
Sequential hydrogen production system from formic acid and H2/CO2 separation under high-pressure conditions†
Masayuki
Iguchi
 a,
Maya
Chatterjee
a,
Naoya
Onishi
b,
Yuichiro
Himeda
b and
Hajime
Kawanami
*a
a,
Maya
Chatterjee
a,
Naoya
Onishi
b,
Yuichiro
Himeda
b and
Hajime
Kawanami
*a
aResearch Institute for Chemical Process Technology, Department of Material and Chemistry, National Institute of Advanced Industrial Science and Technology, Nigatake 4-2-1, Miyagino-ku, Sendai, Miyagi 983-8551, Japan. E-mail: h-kawanami@aist.go.jp
bResearch Institute of Energy Frontier, Department of Energy and Environment, National Institute of Advanced Industrial Science and Technology, Higashi 1-1-1, Tsukuba, Ibaraki 305-8565, Japan
First published on 18th May 2018
Abstract
Hydrogen (H2) production from formic acid (FA) is highly attractive as a sustainable energy source from the interconversion between CO2 and FA. Dehydrogenation of FA at high pressures has advantages over a reaction at atmospheric conditions for the separation of H2 and CO2 due to the reaction and the volumetric energy density of H2. We demonstrated the continuous production of high-pressure H2 by catalytic decomposition of FA, and subsequent separation of H2 and CO2 from FA decomposition gas (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 = 1:1) using the phase change phenomenon at low temperatures while maintaining high pressure. An iridium aqua complex coordinated with a bidentate pyridyl-imidazoline ligand catalyzed the dehydrogenation of FA with high efficiency at a pressure as high as 153 MPa. The Ir catalyst was found to be stable under continuous addition of neat FA at high pressures. The generation time and rate of high-pressure H2 were controlled by feeding neat FA to the aqueous reaction system. Using our combined system, more than 99 mol% of H2 (96 mol% of purity) and 94 mol% of CO2 (99 mol% of purity) were separately obtained from FA as a gas and liquid, respectively, under the high-pressure conditions without any mechanical compression.
:CO2 = 1:1) using the phase change phenomenon at low temperatures while maintaining high pressure. An iridium aqua complex coordinated with a bidentate pyridyl-imidazoline ligand catalyzed the dehydrogenation of FA with high efficiency at a pressure as high as 153 MPa. The Ir catalyst was found to be stable under continuous addition of neat FA at high pressures. The generation time and rate of high-pressure H2 were controlled by feeding neat FA to the aqueous reaction system. Using our combined system, more than 99 mol% of H2 (96 mol% of purity) and 94 mol% of CO2 (99 mol% of purity) were separately obtained from FA as a gas and liquid, respectively, under the high-pressure conditions without any mechanical compression.
Introduction
Sustainable development of our society has been claimed to need to utilize renewable energy and carbon dioxide (CO2), since we face the depletion of fossil fuels and increasing levels of CO2 in the atmosphere. Molecular hydrogen (H2) is considered as a clean alternative energy, but its gaseous nature inhibits direct storage and long distance delivery.1–4 Lately, formic acid (FA, HCO2H) has attracted a lot of attention as a liquid organic hydrogen carrier (LOHC) because of easy handling and transporting to a large extent using our present infrastructures.5–11 FA is a stable liquid at ambient conditions and has a moderately high H2 content among LOHCs such as methylcyclohexane. The production of FA can be made either from biomass or directly by the catalytic conversion of CO2 in solvents.12–14 The FA/CO2 system as a hydrogen carrier promises a carbon-neutral and environmentally benign method (Fig. 1). The H2 production from FA (dehydrogenation, eqn (1)) has a low reaction enthalpy compared to other potential hydrogen carrier materials,15 and the dehydrogenation of FA is a thermodynamically favourable reaction, which can occur at mild temperatures, even under high-pressure conditions, but a side reaction of FA decomposition (dehydration, eqn (2)) is also thermodynamically favourable.15 Hence, an effective catalyst is required for the selective production of H2 from FA, suppressing the side reaction. In addition, effective separation of H2 and CO2 after the reaction is necessary, not only for utilization of H2 but also for CO2, which could be a starting material for a variety of compounds, including FA, through hydrogenation (Fig. 1).| HCO2H(l) → H2(g) + CO2(g) ΔG° = −32.9 kJ mol−1 | (1) |
| HCO2H(l) → H2O(l) + CO(g) ΔG° = −12.4 kJ mol−1 | (2) |
 | ||
| Fig. 1 System of H2 production from FA and storage with CO2. | ||
Mostly, H2 has its application in the transportation sector to generate electricity from fuel cells. Some research groups have examined the application of H2 produced from FA to a proton exchange membrane (PEM) fuel cell.5,16–21 Selective FA dehydrogenation is vital for PEM fuel cell applications because the electro-catalyst involved in the cell can be easily deactivated by CO that is generated from the side reaction of FA decomposition (eqn (2)).22,23 The presence of CO2 can also affect PEM fuel cell performance at high current density due to the formation of CO via the reverse water-gas shift reaction.21–24 Therefore, CO2 removal from H2 is necessary for storage and applications in PEM fuel cells.
