
Cooperative silanetriolate-carboxylate sensitiser anchoring for outstanding stability and improved performance of dye-sensitised photoelectrodes†
Maxime
Fournier
*a,
Dijon A.
Hoogeveen
a,
Shannon A.
Bonke
 ab,
Leone
Spiccia
a and
Alexandr N.
Simonov
*a
ab,
Leone
Spiccia
a and
Alexandr N.
Simonov
*a
aSchool of Chemistry, The ARC Centre of Excellence for Electromaterials Science, Monash University, Victoria 3800, Australia. E-mail: maxime.fournier@monash.edu; alexandr.simonov@monash.edu
bInstitut Nanospektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Kekuléstraße 5, 12489 Berlin, Germany
First published on 28th April 2018
Abstract
Photosensitising dyes require anchoring groups for attachment to a metal oxide support surface. When a dye-sensitised electrode is designed for applications in aqueous media, e.g. for photoelectrocatalytic water splitting, these anchoring groups must be highly stable towards hydrolysis while retaining sufficient electrical conductivity to sustain efficient transfer of photogenerated charges. With this motivation, we introduce herein a cooperative silanetriolate-carboxylate anchoring combination. A ruthenium(II) tris-bipyridine dye has been functionalised with this dual-anchor through a facile peptide coupling reaction. The photoelectrooxidation performance and stability of mesoporous TiO2 electrodes sensitised with this new dye have been systematically examined in solutions with pH 1–9 in the absence and presence of a sacrificial electron donor and/or buffering electrolyte following a specifically designed testing protocol, which involves comprehensive characterisation of the electrodes and electrolyte solutions by ICP-MS and UV-vis spectroscopy. When compared to the state-of-the-art phosphonate anchoring group, the silanetriolate-carboxylate combination enables approximately 4- and 8-fold enhancements in the rate of sulphite oxidation by the Ru(II) (bipyridine)3-sensitised TiO2 photoanodes under 1 sun irradiation after 2 and 24 hours of reaction, respectively. In the absence of a sacrificial electron donor, photoanodes based on carboxylate- and phosphonate-anchored dyes undergo continuous and rapid degradation under all conditions examined, leading to almost completely bleached electrodes within less than an hour of operation. Conversely, silanetriolate-carboxylate anchoring provides quasi-stable operation with typically less than 10% ruthenium loss from the electrode surface under the same conditions. The analysis undertaken here unambiguously attests to the significantly improved long-term performance of the dye-sensitised photoelectrodes provided by the cooperative anchoring system where silanetriolate provides high stability and carboxylate sustains efficient charge transfer. This furnishes a practical pathway towards the synthesis of photosensitisers capable of stable and efficient operation within photoelectrochemical devices in aqueous environments.
Introduction
Inspired by natural systems,1 the concept of artificial photosynthesis has excited the scientific community for over a century2,3 as the only truly sustainable and renewable technology that can satisfy the ever-growing energetic demands of humanity.4 This approach enables the immense solar power reaching the surface of the planet (ca. 90 PW)5 to be harnessed for use in a fuel production/utilisation cycle, which is entirely neutral with respect to the release of CO2 into the atmosphere.6,7 Despite the technical feasibility of existing ideas and technology, much effort is still required to overcome hurdles related to the materials, processing, solar-to-fuel conversion efficiencies, costs and stability, which currently prevent widespread implementation of solar fuel installations by industry.8A simple and straightforward modular approach for solar fuel synthesis uses a photovoltaic cell to power an electrolyser where water is split, forming oxygen (by-product) at the anode and molecular hydrogen (fuel) at the cathode.9–11 A topological variation of this is a photoelectrochemical cell (PEC) configuration,12 where the light absorbing and electrocatalytic functions are combined.13 Notwithstanding the more challenging engineering required to create efficient PEC solar fuel devices, such ‘artificial leaves’ can potentially offer significant advantages in terms of the final product price,8 as well as theoretical maximum efficiency.14 Efficient water splitting via a PEC approach requires a photoelectrode to satisfy a very demanding set of requirements. The absolute positions of the valence/conduction band edges of the light absorbing material need to be more positive/negative than the standard electrode potentials for water oxidation/reduction, and also provide enough overpotential to efficiently drive both electrode processes. Meanwhile, the gap between band edges ideally needs to be small enough to allow absorption of the largest portion of the solar spectrum, i.e. visible light. Equally important is the stability of the photoelectrodes under the harsh conditions of water-splitting. Among a truly broad range of materials studied in this context, the most significant progress has been achieved with photoelectrode systems based on inorganic and comparatively simple light absorbers like oxides, (oxy)nitrides, chalcogenides, silicon- and carbon-based semiconductors, etc.15 Nevertheless, despite more than four decades of intensive research into such monolithic devices, Jacobsson et al.12 have argued that ‘no material performing well in all the fundamental steps has been found nor seems likely to ever be discovered’. This has led to the development of increasingly elaborate, multicomponent PEC devices.16–19
A comparatively young and less developed PEC subclass are dye-sensitised (DS) photoelectrochemical cells (DS-PECs), which recently emerged from extensive research into dye-sensitised solar cells (DSSCs).20,21 Similarly to DSSCs, DS-PECs involve the sensitisation of mesoporous wide-band gap semiconductors with light-harvesting dyes. The major difference is that the photogenerated charges from the dye molecules are used for the bond forming/breaking processes needed to split water into H2 and O2 as opposed to the simple, typically outer sphere, electron transfer involved in the reversible oxidation/reduction of a redox mediator. The bond formation/breaking processes of water splitting introduce significant kinetic hurdles, which necessitate integration of a catalyst into dye-sensitised electrodes,22 a strategy that is also widely implemented in the design of PECs based on inorganic light-harvesters. A key advantage of small organic and organometallic dyes vs. inorganic semiconductors is the facile and broad tunability of their energy levels, which can be achieved via variations in chemical structure. Thereby, dye-sensitised photoelectrodes are more flexible in terms of design, and in some cases are based on potentially cheaper and more abundant materials,23 which expands the range of strategies for cost and performance optimisation.24
Several groups have made tremendous progress developing dye-sensitised photoelectrochemical water splitting systems. Most of these use ruthenium(II) tris-bipyridine derivatives ([Ru(bpy)3]2+; Scheme 1a) as the photosensitiser. It is either co-adsorbed with or linked covalently to a molecular catalyst at the surface of a mesoporous and selective charge transporting electrode (most commonly, n-type TiO2 or p-type NiO). The beneficial optoelectronic characteristics of the [Ru(bpy)3]2+-based dyes have put this class of sensitisers to the forefront of research into dye-sensitised systems for photoinduced water splitting at both anodes and cathodes,25 though the main focus so far has been on the water oxidation reaction. Mallouk and colleagues synthesised a [Ru(bpy)3]2+ dye with phosphonate groups to anchor it to TiO2 (RuP, Scheme 1a), and a malonate group on the opposite side to attach nanoparticulate iridium oxide as a water oxidation catalyst to show ca. 0.03 mA cm−2 photooxidation current density over 60 seconds at pH 5.8.26 Spiccia and co-workers subsequently achieved water oxidation at 93% faradaic efficiency with a photocurrent density of 0.005 mA cm−2 for 120 min at pH 6.5 in 0.1 M Na2SO4 under 100 mW cm−2 white light illumination using a [Ru(bpy)3]2+ dye with a carboxylate anchoring group (RuAc, Scheme 1a).27 In that case the dye was covered with an ionic polymer, Nafion, which suppressed dissolution and contained the manganese oxide catalyst. Mallouk and colleagues then improved upon their system by adding a redox mediator and altering the [Ru(bpy)3]2+ derivative dye, though retaining phosphonate anchoring groups, to demonstrate water oxidation at 85% faradaic efficiency with a photocurrent density of 0.09 mA cm−2 for 100 s at pH 5.8 in silicate buffer.28 Sun and co-workers reported transient water oxidation photocurrents above 0.5 mA cm−2 (70–80% faradaic efficiency) for 100 s under 300 mW cm−2 white light illumination at pH 6.8 when using RuP and a co-adsorbed molecular ruthenium catalyst.29 A multitude of other dye-sensitised systems have also been reported with different [Ru(bpy)3]2+ derivatives, porphyrins and organic dyes combined with a variety of catalysts.30
 | ||
| Scheme 1 (a) Structures (counter-anions are omitted for clarity) and (b) energy levels (vs. normal hydrogen electrode and vacuum) for ruthenium(II) tris-bipyridine dyes with different anchoring groups (data for RuNPhSiSi were obtained with the silatrane version of this sensitiser, viz.RuNPhSiLSiL). | ||
A critical problem that applies to many types of DS-PECs hitherto reported is unsatisfactory stability, which is manifested as persistent photocurrent decays and faradaic efficiencies below 100%. Hoogeveen et al. have shown a rare example that defies this previous statement with a DS-photocathode stably operating at 100% faradaic efficiency on the scale of hours to days.31 However, even very recent publications on DS-PEC water oxidation ubiquitously report declining photocurrents on a relatively short timescale.32 There are many reasons for this, given the complexity of photoelectrodes33,34 and secondary processes occurring upon photoexcitation of the dye. However, it is clearly recognised that one of the major causes is the disruption of bonding between dye molecules and the semiconductor surface.35,36 Weak linkages at the dye|semiconductor interface can be hydrolysed when in contact with aqueous media, and this process is facilitated in the presence of buffer bases like HPO42− and at high pH.36–38 Sensitiser electro-polymerisation,39,40 and the use of hydrophobic polymer41 or inert oxide overlayers42–44 on dye-sensitised electrodes have been examined as promising strategies to improve the robustness of the dye|semiconductor interface. However, even with these modifications, DS-PEC electrodes still suffer degradation, especially when concentrated buffered electrolyte solutions are used, as needed for efficient ion transfer during water splitting.45 In light of the more impressive stability of the PEC systems based on inorganic light-harvesters46–48 and the current industry stability requirements,49 significant improvement in the durability of dye-sensitised photoelectrochemical cells is indispensable to make this attractive technology economically exploitable.
