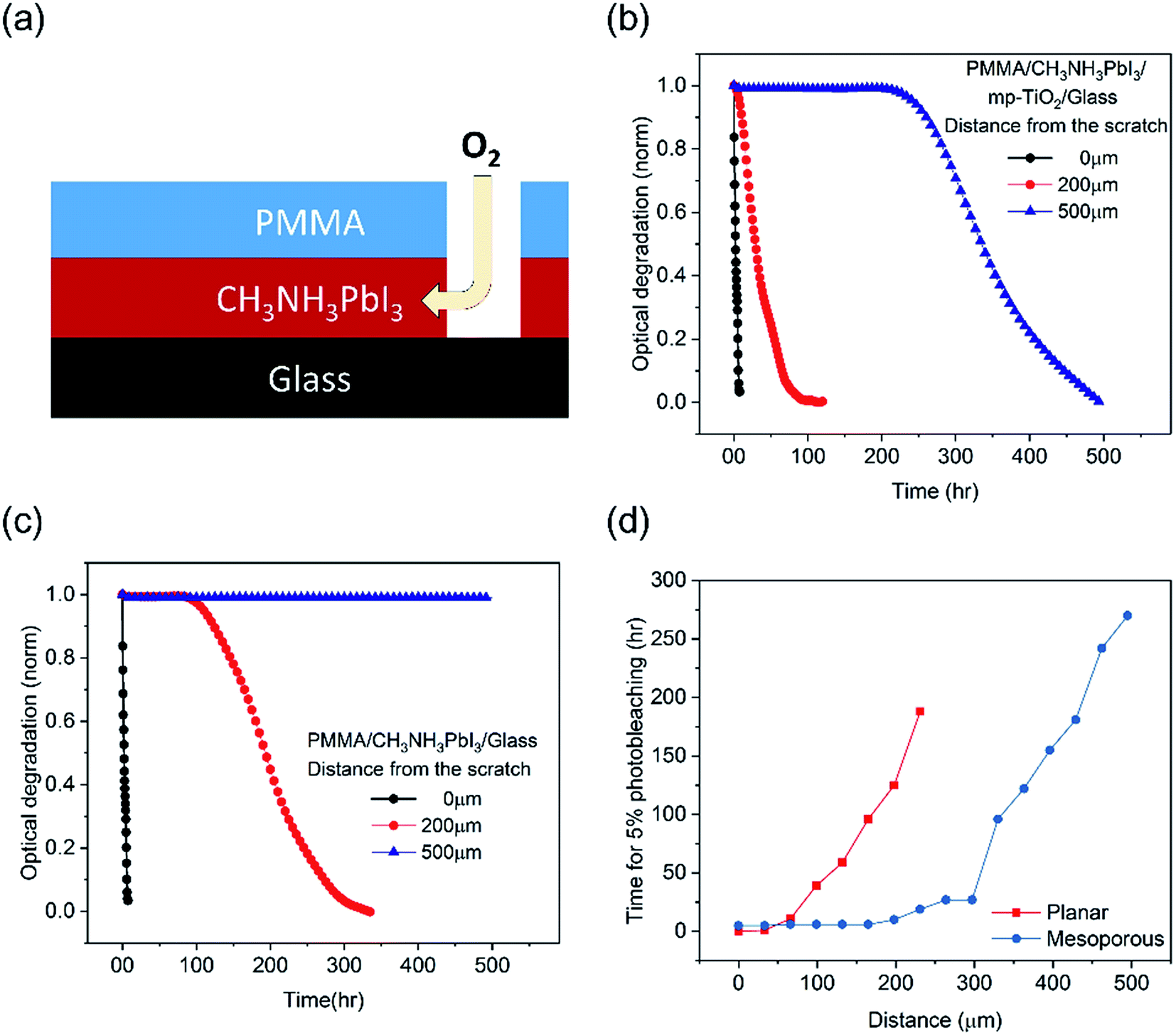 | ||
| Fig. 5 (a) Illustration of oxygen diffusion in the scratched CH3NH3PbI3/PMMA film. Normalized optical degradation of CH3NH3PbI3 at different distances from the scratch on (b) a mesoporous TiO2 scaffold substrate and (c) a planar substrate. (d) The distance from the scratch for 5% photobleaching of CH3NH3PbI3versus time on planar/mesoporous substrates. The measurements were done in dry air and under 1 sun LED illumination. | ||
According to the results above, we can provide some explanation for the varied stability results shown by different research groups, even with similar device configurations or environmental stress.13,14,35 First, we have revealed that the capping layer is critical for oxygen/light stability because it can act as an oxygen barrier. However, the thicknesses of the capping layer and electrode vary between the optimized device fabrication procedures in each group. Second, oxygen diffusion in the lateral direction can be the rate limiting step, particularly in planar structure devices (Fig. 4a). As a result, efficiency losses will depend on the size of the device active area, with larger active area devices likely to result in improved ambient operation stability due to longer distances for lateral oxygen diffusion (at least in the absence of pinholes in the metal electrode). Thirdly, the JV scanned pixel was observed herein to degrade faster than pixels held at VOC, indicating that the oxygen/light stability is dependent on the details of JV device scans. As scan procedures are likely to vary substantially between different stability studies, this may also contribute to variations in observed device stabilities.