The relevant separation methods of H2 and CO2 are adsorption, absorption, use of membranes, and low-temperature distillation.25 High-pressure conditions can enhance the efficiency of H2/CO2 separation, the volumetric energy density of H2, and handling of CO2 for its delivery and storage, and further reaction with H2 to produce FA (hydrogenation of CO2). Furthermore, pressurization and subsequent dehydrogenation of FA in its liquid state avoids the large amount of energy required for generating high-pressure H2 (as much as 10–15% of its energy content).26 Thus, sequential production of H2 from FA and H2/CO2 separation at high pressures can offer an energy-efficient process rather than that at atmospheric pressure. However, a high-pressure process relies on the development of a suitable catalyst to generate high-pressure H2 at mild temperatures.
Since the selective decomposition of FA into H2 and CO2 was performed under mild temperatures,27,28 many homogeneous and heterogeneous catalysts have been developed.5–10 Especially, iridium-based catalysts have shown relatively higher activity and stability in water (turnover number (TON) > 2 × 106).29,30 However, there are few literature references for high-pressure H2 production from FA at less than 100 °C.18,27,31–35 We recently demonstrated high-pressure gas generation up to 123 MPa by dehydrogenation of FA using Cp*IrIII complexes (Cp* = pentamethylcyclopentadienyl, 1 and 2 in Fig. 2) at 80 °C.36,37 The reaction rate was largely decreased with an increase in generated gas pressure by FA decomposition, especially above 10 MPa.38 Some highly active catalysts for dehydrogenation of FA at atmospheric pressure quickly lost their activity at high pressures.39 During the reaction at high pressures, a catalyst needs to be stable, especially under concentrated H2, CO2 and formic acid. Hence, a highly active and durable catalyst is required for the efficient production of high-pressure H2 from FA.
 | ||
| Fig. 2 Time course of high-pressure gas generation by FA decomposition using the catalyst: 1 (cross), 3 (circle); 80 °C and 20 mol L−1 FA aqueous solution (red), 40 °C and 5 mol L−1 FA aqueous solution (blue). Reaction conditions: FA aqueous solution (13 mL), catalyst (2 mmol L−1). | ||
In practical applications, H2 should be produced by the catalytic decomposition of FA on demand. Either reaction temperature or the FA concentration in reaction solution can control the H2 production considering its storage and handling. The decomposition of FA accelerates at higher temperatures; however, the increasing temperature causes catalyst deactivation and a complicated apparatus for evaporation of solvents during the reaction.17,40 The rate of FA dehydrogenation is linearly related to substrate concentration,38 and the addition of substrate can be conducted using a liquid pump. Thus, the reaction time and rate of H2 generation is easier to control by modulating the FA concentration rather than temperature. The addition of neat FA to the catalyst solution allows full use of the hydrogen density of FA. Several studies were reported for H2 production by continuous decomposition of FA at atmospheric pressure,17–19,40–43 but only a few of them focused on the continuous production of H2 from FA at high pressures.31
In our previous work, the Cp*IrIII complex having 2-(2′-pyridyl)imidazoline as the N,N′-bidentate ligand (3 in Fig. 2) was found to show high activity and stability for the catalytic dehydrogenation of FA under atmospheric conditions.44 Herein, we investigated the control of high-pressure H2 generation time and rate by feeding neat FA to the aqueous reaction system using catalyst 3 at mild temperatures. Furthermore, efficient separation of H2 and CO2 after the reaction was studied using the phase change phenomenon of FA decomposition gas (H2:CO2 = 1:1) under high-pressure and low-temperature conditions.