A more direct approach to address instability issues involves the design of stronger dye anchoring groups, viz. anchors that provide a more robust connection between the dye molecules and the charge-transporting semiconductor. From this perspective, a significant body of work has been done in the framework of DSSC research, where anchoring groups based on silanetriolate, hydroxamic acid, pinacolic acid, salicylic acid and phenol derivatives have been investigated,50 although these studies were primarily undertaken in organic media as typically used in DSSCs. Aqueous pH buffered environments are significantly more hostile to the dye|oxide interface, with anchoring groups for these conditions having been recently summarised in an expert review from the groups of Crabtree and Brudvig.51 Silanetriolate groups (Table S1†) have been demonstrated to be the most hydrolytically stable anchoring groups under acidic, neutral and alkaline conditions.52–55 To clarify, silanetriolate is frequently reported in this context as siloxane, but the former term is used herein as is consistent with the IUPAC recommended nomenclature (the distinction is elaborated upon in the ESI†).56 Although providing stable physical attachment, the robust dye–Si–O–semiconductor bond comes at the expense of the electron transfer efficiency between the light-absorber and charge-transfer material, which is less than that provided by other anchoring groups such as phosphonate or carboxylate.
To overcome the limitations of individual anchoring groups in an aqueous environment, we apply herein a ‘division of labour’ approach through the use of two cooperative linkers. We selected a silanetriolate moiety for robust attachment, and a widely used in the DS field carboxylate counterpart to provide intimate electronic coupling and efficient charge transfer.24 This multiple-anchor concept has been recently suggested as an approach to improve the performance of DSSCs based on a dye featuring a combination of two phosphonate and one carboxylate anchors, and functioning in acetonitrile electrolyte solutions.57 However, to the best of our knowledge, a ‘division of labour’ strategy to create a robust dye-sensitised photoelectrode efficiently operating in the presence of water has not been developed until now. To achieve this, we introduce herein a straightforward procedure to functionalise a ruthenium(II) tris-bipyridine core with silanetriolate and carboxylate linkers. Derivatives of [Ru(bpy)3]2+ are amongst the most reliable photoanode dyes owing to their intense metal-to-ligand charge transfer absorption (extinction coefficient ca. 104 M−1 cm−1), excited state quantum yields close to unity, ease of synthesis and energy level suitability and tunability.58,59 These properties combined with reasonable hydrophilicity make this category of sensitisers highly suitable for the purposes of the present study.
To unambiguously demonstrate the advantages of the new dual-anchor sensitiser, we compare it to a representative set of ruthenium(II) tris-bipyridine dyes having only carboxylate, phosphonate or silanetriolate anchoring groups. The first two serve as important references in our analysis as they are ubiquitous in the DS-PEC field.30 This especially is necessary as there is significant variation in DS electrode fabrication techniques (e.g. in the morphology and thickness of mesoporous charge-transfer layers, synthesis, deposition and sensitisation protocols, additive usage, blocking layers, and spacers) and in the conditions for photoelectrocatalytic measurements (including light intensity, pH, the nature of the electrolyte and the use of a sacrificial electron donor) among different laboratories.23,24,30 As a result, direct comparisons between existing reports on the performance of water oxidation DS photoanodes are often impossible and might even be misleading. To overcome this limitation, we introduce herein a methodology based upon UV-visible spectroscopy and inductively coupled plasma mass spectrometry (ICP-MS) that enables discrimination between photoelectrochemical contributions from the dye molecules adsorbed on the electrode and dissolved into electrolyte solution. This analytical strategy allows for a valid comparison of the performance of different dyes under a wide range of conditions.
Experimental
Materials
Reagent or analytical grade chemicals were used as received from commercial suppliers. Thionyl chloride (97%), 2,2′-bipyridine-4,4′-dicarboxylic acid (98%), 4-aminobenzoic acid (≥99%) and acetic acid (ACS reagent grade, ≥ 99.7%) were purchased from Sigma-Aldrich. Ruthenium(III) chloride, metal base anhydrous (99.9%); sodium phosphate, monobasic, anhydrous (98%); sodium phosphate, dibasic, anhydrous (98%); and sodium acetate, anhydrous (99%) were purchased from Acrös Organics. Sodium sulphite, anhydrous (97.0–100.5%) was purchased from Merck. p-Aminophenyl-trimethoxysilane (98%) was purchased from Gelest. Triethanolamine (≥98%) was purchased from Alfa Aesar. Reverse osmosis purified water with a quoted resistivity of 1 MΩ cm at 25 °C was used in all experimental procedures and for the preparation of electrolyte solutions, except for ICP-MS studies where high purity deionised water (Millipore Synergy®UV, 18 MΩ cm) was used to prepare solutions. Glass coated with fluorine-doped SnO2 (FTO; sheet resistance ca. 8 Ω square−1) and 18 nm TiO2 anatase paste were purchased from Dyesol. High-purity argon (99.999%; O2 ≤ 2 ppm) from Air Liquide was used for photoelectrochemical experiments.Synthesis
Dye structures are shown in Scheme 1a, while full synthetic schemes, detailed synthesis procedures, purification methodologies and characterisation of these compounds are provided in the ESI (Schemes S1–S6†). In brief, complexes (4,4′-dicarboxy-2,2′-bipyridine)bis(2,2′-bipyridine)ruthenium(II) dihexafluorophosphate (RuAc·2PF6)60 and (4,4′-dicarboxy-2,2′-bipyridine)bis(2,2′-bipyridine)ruthenium(II) dichloride (RuAc·2Cl)61 were synthesised following established literature procedures using Ru(bpy)2Cl and 4,4′-dicarboxy-2,2′-bipyridine as starting reactants. (4,4′ Diphosphonic acid-2,2′-bipyridine)bis(2,2′-bipyridine)ruthenium(II) dichloride dihydrate (RuP·2Cl) was synthesised following the same protocol using Ru(bpy)2Cl2 and 4,4′-diphosphonic acid-2,2′-bipyridine.61 New dyes (4,4′-(diphenyldicarboxylic acid)-dicarboxamide)bis(2,2′-bipyridine)ruthenium(II) dihexafluorophosphate (RuNPhAcAc·2PF6), (4,4′-(diphenyldisilatrane)-dicarboxamide-2,2′-bipyridine)bis(2,2′-bipyridine)ruthenium(II) dichloride (RuNPhSiLSiL·2PF6), and (4,4′-(phenylsilanetriol)-(phenylcarboxylic acid)-dicarboxamide-2,2′-bipyridine)bis(2,2′-bipyridine)-ruthenium(II) dichloride (RuNPhAcSi·2Cl) were synthesised directly from RuAc·2Cl or RuAc·2PF6, respectively, following a peptide-coupling method inspired by previous reports from our group62 and Brennan and co-workers.52,53RuNPhSiSi was obtained from the protected silanetriolate precursor, RuNPhSiLSiL·2PF6, upon sensitisation onto TiO2 films.Photoanode preparation