Conclusions
We have shown that an inverted structure, organic interlayer planar CH3NH3PbI3 device architecture with a PCBM electron collection layer is more stable under oxygen and light environments than a conventional one employing mesoporous TiO2 and spiro-OMeTAD interlayers. This enhanced stability is attributed to slower oxygen diffusion kinetics, superoxide quenching and PCBM passivation of degradation sites. The PCBM electron collection layer acts as an oxygen barrier and also as a superoxide quencher. This quenching effect is strongly dependent upon the electron affinity of the fullerene employed in the ETL, with high LUMO fullerenes resulting in faster degradation. We note that this observation has important implications for the selection of ETL layer materials; materials with high LUMO levels may result in high device VOC values, but may also result in increased susceptibility to superoxide mediated photodegradation. PCBM may also sterically passivate the surface defect sites, thus inhibiting oxygen penetration into these degradation sites and consequently suppressing oxygen/light induced degradation. Moreover, the planar devices exhibit slower lateral oxygen diffusion than mesoporous scaffold devices, with this lateral oxygen diffusion being sufficiently slow to provide a key determinant of the rate of degradation. This lateral diffusion is shown to be accelerated under J/V cycling, suggesting that the oxygen diffusion kinetics may be enhanced in the presence of ion motion. Herein we demonstrate the origin of stability enhancement under oxygen and light stress in inverted structure devices, and the strategies to stabilize the perovskite layer by selecting a suitable ETL and device configuration. These results therefore provide key new insights into the factors determining the sensitivity of perovskite solar cells to oxygen and light induced degradation, and in particular the potential of PCBM ETLs to enhance device stability, which can aid the design of environmentally stable perovskite solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Welsh government funded Sêr Solar project, as well as the EPSRC Plastic Electronics CDT and CSC for financial support, and Saif Haque and colleagues for helpful discussions.Notes and references
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. IlSeok, Nature, 2014, 517, 476–480 CrossRef PubMed.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef.
- H. Kim, C.-R. Lee, J. Im, K. Lee, T. Moehl, A. Marchioro, S. Moon, R. Humphry-Baker, J. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. IlSeok, Nano Lett., 2013, 13, 1764–1769 CrossRef PubMed.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed. Engl., 2015, 54, 1–6 CrossRef PubMed.
- A. Dualeh, P. Gao, S. IlSeok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014, 26, 6160–6164 CrossRef.
- C. Law, L. Miseikis, S. Dimitrov, P. Shakya-Tuladhar, X. Li, P. R. F. Barnes, J. Durrant and B. C. O'Regan, Adv. Mater., 2014, 26, 6268–6273 CrossRef PubMed.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef.
- E. M. Sanehira, B. J. Tremolet de Villers, P. Schulz, M. O. Reese, S. Ferrere, K. Zhu, L. Y. Lin, J. J. Berry and J. M. Luther, ACS Energy Lett., 2016, 1, 38–45 CrossRef.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- S. Pont, D. Bryant, C.-T. Lin, N. Aristidou, S. Wheeler, X. Ma, R. Godin, S. A. Haque and J. R. Durrant, J. Mater. Chem. A, 2017, 5, 9553–9560 RSC.