Results and discussion
High-pressure gas generation from FA was studied using catalyst 3 in a batch-wise operation (Fig. 3). When 20 mol L−1 FA aqueous solution was used, the gas pressure reached to 153 MPa in 5 h at 80 °C. The final conversion of FA was calculated as 90 mol% at 153 MPa, and the high-pressure gas was composed of equimolar H2 and CO2 with a quite low concentration of CO (<6 vol ppm). Under the same conditions, the attained pressure using 1 was lower (123 MPa)36 than that of 3, which indicates that 3 is more stable and active catalyst than 1 under the high-pressure conditions with H2 and CO2. In our previous work, we suggested that the catalyst deactivation of 1 is related to change in the chelating conformation by rotation around the pyridyl–pyridyl bond of the bipyridine ligand in the complex.37,39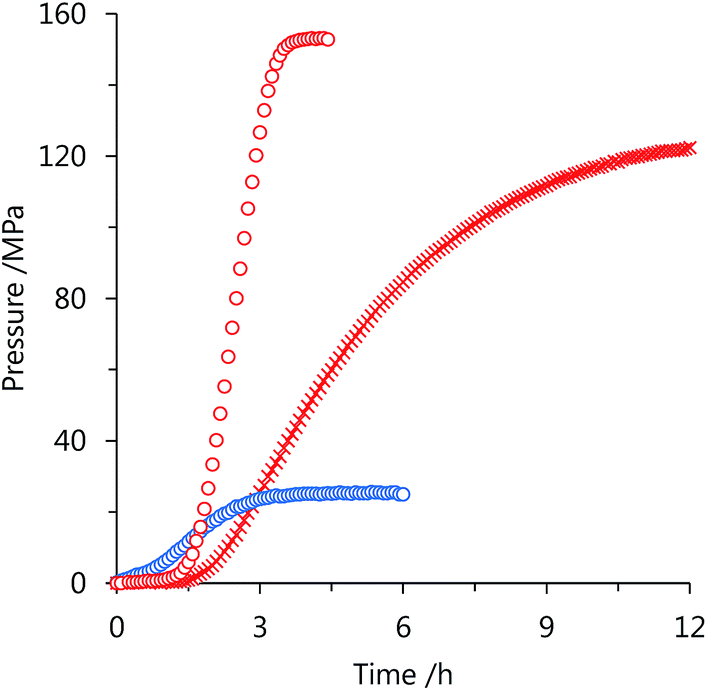 | ||
| Fig. 3 Time course of high-pressure gas generation by FA decomposition using the catalyst: 1 (cross), 3 (circle); 80 °C and 20 mol L−1 FA aqueous solution (red), 40 °C and 5 mol L−1 FA aqueous solution (blue). Reaction conditions: FA aqueous solution (13 mL), catalyst (2 mmol L−1). | ||
In the case of catalyst 3, the chelating conformation is maintained in the complex when the imidazoline moiety rotates around the pyridyl-imidazoline bond. Therefore, the catalytic activity of catalyst 3 can be retained at a pressure as high as 153 MPa. At 40 °C, 92 mol% of FA conversion with a gas pressure of 25 MPa was obtained from 5 mol L−1 FA aqueous solution. Although the presence of bases such as amine can accelerate the rate of FA dehydrogenation, only 44 mol% of FA was converted to H2 and CO2 in the presence of trimethylamine as a co-catalyst, and the attained pressure reduced to 4 MPa under this same condition.45 This significant reduction of FA conversion and the attained pressure may be attributed to the formation of FA-base complexes with trimethylamine, which causes an increase in the Gibbs energy of reaction.46 However, generation of high-pressure H2 by FA decomposition with high conversion can be achieved in the absence of bases.
The dehydrogenation rates of FA were compared among the catalysts 1, 2, and 3 at 40 MPa of the gas pressure (Table 1). For all the catalysts, the dehydrogenation rate remained constant at the beginning of the reaction, whereas it decreased with time as the FA concentration decreased (Fig. S1 in ESI†). For catalyst 2, the FA dehydrogenation was slightly faster, but took a longer time to reach equilibrium than 1 due to the precipitation of the catalyst (entries 1 and 2 in Table 1), which was caused by an increase in pH of the solution during the reaction.37 Using catalyst 3, the reaction was completed quickly compared to the other catalysts, and a high TOF value of 3130 h−1 was obtained at 60 °C and 40 MPa (entry 4 in Table 1). Interestingly, regardless of the catalyst, the reaction rate under the high-pressure condition of 40 MPa decreased to about 1/5 from that of atmospheric pressure. This trend indicates that the effect of gas pressure on the reaction rate is similar among the studied catalysts. The activation energy (Ea) of 3 was determined from the Arrhenius plot under high-pressure conditions (Fig. S2 and Table S1 in the ESI†). The calculated value of Ea at 40 MPa (74 kJ mol−1) is almost the same as that at atmospheric condition (72 kJ mol−1), which agrees with the results of 1.38 The pressure of generated gas by FA decomposition barely affects the catalytic reaction mechanism under the applied conditions.