FTO slides were laser engraved (Universal Laser Systems VLS3.50) to provide a 5 mm2 square-shaped electroactive surface and then cleaned by successive 30 minute sonications (Elma S 300H, Elmasonic) in aqueous surfactant solution (Hellmanex III, Sigma-Aldrich), water and ethanol (96%). A commercial semi-automatic screen-printer (Keywell), with a 90T medium thread diameter screen, was used to print square films of TiO2 with a geometric area of 0.16 cm2 inside the laser-engraved regions on the FTO surface. Two successive printing cycles produced titania films with a thickness of ca. 2.5 μm as measured using a stylus profilometer (Veeco Dektak 6M). Between each printing cycle, the TiO2/FTO slides were warmed on a large hotplate (Präzitherm) set at 121 °C for 15 min. The slides were then sintered at 450 °C for 30 min and at 500 °C for 15 min, allowed to cool down under ambient conditions and cut into individual electrodes.Each electrode was UV–ozone treated at 1000 mtorr (Harrick Plasma) for 5 min immediately before being immersed into 5 mL of a dye soaking solution for sensitisation. Conditions for dye adsorption were as follows: RuAc, RuP,RuNPhAcAc and RuNPhAcSi – 0.1 mM solution in methanol, 16 h at room temperature; RuNPhSiLSiL – 0.05 mM solution in 1 vol% (C2H5)3N in dimethylsulphoxide, 72 h at 80 °C. Dye solutions were always used and stored in the dark in leak-proof screw-cap vials to prevent photodegradation (glassware was covered with dense layers of aluminium foil). Upon sensitisation, the electrodes were sonicated in the dark for 30 min in methanol to remove physisorbed dye. A dye-sensitised photoanode is exemplified photographically in Fig. S1.†
Photoelectrochemical measurements
All photoelectrochemical experiments were performed with a Bio-Logic VSP electrochemical workstation in a three-electrode configuration using custom-made ‘Saubrigues’ cells (Fig. S2†). Smaller (total volume = 400 μL, head space = 150 μL) and larger (total volume = 2000 μL, head space = 800 μL) cells were used for experiments in the absence and in the presence of Na2SO3, respectively. The very low volume of the electrolyte solution was used to facilitate ICP-MS analysis of dissolved ruthenium after experiments. A platinum wire with ca. 3 cm2 geometric surface area was used as an auxiliary electrode. A ‘leak-free’ Ag|AgCl|KCl (3.4 M) LF-1 reference electrode (Innovative Instruments, Inc.) was used throughout, but the potentials are reported versus the reversible hydrogen electrode (RHE) according to the relationship E vs. RHE = E vs. Ag|AgCl + 0.205 + 0.059 pH. Dye-sensitised TiO2/FTO films were used as working electrodes. All current densities are reported normalised to the geometric area of the screen-printed TiO2 layers (4 mm2) or to the amount of adsorbed dye, as indicated in the text.A solar simulator (Newport, 67005) equipped with a 150 W xenon lamp with a spectral distribution of AM1.5G and intensity of 100 mW cm−2 was used to provide irradiation during experiments. A reference solar cell (CalLab, Frauenhofer ISE; serial no. 010-2010, calibration mark: 1010-2010014ISE0311) was used to adjust the neutral density filters to reach the stated current density–voltage profile. A λ > 400 nm filter was positioned in front of the beam, blocking UV irradiation and thereby preventing direct band-gap excitation of TiO2. Irradiation of the photoelectrodes in all experiments was always from the back (through the FTO-covered glass support), though control experiments confirmed that irradiating from the front does not change the results.
The cell was assembled in the dark with outlet- and inlet-gas needles, and a syringe containing an electrolyte solution (via a needle). After the latter was injected into the cell, the syringe was removed and the solution was deoxygenated by bubbling high purity argon (flow rate 1 mL min−1; Bronkhorst EL Flow Select mass flow controller). In all experiments undertaken in the absence of Na2SO3, deoxygenation continued for exactly 2 minutes and photoelectrochemical measurements were launched exactly 30 seconds after the deoxygenation step, which were needed to adjust the inlet-gas needle above the solution and allow argon to flow into the headspace at the same flow rate, leaving the solution quiescent. Knowing that hydrolysis of the phosphonate and carboxylate anchors attached to titania commences immediately upon contact with water, the duration of the Ar pre-purging stage prior to the photoelectrochemical measurement was kept unchanged at 2.5 min for every sample. In the experiments for oxidation of Na2SO3, the electrolyte solutions were deoxygenated for exactly 1 hour immediately after being injected into the cell, and then photoelectrochemical measurements commenced with leaving Ar flowing through the solution to intensify mass-transport of the sulphite anions. After each experiment, the electrolyte solution was extracted and the cell was washed with two 100 μL aliquots of ultra-pure water, which were collected and subsequently analysed. All experiments were performed 3 times.
UV-vis spectroscopy
UV-vis spectra were recorded with a LAMBDA 950 spectrophotometer equipped with an integrating sphere (Perkin-Elmer). Solutions were analysed using quartz cuvettes with a 1 mm optical path. Electrodes were fixed during measurements using a homemade holder that was attached in front of the integrated sphere chamber detector, minimising the effects of light scattering at the TiO2.Inductively coupled plasma-mass spectroscopy
ICP-MS analysis was undertaken with a GBC Optimass 9500 ICP-TOF-MS instrument. Aqueous 2 wt% HNO3 was used as the carrier solution and also to dilute analysed samples. Rh was spiked into each sample at 25 ppb as an internal standard (diluted from commercial solution, Fluka). Ru calibration solutions were prepared by diluting a commercial stock solution (EM Science). Analysis of each sample was performed ten consecutive times. Photoanodes were digested in aqueous 0.5 wt% HCl + 2 wt% HNO3 (RuAc and RuNPhAcAc) or 0.5 wt% HCl in H2SO4 (96%): H2O2 (30%) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 vol) (RuP, RuNPhSiSi and RuNPhAcSi) for one week.
:3 vol) (RuP, RuNPhSiSi and RuNPhAcSi) for one week.
Attenuated total reflectance infrared spectroscopy
ATR-FTIR analysis on dye powder and FTO/TiO2 dye films was performed using Bruker Vector 22 FTIR spectrometer with ATR accessory.Results and discussion
Photoanode assembly and testing protocol
The present study focuses on a dye-sensitised anode configuration based on an n-type semiconductor support. Considering its modest cost, visible light transparency, good electrical conductivity, sufficiently wide band gap and appropriate energy of the conduction band edge,63,64 titanium(IV) oxide (anatase phase) remains the most used n-type semiconductor for DS-PEC water-splitting anodes and was therefore used herein. Typically, the best performing dye-sensitised photoanodes require comparatively thick (7–25 μm),27–29,65 highly porous electron-transporting layers and post-necking treatment. However, the present work aims to assess the stability of the dye-support linkage, and therefore uniform exposure of the dye film to the electrolyte solution is preferred. Ideally, this can be achieved with a flat support, though quantitative analysis in this case would be severely impeded due to very low photocurrents and concentrations of adsorbed and dissolved dyes. As a compromise, we have employed 2.5 μm thick mesoporous TiO2 films (18 nm particle size), which also facilitates UV-vis characterisation compared to thicker films that scatter more strongly.The photosensitisers use a [Ru(bpy)3]2+ core functionalised with carboxylate, phosphonate and/or silanetriolate moieties (Scheme 1a). RuAc and RuP contain two carboxylate and phosphonate linkers, respectively, that are directly attached to the bipyridine. These dyes have been widely used in DS-PEC applications, and are therefore used as reference materials herein. The silanetriolate linkers have an amide-benzyl bridge connecting them to the bipyridine ligand, with RuNPhSiSi containing two silanetriolate anchors, whereas RuNPhAcSi contains one carboxylate and one silanetriolate anchor. A further reference RuNPhAcAc was synthesised, containing two carboxylate groups connected via amide–benzyl bridges.