- Q. Sun, P. Fassl, D. Becker-Koch, A. Bausch, B. Rivkin, S. Bai, P. E.Hopkinson, H. J. Snaith and Y. Vaynzof, Adv. Energy Mater., 2017, 7, 1700977 CrossRef.
- A. J. Pearson, G. E. Eperon, P. E. Hopkinson, S. N. Habisreutinger, J. T. W. Wang, H. J. Snaith and N. C. Greenham, Adv. Energy Mater., 2016, 6, 1–10 Search PubMed.
- F. T. F. O'Mahony, Y. H. Lee, C. Jellett, S. Dmitrov, D. T. J. Bryant, J. R. Durrant, B. C. O'Regan, M. Graetzel, M. K. Nazeeruddin and S. A. Haque, J. Mater. Chem. A, 2015, 3, 7219–7223 RSC.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- F. Xu, T. Zhang, G. Li and Y. Zhao, J. Mater. Chem. A, 2017, 5, 11450–11461 RSC.
- F. Hao, C. C. Stoumpos, Z. Liu, R. P. H. Chang and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 16411–16419 CrossRef PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. DeAngelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef PubMed.
- Z. Wang, D. P. McMeekin, N. Sakai, S. vanReenen, K. Wojciechowski, J. B. Patel, M. B. Johnston and H. J. Snaith, Adv. Mater., 2017, 29, 1604186 CrossRef PubMed.
- J. Y. Jeng, Y. F. Chiang, M. H. Lee, S. R. Peng, T. F. Guo, P. Chen and T. C. Wen, Adv. Mater., 2013, 25, 3727–3732 CrossRef PubMed.
- M. Kaltenbrunner, G. Adam, E. D. Głowacki, M. Drack, R. Schwödiauer, L. Leonat, D. H. Apaydin, H. Groiss, M. C. Scharber, M. S. White, N. S. Sariciftci and S. Bauer, Nat. Mater., 2015, 14, 1032–1039 CrossRef PubMed.
- K. Domanski, E. A. Alharbi, A. Hagfeldt, M. Grätzel and W. Tress, Nat. Energy, 2018, 3, 61–67 CrossRef.
- H. Back, G. Kim, J. Kim, J. Kong, T. K. Kim, H. Kang, H. Kim, J. Lee, S. Lee and K. Lee, Energy Environ. Sci., 2016, 9, 1258–1263 RSC.
- R. Brenes, D. Guo, A. Osherov, N. K. Noel, C. Eames, E. M. Hutter, S. K. Pathak, F. Niroui, R. H. Friend, M. S. Islam, H. J. Snaith, V. Bulović, T. J. Savenije and S. D. Stranks, Joule, 2017, 1, 155–167 CrossRef.
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. McDowell, P. Kanjanaboos, J.-P. Sun, X. Lan, L. N. Quan, D. H. Kim, I. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 7081 CrossRef PubMed.
- E. T. Hoke, I. T. Sachs-Quintana, M. T. Lloyd, I. Kauvar, W. R. Mateker, A. M. Nardes, C. H. Peters, N. Kopidakis and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1351–1357 CrossRef.
- P. Calado, A. M. Telford, D. Bryant, X. Li, J. Nelson, B. C. O'Regan and P. R. F. Barnes, Nat. Commun., 2016, 7, 13831 CrossRef PubMed.
- Y. Zhao, W. Zhou, W. Ma, S. Meng, H. Li, J. Wei, R. Fu, K. Liu, D. Yu and Q. Zhao, ACS Energy Lett., 2016, 1, 266–272 CrossRef.
- M. Klinger, L. P. Tolbod, K. V. Gothelf and P. R. Ogilby, ACS Appl. Mater. Interfaces, 2009, 1, 661–667 CrossRef PubMed.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- S. Shoaee and J. R. Durrant, J. Mater. Chem. C, 2015, 3, 10079–10084 RSC.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00095f |
| This journal is © The Royal Society of Chemistry 2018 |