| Entry | Catalyst | Catalyst concentration (mmol L−1) | TOFb at 40 MPa (h−1) | Reaction time (h) | Pressure effectc (—) |
|---|---|---|---|---|---|
|
a Reaction conditions: 60 °C, generated gas pressure: 40 MPa (H2:CO2 = 1:1), FA aqueous solution (16 mol L−1, 40 ml).
b Turnover frequency (TOF) is defined as the number of FA that convert into H2 by one catalyst per hour, which was calculated from the gas generation rate.
c Pressure effect represents the ratio of TOF at 40 MPa to that at atmospheric pressure.
|
|||||
| 1 | 1 | 2.0 | 450 | 10 | 0.22 |
| 2 | 2 | 2.0 | 590 | 13 | 0.20 |
| 3 | 2 | 0.4 | 630 | 36 | 0.21 |
| 4 | 3 | 0.4 | 3130 | 7 | 0.24 |
Using catalyst 3, the continuous production of high-pressure H2 by FA decomposition was performed at mild temperatures. After stopping the gas generation, neat FA was added to the reaction solution at a constant rate by a high-pressure liquid pump (Fig. S3 in the ESI†). The added FA was continuously and selectively decomposed into H2 and CO2 under the high-pressure conditions (Fig. 4 and 5). The generation rate of high-pressure gas can be controlled by the FA feeding rate, and stop-and-flow gas production experiments can be successfully demonstrated by the injection of FA and keeping the high-pressure (Fig. 4 and Table S3 in the ESI†). Furthermore, when FA was added to the reaction system over 50 h at 40 MPa, high-pressure H2 was continuously generated with a low amount of CO (below 12 vol ppm) at 60 °C (Fig. 5). The generation rate of high-pressure gas remained constant until an initial 30 h, but the rate gradually decreased with duration of the FA addition and the gas generation continued even after FA feeding was stopped. The catalytic activity of 3 appears to be lost very slowly under the high-pressure conditions. The gas generation rate decreased very slowly with catalyst 3, during the addition of high-pressure FA as compared to the catalyst 1 (Fig. S4 in the ESI†).
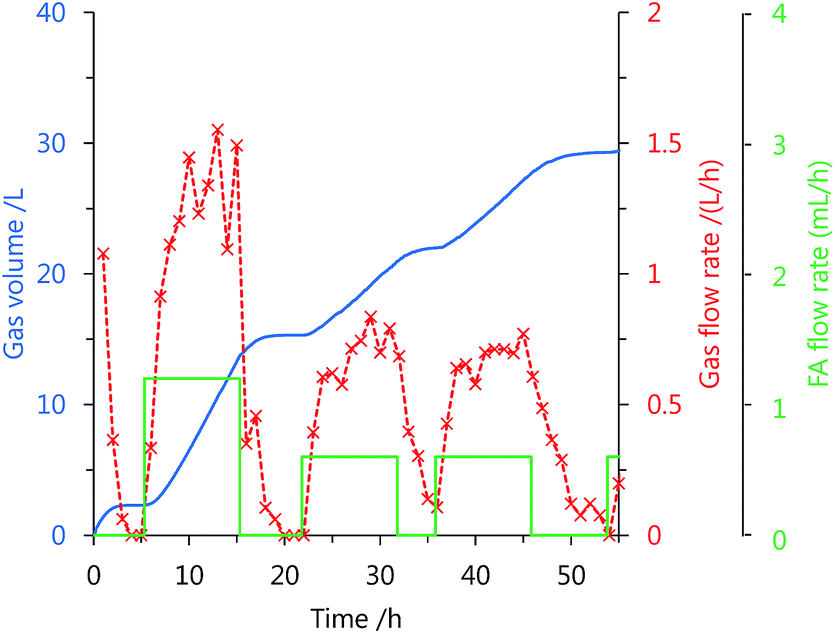 | ||
| Fig. 4 Generation rate and time of high-pressure gas controlled by neat FA addition: gas volume (blue; solid line), gas rate (red; cross), FA addition rate (green; solid line). Reaction condition: 50 °C, 20 MPa, initial FA aqueous solution (5 mol L−1, 40 mL), FA addition rate (0.6–1.2 mL h−1), catalyst 3 (16 μmol). Time starts after reaching the pressure. | ||
 | ||
| Fig. 5 Continuous decomposition of FA at high pressure: gas volume (blue; solid line), gas rate (red; cross). Reaction condition: 60 °C, 40 MPa, initial FA aqueous solution (8 mol L−1, 40 mL), FA addition (1.2 mL h−1, 50 h), catalyst 3 (16 μmol). Time starts after reaching the pressure. | ||
To further examine the stability of catalyst 3 at high pressures, the constant addition of high-pressure FA over 10 h was repeated for several times after stopping the gas generation by FA decomposition at 50 °C (Tables 2 and S3 in the ESI†). When the rate of FA addition was set to 0.6 mL h−1 at 20 MPa, the gas generation rate remained constant until 3 times of FA addition (entry 1 in Table 2). However, CO was observed in the gas generated at the fourth time of FA addition. Catalyst 3 was stable over 30 h under the applied conditions. In the presence of sodium formate (SF), the high-pressure gas was generated slower than in the absence of SF and CO was formed in the gas generated even at the first time of FA addition (entry 2 in Table 2). The reaction temperature barely affects the catalyst stability under the applied conditions, but higher temperature accelerated the formation of CO (entry 3 in Table 2). We therefore hypothesized that CO may be formed by the thermal decomposition of FA due to some catalyst's deactivation. When the FA addition rate was changed from 0.6 to 1.2 mL h−1, the gas generation rate increased about two times compared to the previous one, though the catalyst stability decreased with the increased rate of FA addition (entry 4 in Table 2). The TOF value