The inclusion of the amide-benzyl bridge allowed a facile synthesis via peptide-coupling.52,53,62 Its presence induced notable but not substantial changes in the absorption spectra of the dye, in particular a peak at 460 nm was red-shifted by approximately 30 nm and absorption at wavelengths shorter than 420 nm was enhanced (Fig. S3†). The energy levels of the dyes derived from cyclic voltammetric, UV-vis and photoluminescence measurements were only slightly altered by the chemical nature of the anchoring groups (Scheme 1b). Notably, introduction of the amide–benzyl connection results in significantly reduced solubility in common solvents like methanol, ethanol and acetonitrile. The presence of silanetriolate groups accentuates this effect, and RuNPhSiLSiL·2PF6 (L = triethanolamine protective group) is essentially insoluble in the aforementioned solvents and can be only dissolved in acidified water (pH = 1–3) or 1 vol% (C2H5)3N in dimethylsulphoxide. Removal of the protective group (L) from the silanetriolate anchors decreases the solubility even further, consistent with previous reports.54,66RuNPhAcSi·2Cl is moderately soluble in alcohols and acidic aqueous solutions, but remains insoluble in acetonitrile.
Moderate levels of variability in the amounts of dye adsorbed onto the titania surface were found to be unavoidable, especially between the sensitisers with different anchoring groups, as inferred from the ICP-MS analysis on the solutions derived from digesting freshly prepared electrodes (Table S2†). Preferential chemisorption as opposed to physisorption of dyes onto the TiO2 surface was confirmed by ATR-FTIR (Fig. S4†). Typically, the surface concentrations of the dyes followed the sequence Ac ≈ NPhAcAc > P > NPhAcSi > NPhSiSi. Suppressed adsorption of the complexes containing silanetriolate anchors presumably originates from steric hindrance60 and/or partial polymerisation of the molecules. Obviously, differences in the amount of adsorbed dye significantly affect the performance. This further emphasises the difficulties in directly comparing literature results based solely on geometric area current densities, and is addressed herein by comparing the photoelectrochemical data in terms of the current densities normalised to the amount of dye initially adsorbed on the electrode as precisely measured by ICP-MS.
The overall protocol employed herein for characterisation of the dye-sensitised photoanodes is summarised in Fig. 1. The effect of variability in dye loading on the UV-vis spectra and photoelectrochemical properties measured in a potentiostatic mode (chronoamperometry) is exemplified for three similarly prepared and tested RuAc/TiO2/FTO electrodes in steps 1 and 2 (green, orange and wine data in Fig. 1). UV-vis analysis of the electrodes after photoelectrochemical measurements indicates the integrity of any remaining dye (Fig. 1, step 3). Subsequent ICP-MS analysis cannot distinguish between intact and degraded dye molecules, but facilitates quantification of the amounts of ruthenium remaining on the electrode surface after testing as well as that which was dissolved into the electrolyte solution. Taken together, these ICP-MS derived quantities yield a precise value of the absolute amount of dye prior to photoelectrochemical measurements. Note that UV-vis analysis of the films was not as accurate and reliable between samples (e.g. compare ICP-MS loading and UV-vis peak absorbance data in Tables S3, S4, and Fig. S5†), which emphasises that absorption spectroscopy on highly scattering and unavoidably heterogeneous films should be used with caution when precise quantification of the surface dye concentration is pursued. As can be observed with representative RuAc/TiO2/FTO electrodes (Fig. 1), the oxidative photocurrent densities recorded after approximately 20–30 min of testing perfectly align when normalised to the absolute amount of the dye determined by ICP-MS (Fig. 1, step 4). Importantly, this holds true for the similarly prepared photoanodes (green, orange and wine data in Fig. 1) as well as those with a significantly lower dye loading (navy data in Fig. 1) or with a much thicker TiO2 layer (ocher data in Fig. 1). Thus, this testing protocol enables reliable comparisons of both the photoelectrochemical efficiency and the stability of the dye-sensitised anodes by eliminating the effects of commonly observed variability in the amount of adsorbed sensitiser.
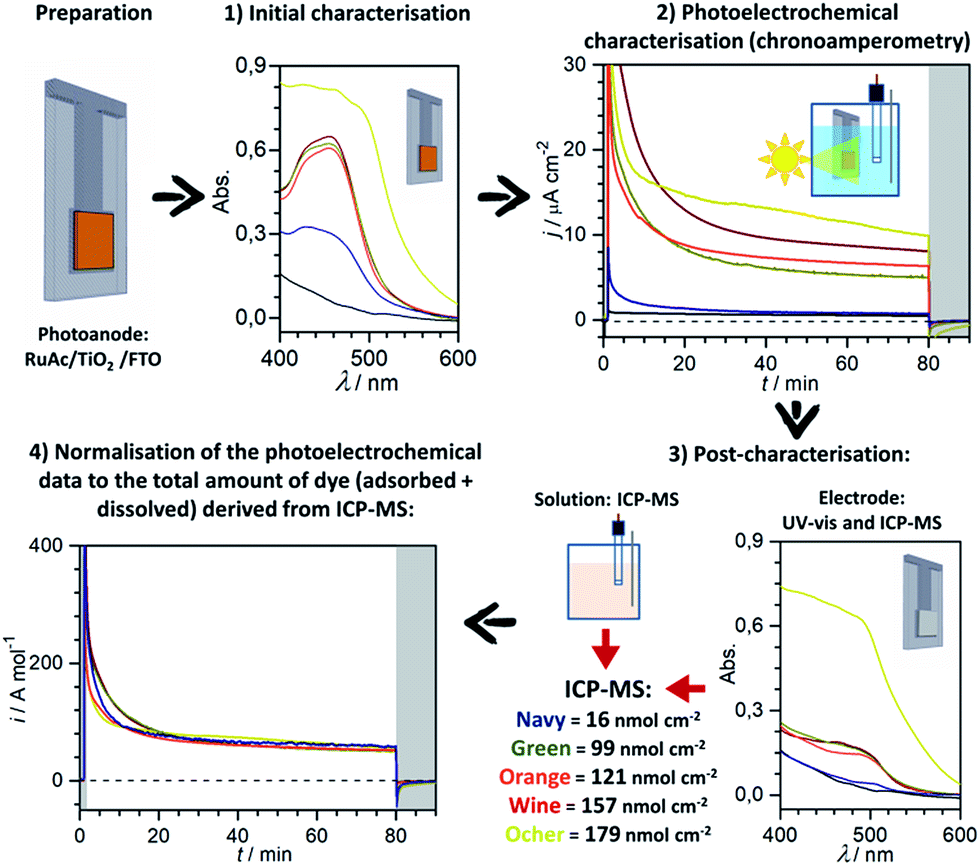 | ||
| Fig. 1 Experimental protocol for characterisation of the dye-sensitised electrodes. UV-vis and photoelectrochemical data are exemplified for three similarly prepared RuAc/TiO2 (2.5 μm)/FTO electrodes (green, orange, wine), RuAc/TiO2 (2.5 μm)/FTO with very low sensitiser loading (navy), and RuAc/TiO2 (10 μm)/FTO photoanode (ocher). Grey data were obtained with an unsensitised electrode. Photoelectrochemical experiments were undertaken in aqueous 0.5 M NaClO4 at pH 3. | ||
Effect of the anchoring group on the intrinsic photoelectrochemical performance
In all experiments, photoanodes were irradiated with the visible portion (λ > 400 nm) of simulated 100 mW cm−2 AM1.5G irradiation (hereinafter, 1 sun) to avoid direct band-gap excitation of the TiO2 support.64,67 Chronoamperograms were recorded at potentials corresponding to the dark open-circuit values measured prior to illumination (Eapplied; values provided in the corresponding figure captions) to avoid any undesired electrochemical transformations contributing to the measurements. Typically, Eapplied ranged from 0.42 to 0.72 V vs. the reversible hydrogen electrode (RHE), did not show systematic dependence on the nature of the dye or electrolyte and were always located within a region where photocurrents were essentially invariable with respect to the applied potential (Fig. S6†). The latter was critically important from the perspective of quantitative comparisons of photoelectrochemical performance of different dyes studied at different potentials.Comparisons of the intrinsic photoelectrochemical performance of the dye-sensitised electrodes were undertaken in phosphate buffered (1.0 M, pH 7.0) solutions of 1 M Na2SO3 (Fig. 2). The latter undergoes a thermodynamically and kinetically facile two-electron oxidation, with a standard reversible potential of −1.12 V vs. the normal hydrogen electrode.68 Chronoamperograms were recorded under a continuous flow of Ar through the solution for a total of 50 hours, including two 24 hour-long irradiation periods separated by 2 hours in the dark, during which the solution in the cell was replaced with a fresh portion.