from the gas generation rate was calculated as 1700 h−1, which is about 7 times higher compared to the Ru complex catalyst tested under the milder temperature (230 h−1 at 100 °C).31 The catalytic activity at 20 MPa started to decrease when the turnover number (TON) exceeded 40000–50000. Under atmospheric conditions, FA was selectively dehydrogenated over 100 h at a constant rate, which corresponded to the TON value of 200 thousand (entry 5 in Table 2). When the gas pressure was 40 MPa, the generation rate of CO-free gas remained constant over 20 h (entry 6 in Table 2). Thus, gas pressure seems to affect the catalyst stability of 3. When the catalyst was exposed to the gas product at high pressures, then catalytic activity was gradually lost with the time of high-pressure gas generation (Fig. S5 in the ESI†). After completion of the reaction, no formation of insoluble compounds was observed and N,N′-bidentate ligand was detected in the solution (Fig. S6 and S7 in the ESI†). Ligand elimination from the complex might cause a decrease in the catalytic activity at high pressures.39 Further investigations on catalyst deactivation are under way.
| Entry | Pressure (MPa) | FA flow rateb (mL h−1) | Stabilityc (h) | Gas generation rated (L h−1) | CO (vol ppm) | TONe (—) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 50 °C, initial FA aqueous solution (5 mol L−1 for 20 MPa and 8 mol L−1 for 40 MPa, 40 mL), catalyst (16 μmol). b FA was continuously added over 10 h and then stopped for several hours. c Time lapsed from the beginning until a decrease in the gas generation rate. d Average value during the continuous gas generation. e Turnover number (TON) is defined as the number of FA that converts into H2 by one catalyst, which was calculated from the total volume of gas release. f Sodium formate was added to the initial FA solution (FA/SF = 10/1 mol mol−1). g Temperature was 70 °C. h Below the detection limit (<2 vol ppm). | ||||||
| 1 | 20 | 0.6 | 30 | 0.65 ± 0.14 | n.d.h | 35400 |
| 2f | 20 | 0.6 | <10 | 0.53 ± 0.09 | 13 ± 6 | 17200 |
| 3g | 20 | 0.6 | 30 | 0.68 ± 0.14 | 46 ± 23 | 36900 |
| 4 | 20 | 1.2 | 20 | 1.36 ± 0.14 | n.d. | 45000 |
| 5 | 0.1 | 1.2 | ≥100 | 1.58 ± 0.24 | n.d. | 198500 |
| 6 | 40 | 0.6 | 20 | 0.54 ± 0.11 | n.d. | 30500 |
High-pressure H2 can be produced by FA decomposition in the presence of catalyst; however, separation of H2 and CO2 after the reaction is required for hydrogen storage using CO2 and the application to PEM fuel cells. When high-pressure H2 is released, it should be cooled in advance to prevent it from igniting due to the negative value of the Joule–Thomson coefficient, as in hydrogen fuelling stations.47 Therefore, a gas cooling unit is inevitable in the application of high-pressure H2. The decrease in gas temperature during depressurization can reduce the energy required for cooling the product gases from the reaction temperature (Table S3 and Process simulation in the ESI†). According to the phase diagram of a H2 and CO2 gas mixture, the vapor–liquid phase separation occurs at high pressures and low temperatures.48,49 The vapor phase mainly consists of H2 whereas the liquid contains more CO2 than H2 under the conditions applied. Previously, we obtained 85 mol% of H2 gas from FA using the vapor–liquid phase separation of FA decomposition gas (H2:CO2 = 1:1) at a low temperature.36 The content of H2 in the gas mixture increased with a decrease in the separator temperature. In this work, the FA decomposition gas was cooled to low temperatures where CO2 forms a solid aiming for the production of highly concentrated H2 gas and the recovery of CO2 under high-pressure conditions (Table 3). The high-pressure gas was generated by FA decomposition products using 3 at 60 °C, and then transferred to the separator cooled to −78 °C, while maintaining the pressure. A pressure drop was observed from 13 to 12 MPa when cooling the separator from −60 to −70 °C (Fig. S8 in the ESI†), which indicates the formation of a solid state. The gas composition was analyzed during depressurization from 11 MPa to atmospheric pressure. More than 95 mol% H2 gas was continuously obtained during the depressurization while keeping the separator cool (entry 3 in Table 3 and Fig. S9 in the ESI†). After depressurizing to atmospheric pressure, the separator was closed and heated to room temperature. The separator pressure increased to 5.2 MPa at 16 °C. The high-pressure gas in the separator was composed of >99 mol% CO2 with less than 1 mol% H2. The attained pressure in the separator corresponds to the saturated pressure of CO2 at 16 °C.50 Thus, when the separator temperature was −78 °C, the FA decomposition gas was separated into H2 gas and CO2 solid with its concentration above 90 mol% in both phases. CO2 can be recovered as a liquid by heating to room temperature without any mechanical compression. Furthermore, both H2 and CO2 were separately recovered from FA with >99 mol% and 94% yield as a gas and liquid, respectively. When the gas pressure was increased from 11 to 27 MPa, a pressure drop occurred at the mild temperature (Fig. S8b in the ESI†), and the CO2 recovery yield decreased to 69 mol% due to an increase of the CO2 concentration in the vapor phase at 27 MPa.48
| Entry | Pressure (MPa) | Separator temperature (°C) | Composition (mol%) | Recoveryc (mol%) | ||
|---|---|---|---|---|---|---|
| H2 in vapor phase | CO2 in liquid phaseb | H2 as gas | CO2 as liquid | |||
|
a Reaction conditions: 60 °C, FA aqueous solution (8 mol L−1, 40 mL), catalyst 3 (0.2 mmol L−1).
b Gas was obtained by heating the separator to room temperature after the high-pressure gas release during the separation was cooled.
c Recovery is defined as the rate of H2 as a gas and CO2 as a liquid recovered from FA decomposition gas (H2:CO2 = 1:1).
d Data was taken from previous work.36
|
||||||
| 1 | 40 | rt | 49 ± 2 | — | — | — |
| 2d | 30 | −15 | 69 | — | — | — |
| 3 | 11 | −78 | 96 ± 1 | >99 | >99 | 94 ± 6 |
| 4 | 27 | −78 | 81 ± 1 | 99 ± 1 | >99 | 69 ± 5 |
Conclusions
In conclusion, we investigated the sequential production of high-pressure H2 by catalytic decomposition of FA and H2/CO2 separation towards the development of FA/CO2 system as a hydrogen carrier and applications in PEM fuel cells. Using an Ir aqua complex 3, the FA added to the reaction system was continuously and selectively decomposed into H2 and CO2 under high-pressure conditions. Neat FA feeding is capable of controlling the generation time and rate of high-pressure H2 for on-demand production. Furthermore, well purified H2 gas (96 mol%) and CO2 liquid (>99 mol%) were obtained by cooling the gas generated from FA decomposition after the reaction while maintaining the pressure. The recovery of CO2 in the liquid state without any mechanical compression gives advantages for delivery and storage, which could be a source of hydrogen carrier. This work suggests that FA decomposition at high pressures can lead to effective separation of H2 and CO2 after the reaction as well as increasing volumetric energy density of H2.Methods
General
The Ir aqua complexes, [Cp*Ir(4,4′-dihydroxy-2,2′-bipyridine)(H2O)][SO4] (1),51 [Cp*Ir(4,7-dihydroxy-1,10-phenanthroline)(H2O)][SO4] (2)52 and [Cp*Ir(2-(2′-pyridinyl)-4,5-dihydro-1H-imidazole)(H2O)][SO4] (3),53 were prepared according to the literature. Formic acid (FA, >99.0%) and sodium formate (SF, >99.0%) were used as received from Wako Pure Chemical Industries, Ltd. Deionized water was purified through a filtration system (EMD Millipore Corp., ZFSQ240P4) and water distillation system (Toyo Roshi Kaisha, Ltd., GS-590). All reagents were degassed by bubbling helium gas before use.The decomposition of FA and separation of the generated gas under high-pressure conditions was carried out using the same apparatus described in our previous work except for the valve between the reactor and the separator.36 