 | ||
| Fig. 2 Chronoamperometric photoelectrooxidation (1 sun, λ > 400 nm) of aqueous 1 M Na2SO3 buffered with phosphate (1.0 M, pH 7.0) by RuAc/TiO2/FTO (orange; Eapplied = 0.463 V vs. RHE), RuP/TiO2/FTO (green; Eapplied = 0.543 V vs. RHE), RuNPhAcAc/TiO2/FTO (purple; Eapplied = 0.423 V vs. RHE); RuNPhSiSi/TiO2/FTO (grey; Eapplied = 0.585 V vs. RHE); RuNPhAcSi/TiO2/FTO (cyan; Eapplied = 0.713 V vs. RHE). Measurements were undertaken under continuous purging of argon through solutions. Currents are normalised to (a) the geometric surface area of the electrodes and (b) the amount of dye initially adsorbed. Shaded parts of the plots indicate time periods of no illumination. Electrolyte solution was replaced with a fresh portion after 25 hours in each experiment. | ||
During the initial few minutes of illumination, RuP/TiO2/FTO demonstrated a photocurrent density (ihv) of up to 3.9 × 103 A mol−1 (jhv 180 μA cm−2), however it rapidly degraded to ca. 0.75 × 103 A mol−1 (35 μA cm−2) after only two hours. Simultaneous colouration of the solution indicated RuP solubilisation, which appeared to be essentially complete after 2–3 hours when the photocurrent adopted a slower decay regime, eventuating in ihv below 0.20 × 103 A mol−1 (<10 μA cm−2) after 24 hours of testing. At the end of the measurement, RuP visibly appeared to be completely absent from the TiO2 surface, and its characteristic absorption peaks were barely observable in the UV-vis spectrum (Fig. S7†). On a quantitative level, 99% of the dye was lost from the electrode, with ICP-MS analysis confirming that 94% of ruthenium was dissolved and only 6% was still present on the titania surface (Table S5†). The photoanodes with acetate-anchored sensitisers, RuAc and RuNPhAcAc, followed similar behaviour, though with significantly lower photocurrent densities observed from the onset. This is likely due to faster desorption, which could be visually identified even prior to illumination. The performance of the electrodes sensitised with water-soluble RuAc and RuP dyes was also found to strongly depend on both convection and the solution volume, i.e. the use of small volumes and quiescent conditions slows the rate of sensitiser desorption, which allows for higher transient photocurrents. In addition to desorption, light-induced degradation leading to the formation of Ru μ-oxo species has been observed in the presence of sulphur based sacrificial electron donors.69
Among the examined photoanodes, RuNPhSiSi/TiO2/FTO demonstrated the most stable photocurrent with little deterioration during long-term oxidation of Na2SO3, although the quasi-steady state photocurrent density was rather low at ca. 0.45 × 103 A mol−1 (4 μA cm−2). This low performance is consistent with the expected poor electron transfer through the silanetriolate anchors. The highest oxidative photocurrent densities over the whole period of the test were achieved with RuNPhAcSi/TiO2/FTO, which provided ca. 1.5 × 103 A mol−1 (31 μA cm−2) and 1.3 × 103 A mol−1 (25 μA cm−2) at t = 24 and 48 h, respectively. This demonstrates the improved electron transfer of the silanetriolate-carboxylate combination, compared to the dual-silanetriolate anchor. Furthermore, the intrinsic photoelectrooxidation capacity of RuNPhAcSi is no worse than that of the benchmark RuP under the employed conditions, as concluded from comparison of the photocurrent densities during the very first minutes of the tests (Fig. 2b), i.e. prior to dissolution of RuP. Deterioration in the performance of RuNPhAcSi/TiO2/FTO can be attributed to the loss of ca. 30% of the dye from the electrode surface, as inferred from UV-vis after the 50 h experiment (Fig. S7 and Table S5†). Ruthenium loss from the electrode was very similar in this case (31% by ICP-MS), pointing to the dissolution of the sensitiser as one of the major causes for decay in the photoelectrooxidation capacity of the photoanode in the presence of Na2SO3. However, injection of a fresh portion of Na2SO3 solution after 25 hours appreciably enhances the photocurrent density measured with the RuNPhAcSi/TiO2/FTO anodes (Fig. 2), suggesting that depletion of the sulphite anion also notably contributes to the deterioration of the photoelectrochemical performance in these experiments. Notwithstanding this instability of the electron donor on a longer timescale, the results obtained can be used for unequivocal comparisons of the intrinsic photooxidation capacities of the examined dyes, since all experiments were undertaken under exactly the same conditions with freshly prepared Na2SO3 solutions. Importantly, examination of the data in Fig. 2b leads to an unambiguous conclusion that the rate of sulphite photoelectrooxidation sustained by RuNPhAcSi is well in excess of the aggregate of the rates for RuNPhSiSi and RuNPhAcAc (or RuAc). This observation confirms the cooperative action of the silanetriolate-carboxylic anchoring of the developed sensitiser, where one linker provides robust attachment and another sustains efficient charge transport.
Stability under stress conditions
To further demonstrate the very significant differences in stability of the dye-sensitised electrodes provided by different anchoring groups, a set of experiments was undertaken under ‘stress’ conditions, viz. in the absence of readily available electron donors. These conditions represent an extreme case of a functioning water-oxidising photoanode, where charge transfer between the photoexcited dye molecules and catalyst species (electron donor) becomes fully prohibited. In a more realistic scenario of a properly designed system, the portion of adsorbed dye molecules failing to be quickly reduced upon photon absorption should of course be very low, but can hardly be completely eliminated. This can promote chemical degradation of the dyes,70 which is another major mechanism contributing to the notorious deteriorating performance of dye-sensitised electrodes along with desorption.In the case of the [Ru(bpy)3]2+-based dyes, oxidative quenching of the sensitiser by TiO2 following photoexcitation leads to the formation of a ruthenium(III) complex, which is known to be unstable in aqueous solutions.38,71,72 At higher pH, the postulated decomposition mechanism involves an attack by OH− on the α-carbon atom of one of the bipyridine ligands, which initiates a cascade of oxidative decomposition reactions.71,72 At lower pH, substitution of the bipyridine ligand(s) with H2O or supporting electrolyte anion is instead thought to be the primary mechanism.38 The presence of buffering anions is also known to promote degradation of [Ru(bpy)3]3+, although the relevant mechanism remains unclear.73
Thus, under the photoelectrooxidation ‘stress’ conditions employed herein, there are no easily-oxidisable species, which enhances the amount of time the dye spends in the Ru(III) state and facilitates assessment of the chemical stability of the immobilised dyes. Furthermore, to fully explore the aqueous stability of the dyes, measurements were undertaken over a wide pH range, as well as with solutions containing the common buffering anions CH3CHOO− and PO43−, the latter of which is known to promote hydrolysis of adsorbed dyes.74 Chronoamperograms were recorded for a total of 90 minutes under 1 sun irradiation, preceded by 1 and followed by 10 minutes in the dark (Fig. 3 and S8–S10†). The nature of the measured photocurrents cannot be easily established, due to their very low absolute values, but they are expected to be a combination of the oxidation of either water, unknown admixtures in solution or the dye complex itself, resulting in degradation.