For the appropriate evaluation of H2/CO2 gas separation, it is necessary to prevent line blocking by solid CO2 or overflowing at the separator due to the transfer of high-pressure gas. As a solution, we incorporated a new valve, which can control the high-pressure gas transfer rate from the batch reactor to the separator. The aqueous solutions of the catalyst and FA were loaded into a reactor (50 mL) at room temperature, then the reaction solution started stirring and was heated to the desired temperature. Pressure of the generated gas was monitored by a pressure sensor (PGM-500KH, Kyowa Electronic Instruments Co., Ltd.) and the gas pressure was controlled by a back-pressure regulator (JASCO Corp., BP-2080). The volume of gas release was measured by a wet gas meter (Shinagawa Co., Ltd., W-NK-0.5A), and the gas composition was monitored using a GC-μTCD system (Agilent Technologies, 3000A Micro GC). A TOF value was determined from an average rate of gas generation using the ideal gas law. In the GC analysis, the detection limits and quantification were calculated from uncertainty measurements with the coverage factor k = 3 and 10, respectively. For testing the continuous gas generation, neat FA was constantly introduced to the reaction solution by a liquid pump (JASCO Corp., PU-980) under high-pressure conditions (Fig. S3 in the ESI†). After completion of the reaction, the reactor was cooled and then depressurized carefully to atmospheric pressure. A HPLC-UV system on an ion exclusion column (Showa Denko K. K., KC-811; 0.02 mol L−1 phosphoric acid aqueous solution) was used to determine the FA concentration in a reaction solution. Conversion of FA was reported as the mean value of the residual FA concentration in the reaction solution and the volume of gas generated by FA decomposition. Analysis of the reaction solution was measured with an electrospray ionization mass spectrometer (ESI-MS) in a positive ion mode (Agilent Technologies, 6224 TOF LC/MS; methanol/water = 1/1 v/v).
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the financial support of Japan Science and Technology Agency (JST), CREST (No. JPMJCR1342), and the International Joint Research Program for Innovative Energy Technology of the Ministry of Economy, Trade, and Industry (METI) of Japan.References
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef PubMed.
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC.
- S. Niaz, T. Manzoor and A. H. Pandith, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 Search PubMed.
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 Search PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef PubMed.
- H. Kawanami, Y. Himeda and G. Laurenczy, in Advances in Inorganic Chemistry, ed. E. Rudi van and D. H. Colin, Academic Press, 2017, vol. 70, pp. 395–427 Search PubMed.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef PubMed.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef PubMed.
- D. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef PubMed.
- P. G. Jessop, in The Handbook of Homogeneous Hydrogenation, ed. J. G. d. Vries and C. J. Elsevier, Wiley-VCH Verlag GmbH, Weinheim, 2008, ch. 17, pp. 489–511, DOI:10.1002/9783527619382.ch17.
- A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef PubMed.
- A. Majewski, D. J. Morris, K. Kendall and M. Wills, ChemSusChem, 2010, 3, 431–434 CrossRef PubMed.
- M. Czaun, J. Kothandaraman, A. Goeppert, B. Yang, S. Greenberg, R. B. May, G. A. Olah and G. K. S. Prakash, ACS Catal., 2016, 6, 7475–7484 CrossRef.
- I. Mellone, F. Bertini, M. Peruzzini and L. Gonsalvi, Catal. Sci. Technol., 2016, 6, 6504–6512 Search PubMed.
- L. Piola, J. A. Fernández-Salas, F. Nahra, A. Poater, L. Cavallo and S. P. Nolan, Mol. Catal., 2017, 440, 184–189 CrossRef.
- I. Yuranov, N. Autissier, K. Sordakis, A. F. Dalebrook, M. Grasemann, V. Orava, P. Cendula, L. Gubler and G. Laurenczy, ACS Sustainable Chem. Eng., 2018, 6, 6635–6643 CrossRef.
- T. Tingelöf, L. Hedström, N. Holmström, P. Alvfors and G. Lindbergh, Int. J. Hydrogen Energy, 2008, 33, 2064–2072 CrossRef.
- N. Zamel and X. Li, Prog. Energy Combust. Sci., 2011, 37, 292–329 CrossRef.
- M. A. Díaz, A. Iranzo, F. Rosa, F. Isorna, E. López and J. P. Bolivar, Energy, 2015, 90, 299–309 CrossRef.
- M. Voldsund, K. Jordal and R. Anantharaman, Int. J. Hydrogen Energy, 2016, 41, 4969–4992 CrossRef.
- M. Felderhoff, C. Weidenthaler, R. von Helmolt and U. Eberle, Phys. Chem. Chem. Phys., 2007, 9, 2643–2653 RSC.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef PubMed.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef PubMed.
- Z. Wang, S.-M. Lu, J. Li, J. Wang and C. Li, Chem.–Eur. J., 2015, 21, 12592–12595 CrossRef PubMed.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef PubMed.
- C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem.–Eur. J., 2009, 15, 3752–3760 CrossRef PubMed.
- M. Czaun, A. Goeppert, J. Kothandaraman, R. B. May, R. Haiges, G. K. S. Prakash and G. A. Olah, ACS Catal., 2014, 4, 311–320 CrossRef.
- G. Papp, G. Olveti, H. Horvath, A. Katho and F. Joo, Dalton Trans., 2016, 45, 14516–14519 RSC.
- C. Guan, D.-D. Zhang, Y. Pan, M. Iguchi, M. J. Ajitha, J. Hu, H. Li, C. Yao, M.-H. Huang, S. Min, J. Zheng, Y. Himeda, H. Kawanami and K.-W. Huang, Inorg. Chem., 2017, 56, 438–445 CrossRef PubMed.
- H. Zhong, M. Iguchi, F.-Z. Song, M. Chatterjee, T. Ishizaka, I. Nagao, Q. Xu and H. Kawanami, Sustainable Energy Fuels, 2017, 1, 1049–1055 Search PubMed.
- M. Iguchi, Y. Himeda, Y. Manaka, K. Matsuoka and H. Kawanami, ChemCatChem, 2016, 8, 886–890 CrossRef.
- M. Iguchi, Y. Himeda, Y. Manaka and H. Kawanami, ChemSusChem, 2016, 9, 2749–2753 CrossRef PubMed.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem.–Eur. J., 2017, 23, 17017–17021 CrossRef PubMed.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem.–Eur. J., 2017, 23, 17788–17793 CrossRef PubMed.
- P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172–1176 CrossRef PubMed.
- A. Boddien, B. Loges, H. Junge, F. Gärtner, J. R. Noyes and M. Beller, Adv. Synth. Catal., 2009, 351, 2517–2520 CrossRef.
- T. H. Oh, Energy, 2016, 112, 679–685 CrossRef.
- V. Henricks, I. Yuranov, N. Autissier and G. Laurenczy, Catalysts, 2017, 7, 348 CrossRef.
- N. Onishi, M. Z. Ertem, S. Xu, A. Tsurusaki, Y. Manaka, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2016, 6, 988–992 Search PubMed.
- K. Sordakis, M. Beller and G. Laurenczy, ChemCatChem, 2014, 6, 96–99 CrossRef.
- C. Fink, S. Katsyuba and G. Laurenczy, Phys. Chem. Chem. Phys., 2016, 18, 10764–10773 RSC.
- J. Alazemi and J. Andrews, Renewable Sustainable Energy Rev., 2015, 48, 483–499 CrossRef.
- C. Y. Tsang and W. B. Street, Chem. Eng. Sci., 1981, 36, 993–1000 CrossRef.
- O. Fandiño, J. P. M. Trusler and D. Vega-Maza, Int. J. Greenhouse Gas Control, 2015, 36, 78–92 CrossRef.
- R. Span and W. Wagner, J. Phys. Chem. Ref. Data, 1996, 25, 1509–1596 CrossRef.
- Y. Himeda, N. Onozawa-Komatsuzaki, S. Miyazawa, H. Sugihara, T. Hirose and K. Kasuga, Chem.–Eur. J., 2008, 14, 11076–11081 CrossRef PubMed.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef.
- S. Xu, N. Onishi, A. Tsurusaki, Y. Manaka, W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Eur. J. Inorg. Chem., 2015, 2015, 5591–5594 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00087e |
| This journal is © The Royal Society of Chemistry 2018 |