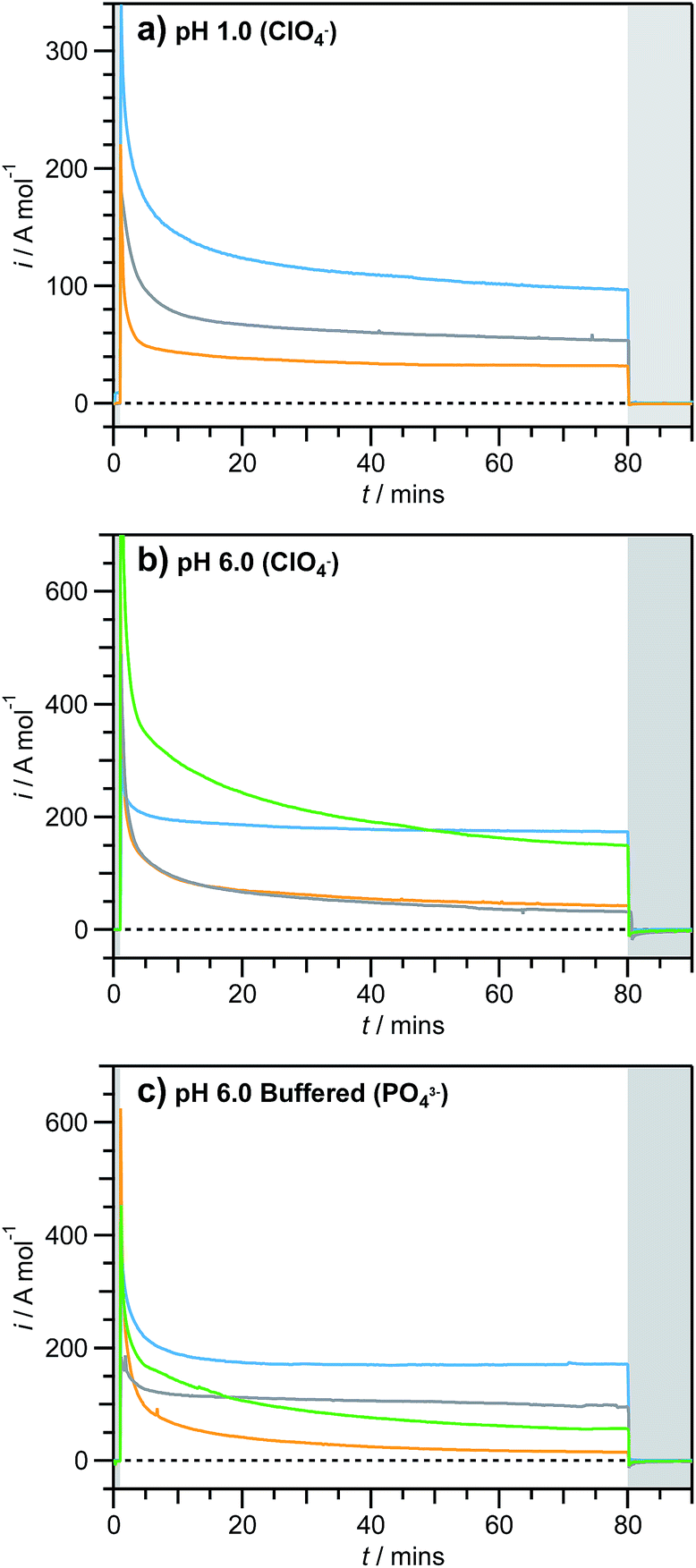 | ||
| Fig. 3 Chronoamperograms for photoelectrooxidation (1 sun, λ > 400 nm) of aqueous 0.5 M NaClO4 at pH 1.0 (a) and 6.0 (b) and 0.1 M phosphate buffer at pH 7.0 (c) by RuAc/TiO2/FTO (orange; Eapplied = (a) 0.517, (b) 0.576, (c) 0.563 V vs. RHE); RuP/TiO2/FTO (green; Eapplied = (b) 0.581, (c) 0.635 V vs. RHE); RuNPhSiSi/TiO2/FTO (grey; Eapplied = (a) 0.589, (b) 0.659, (c) 0.533 V vs. RHE); RuNPhAcSi/TiO2/FTO (cyan; Eapplied = (a) 0.529, (b) 0.593, (c) 0.632 V vs. RHE). Measurements were undertaken in quiescent argon-saturated solutions. Currents are normalised to the amount of dye initially adsorbed on the electrode surface. Shaded parts of the plots indicate time periods absent illumination. | ||
During the initial minutes of these tests, photocurrents always showed a sharp decrease followed by further slower decreases that were dependent on both the anchoring group and the electrolyte conditions (Fig. 3, Tables 1, 2 and S6–S8†). Photoanodes sensitised with silanetriolate-anchored dyes reached quasi-stabilised photocurrent densities in all but a few harsh conditions (vide infra), though persistent deterioration in performance was always observed for the RuP and RuAc dyes (Fig. 3 and S8–S10†). These trends are qualitatively consistent with the results obtained for the photoelectrooxidation of the sulphite anion (Fig. 2). As above, the origins of the deterioration of the photoelectrochemical performance were distinguished through the aforementioned UV-vis and ICP-MS analysis protocol (Tables 1, 2 and S6, Fig. 4 and S11–S13†). Consequently, the decrease in the amount of adsorbed dye during the experiments (UV-vis before and after), the amount of ruthenium dissolved into solution (ICP-MS), and the amount of chemically degraded dye to produce Ru-based species on the electrode surface (UV-vis vs. ICP-MS) can be separated.
| Dye | Electrolyte | pH | Dye loadinga/nmol cm−2 | i hv /A mol−1 | Ru loss/% (ICP-MS) | Dye loss/% (UV-visc) | |
|---|---|---|---|---|---|---|---|
| 10 min | 80 min | ||||||
| a Determined by ICP-MS. b Derived from chronoamperograms measured under 1 sun irradiation in quiescent Ar-saturated solutions; currents are normalised to the amount of dye initially adsorbed. c Calculated as a ratio of absorbance at λmax (RuAc – 458 nm; RuP – 447 nm) before and after photoelectrochemical testing. | |||||||
| RuAc | NaClO4 (0.5 M) | 1.0 | 99 | 43 | 32 | 95 | 88 |
| 2.0 | 114 | 64 | 45 | 87 | 86 | ||
| 3.0 | 99 | 120 | 52 | 81 | 70 | ||
| 4.0 | 104 | 102 | 49 | 95 | 86 | ||
| 5.0 | 70 | 95 | 43 | 94 | 92 | ||
| 6.0 | 55 | 89 | 43 | 94 | 92 | ||
| 7.0 | 80 | 96 | 43 | 95 | 89 | ||
| 8.0 | 77 | 66 | 36 | 93 | 92 | ||
| Acetate buffer (0.1 M) | 4.0 | 44 | 68 | 22 | 81 | 81 | |
| 5.0 | 70 | 17 | 7 | 91 | 86 | ||
| 5.6 | 56 | 23 | 11 | 92 | 95 | ||
| Phosphate buffer (0.1 M) | 6.0 | 39 | 64 | 16 | 71 | 90 | |
| 7.0 | 47 | 42 | 13 | 87 | 91 | ||
| 8.0 | 33 | 29 | 0 | 91 | 88 | ||
| RuP | NaClO4 (0.5 M) | 6.0 | 99 | 43 | 32 | 95 | 86 |
| Phosphate buffer (0.1 M) | 6.0 | 114 | 64 | 45 | 87 | 96 | |
| Electrolyte | pH | Dye loadinga/nmol cm−2 | i hv /A mol−1 | Ru loss/% (ICP-MS) | Dye loss/% (UV-visc) | |
|---|---|---|---|---|---|---|
| 10 min | 80 min | |||||
| a Determined by ICP-MS. b Derived from chronoamperograms measured under 1 sun irradiation in quiescent Ar-saturated solutions; currents are normalised to the amount of dye initially adsorbed. c Calculated as a ratio of absorbance at λmax = 482 nm before and after photoelectrochemical testing. | ||||||
| NaClO4 (0.5 M) | 1.0 | 31 | 180 | 121 | 22 | 25 |
| 2.0 | 38 | 201 | 141 | 9 | 16 | |
| 3.0 | 36 | 193 | 152 | 7 | 21 | |
| 4.0 | 31 | 187 | 167 | 7 | 23 | |
| 5.0 | 37 | 255 | 169 | 7 | 35 | |
| 6.0 | 47 | 193 | 174 | 7 | 18 | |
| 7.0 | 38 | 207 | 163 | 6 | 24 | |
| 8.0 | 32 | 172 | 145 | 8 | 28 | |
| 9.0 | 33 | 137 | 116 | 11 | 27 | |
| Acetate buffer (0.4 M) | 4.0 | 41 | 170 | 144 | 7 | 28 |
| 5.0 | 38 | 140 | 142 | 6 | 14 | |
| 5.6 | 23 | 166 | 142 | 5 | 5 | |
| Phosphate buffer (0.1 M) | 6.0 | 31 | 189 | 171 | 7 | 17 |
| 7.0 | 25 | 178 | 170 | 7 | 28 | |
| 8.0 | 29 | 177 | 170 | 7 | 28 | |
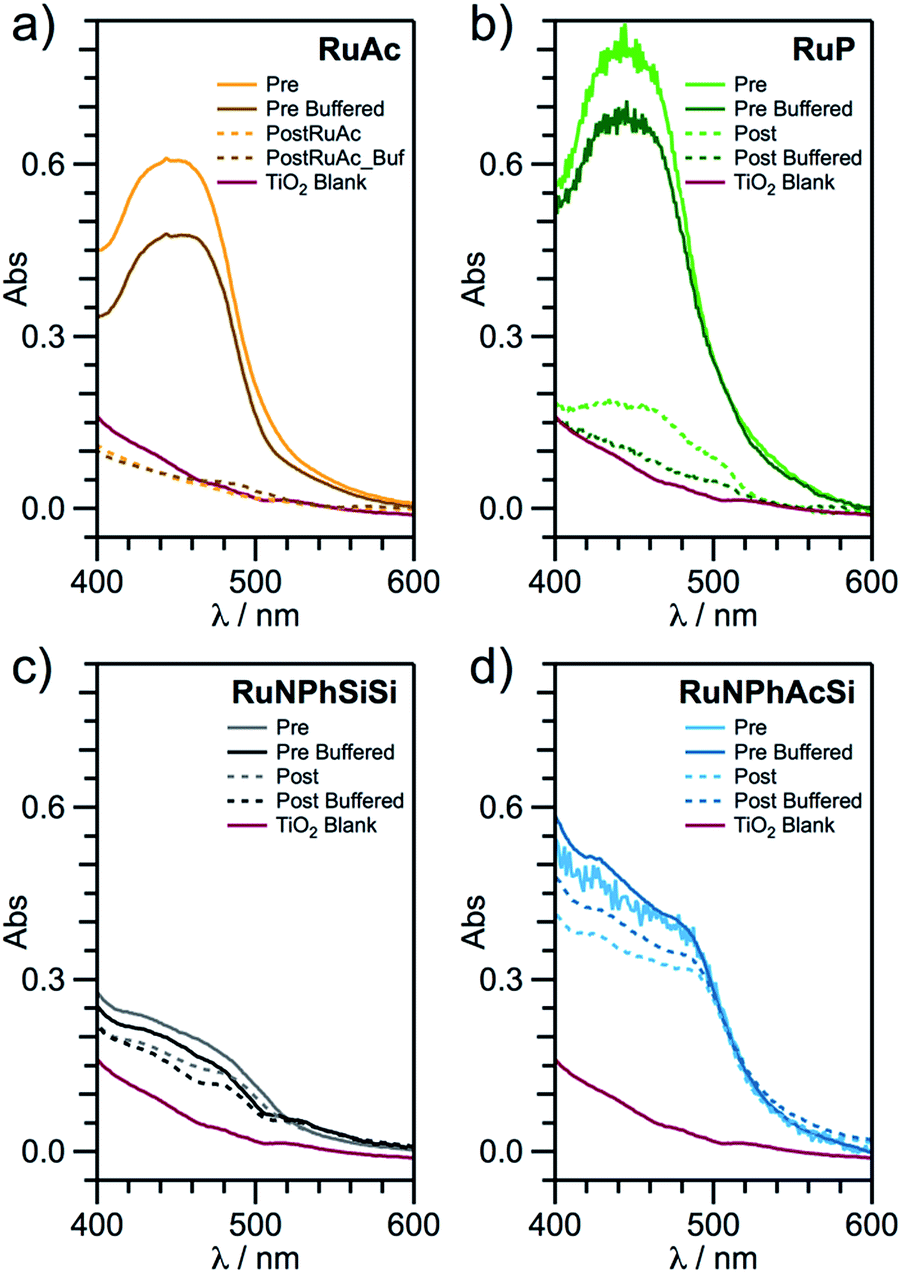 | ||
| Fig. 4 UV-vis absorption spectra of (a) RuAc/TiO2/FTO (orange); (b) RuP/TiO2/FTO (green); (c) RuNPhSiSi/TiO2/FTO (grey); and (d) RuNPhAcSi/TiO2/FTO (cyan), before (solid lines) and after (dashed lines) photoelectrochemical oxidation of aqueous 0.5 M NaClO4, pH 6.0 (bright traces) or 0.1 M phosphate buffer, pH 6.0 (dark traces). Purple curves show background spectra for TiO2/FTO. | ||
A very significant loss of the ruthenium dye (typically, > 80%) within only 80 min of illumination under all examined electrolyte conditions occurred with the RuAc/TiO2/FTO and RuP/TiO2/FTO electrodes (Table 1 and Fig. 4; full set of spectra for RuAc in Fig. S11–S13†). Juxtaposition of the ICP-MS and UV-vis data clearly shows that these losses are, in the first instance, due to dissolution of the sensitisers rather than their decomposition with the formation of other ruthenium species on the electrode surface. Indeed, both methods produced very consistent data, attesting to the unsatisfactory hydrolytic stability of both the carboxylate and phosphonate anchoring groups under aqueous conditions (Table 1). Importantly, the degree of deterioration in ihv values over the course of the experiments was typically less significant when compared to the dye loss from the electrode (Table 1). Moreover, visual inspection of the RuAc/TiO2/FTO and RuP/TiO2/FTO electrodes indicated that severe desorption/degradation of these dyes occurred within the initial several minutes of tests under all conditions examined. These observations further suggest that a significant portion of the photocurrent measured with these photoanodes might originate from dissolved dye molecules, rather than those adsorbed on TiO2.
By comparison, the RuNPhSiSi/TiO2/FTO electrodes demonstrate vastly improved stability with respect to desorption, with ≤7% ruthenium loss in both buffered and unbuffered media (Table S6†). However, photoinduced degradation of the dye complex occurred in this case, as determined by UV-vis (Fig. 4 and Table S6;† full set of spectra for RuNPhSiSi in Fig. S14–S16†), and this is identified as the major mechanism for the slow photocurrent decline (Fig. 3, S9 and Table S6†). The rate of this degradation is variable across the examined pH range, with remarkably little or essentially no degradation at pH 3–5 in unbuffered electrolytes. Interestingly, compared to unbuffered solutions, addition of buffering anions appears to provide increased stability at pH 5–6. Taken together, the photoelectrochemical, ICP-MS and UV-vis data demonstrate exceptional stability of the RuNPhSiSi/TiO2/FTO photoanodes at pH within the range 3–6.
The replacement of one silanetriolate linker with a carboxylate equivalent produces only small changes in the photoanode stability. This is reflected by only 5–7% ruthenium loss for the RuNPhAcSi/TiO2/FTO electrodes under all electrolyte conditions studied except for the most extreme ones (Table 2). Sensitiser photodegradation for both RuNPhSiSi and RuNPhAcSi occurs to a similar extent across the measured pH range with no obvious trend (cf. Table S6† and 2). The most impressive stability of RuNPhAcSi/TiO2/FTO is found at pH 5.6 in the presence of acetate buffer, where less than 10% of the dye is desorbed/degraded over 80 min of experiment and no deterioration of the photooxidative current occurs within the 10 to 80 min timeframe (Table 2 and Fig. S10;† full set of UV-vis spectra for RuNPhAcSi in Fig. S17–S19†).
Overall, the data presented and discussed above demonstrate that the silanetriolate-carboxylate combination of anchoring groups provides vastly superior performance compared to the widely employed phosphonate linker (Tables 1, 2 and S5†). Notwithstanding the higher photocurrent densities provided by photoanodes with RuP compared to RuNPhAcSi under some conditions (Fig. 2 and 3), the stability of the former is unsatisfactory. Finally, it is important to note that the ‘increased stress’ conditions, with no suitable electron donor available are expected to significantly promote photodegradation of RuNPhAcSi. Indeed, the strongly oxidising and unstable Ru3+ state of the sensitiser cannot be rapidly reduced to the robust Ru2+ ground-state under these conditions, which substantially enhances the probability of decomposition. It is anticipated that the presence of a suitable water-oxidation catalyst in the system will significantly suppress the photodegradation of the sensitiser and further improve the long-term operation of the photoanode.
Conclusions
The combination of the silanetriolate and carboxylate anchoring groups provides outstanding stability in the linkage between the ruthenium(II) tris-bipyridine dye molecules and the mesoporous titania support, while also maintaining efficient charge-transfer in a dye-sensitised photoanode. Systematic examination of the photoelectrooxidation properties, stability and degradation mechanisms for the RuNPhAcSi, RuNPhAcAc and RuNPhSiSi sensitisers, described for the first time herein, was undertaken under a wide range of ‘stress’ electrolyte conditions and demonstrated that the silanetriolate and carboxylate linkers act cooperatively in the RuNPhAcSi configuration. Comparisons with the reference RuAc and RuP dyes with carboxylate and phosphate anchoring groups, respectively, attest to the vastly increased resistance of RuNPhAcSi towards desorption. Importantly, this improved attachment stability does not come at the expense of the intrinsic photoelectrochemical performance. We anticipate that the clear advantages provided by the bifunctional silanetriolate-carboxylate anchoring can significantly advance the currently unsatisfactory long-term performance of dye-sensitised photoelectrochemical systems when operating in aqueous solutions. Moreover, functionalisation of the dyes with this anchor combination was achieved herein via a simple peptide-coupling reaction, which facilitates its application to other dyes absent high synthetic complexity.Another important feature of the present work is the introduction of a comparatively simple yet highly informative photoelectrode testing protocol. Comprehensive characterisation of the photoelectrodes and electrolyte solutions, before and after measurements, enables fair comparisons of the stability and intrinsic photoelectrochemical properties of different dye molecules. This approach provides a guide for the reliable assessment of the long-term performance of dye-sensitised electrodes, as needed for the development of high-performing DS-PEC systems for solar-powered fuel synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We dedicate this work to our mentor, colleague and friend Professor Leone Spiccia, who sadly passed away in late 2016. Leone was among the pioneers of photoelectrocatalytic water splitting with dye-sensitised electrodes. The authors acknowledge the Australian Research Council for funding this work through the ARC Centre of Excellence for Electromaterials Science (Grant No. CE140100012). A sincere thank you to Dr Kate Nairn for her diligent proofreading of this paper.References
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. a Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- G. Ciamician, Science, 1912, 36, 385–394 CAS.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- International Energy Agency, Executive Summary, World Energy Outlook, 2016, Paris, 2016 Search PubMed.
- N. Armaroli and V. Balzani, in Energy for a Sustainable World: From the Oil Age to a Sun-Powered Future, Wiley-VCH, Weinheim, Germany, 2010, pp. 157–165 Search PubMed.
- IPCC, Climate Change 2013, The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2013 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 CAS.
- L. Jingshan, I. Jeong-Hyeok, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, P. Nam-Gyu, S. D. Tilley, F. Hong Jin and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef PubMed.
- S. A. Bonke, M. Wiechen, D. R. MacFarlane and L. Spiccia, Energy Environ. Sci., 2015, 8, 2791–2796 CAS.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 77, 13237 CrossRef PubMed.
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2014, 7, 2056–2070 CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- K. Zhang, M. Ma, P. Li, D. H. Wang and J. H. Park, Adv. Energy Mater., 2016, 6, 1–16 Search PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, J. Am. Chem. Soc., 2011, 133, 1216–1219 CrossRef CAS PubMed.
- T. J. Jacobsson, V. Fjällström, M. Sahlberg, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2013, 6, 3676–3683 CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- K. Fan, F. S. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. C. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- J. W. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- P. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58 CrossRef CAS.
- D. A. Hoogeveen, M. Fournier, S. A. Bonke, X.-Y. Fang, A. J. Mozer, A. Mishra, P. Bäuerle, A. N. Simonov and L. Spiccia, Electrochim. Acta, 2016, 219, 773–780 CrossRef CAS.
- B. D. Sherman, Y. Xie, M. V Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 10974–10982 CrossRef CAS PubMed.
- N. S. McCool, J. R. Swierk, C. T. Nemes, T. P. Saunders, C. A. Schmuttenmaer and T. E. Mallouk, ACS Appl. Mater. Interfaces, 2016, 8, 16727–16735 CAS.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 4–11 Search PubMed.
- H. S. Uam, Y. S. Jung, Y. Jun and K. J. Kim, J. Photochem. Photobiol., A, 2010, 212, 122–128 CrossRef CAS.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- J. T. Hyde, K. Hanson, A. K. Vannucci, A. M. Lapides, L. Alibabaei, M. R. Norris, T. J. Meyer and D. P. Harrison, ACS Appl. Mater. Interfaces, 2015, 7, 9554–9562 CAS.
- A. M. Lapides, D. L. Ashford, K. Hanson, D. A. Torelli, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 15450–15458 CrossRef CAS PubMed.
- D. L. Ashford, A. M. Lapides, A. K. Vannucci, K. Hanson, D. A. Torelli, D. P. Harrison, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6578–6581 CrossRef CAS PubMed.
- K. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514 CrossRef CAS PubMed.
- K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2013, 25, 3–5 CrossRef CAS.
- H. Son, C. Prasittichai, J. E. Mondloch, L. Luo, J. Wu, D. W. Kim, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 11529–11532 CrossRef CAS PubMed.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- S. A. Bonke, A. M. Bond, L. Spiccia and A. N. Simonov, J. Am. Chem. Soc., 2016, 138, 16095–16104 CrossRef CAS PubMed.
- S. S. Gujral, A. N. Simonov, M. Higashi, X.-Y. Fang, R. Abe and L. Spiccia, ACS Catal., 2016, 6, 3404–3417 CrossRef CAS.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- R. Sathre, C. D. Scown, W. R. Morrow, J. C. Stevens, I. D. Sharp, J. W. Ager, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3089–3444 Search PubMed.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- B. J. Brennan, A. E. Keirstead, P. A. Liddell, S. A. Vail, T. A. Moore, A. L. Moore and D. Gust, Nanotechnology, 2009, 20, 505203–505213 CrossRef PubMed.
- B. J. Brennan, M. J. Llansola Portolés, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605–16614 RSC.
- K. L. Materna, B. J. Brennan and G. W. Brudvig, Dalton Trans., 2015, 44, 20312–20315 RSC.
- K. L. Materna, B. Rudshteyn, B. J. Brennan, M. H. Kane, A. J. Bloomfield, D. L. Huang, D. Y. Shopov, V. S. Batista, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2016, 6, 5371–5377 CrossRef CAS.
- N. Connelly, T. Damhus, R. Hartshorn and A. Hutton, in Nomenclature of Inorganic Chemistry: IUPAC Recomendations 2005, 2005, vol. 107 Search PubMed.
- D. G. Brown, P. A. Schauer, J. Borau-Garcia, B. R. Fancy and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1692–1695 CrossRef CAS PubMed.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- K. Szpakolski, K. Latham, C. Rix, R. A. Rani and K. Kalantar-Zadeh, Polyhedron, 2013, 52, 719–732 CrossRef CAS.
- P. D. Beer, J. Am. Chem. Soc., 1997, 119, 11864–11875 CrossRef CAS.
- N. Nickita, M. J. Belousoff, A. I. Bhatt, A. M. Bond, G. B. Deacon, G. Gasser and L. Spiccia, Inorg. Chem., 2007, 46, 8638–8651 CrossRef CAS PubMed.
- S. Chen and L. W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- K. Takijiri, K. Morita, T. Nakazono, K. Sakai and H. Ozawa, Chem. Commun., 2017, 53, 3042–3045 RSC.
- B. J. Brennan, D. Gust and G. W. Brudvig, Tetrahedron Lett., 2014, 55, 1062–1064 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- P. Vanýsek, in CRC Handbook of Chemistry and Physics, 87th edn, 2005, pp. 80–89 Search PubMed.
- U. S. Akhtar, E. L. Tae, Y. S. Chun, I. C. Hwang and K. B. Yoon, ACS Catal., 2016, 6, 8361–8369 CrossRef CAS.
- E. Bae, W. Choi, J. Park, H. S. Shin, S. B. Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093–14101 CrossRef CAS.
- B. Limburg, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5273–5284 CrossRef CAS.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and S. Norman, J. Am. Chem. Soc., 1984, 106, 4772–4783 CrossRef CAS.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- P. A. Connor and A. J. McQuillan, Langmuir, 1999, 15, 2916–2921 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic procedures for the dyes; photograph of a photoanode; description of photoelectrochemical cells; UV-vis spectra of dyes in solution; concentration of dyes on TiO2/FTO; linear sweep voltammetry; UV-vis spectra of electrodes before and after photoelectrochemical experiments; chronoamperograms; tabulated performance and stability of RuAc/FTO/TiO2, RuP/FTO/TiO2, RuNPhAcAc/FTO/TiO2, RuNPhSiSi/FTO/TiO2 and RuNPhAcSi/FTO/TiO2. See DOI: 10.1039/c8se00056e |
| This journal is © The Royal Society of Chemistry 2018 |