Bi-functional heterogeneous iron complexes for catalytic conversion of epoxides to cyclic carbonates and their application in the synthesis of polyurethane†
Received
12th January 2018
, Accepted 8th March 2018
First published on 23rd April 2018
Abstract
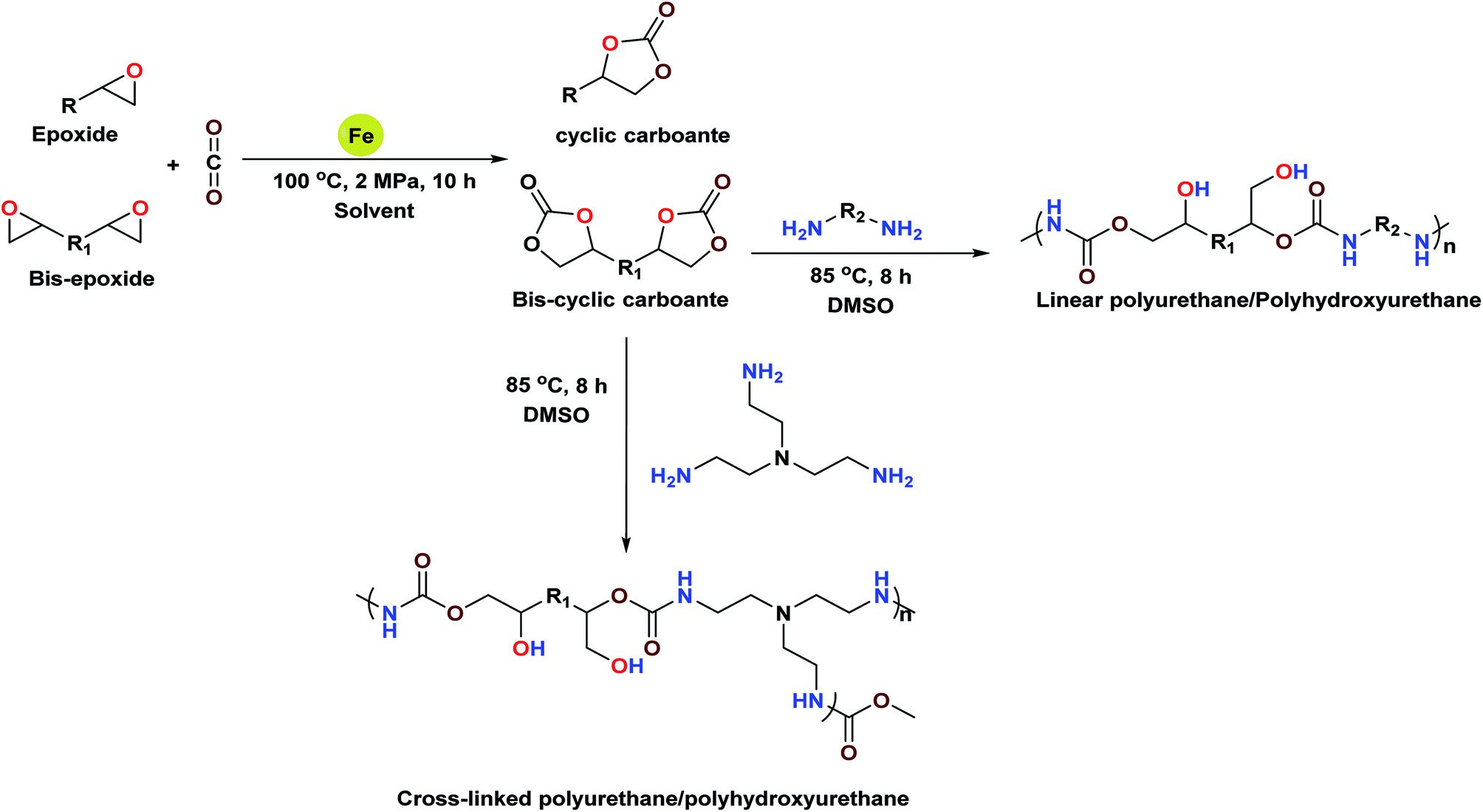
The development of a sustainable heterogeneous catalyst for direct conversion of CO2 to value-added chemicals is of great interest. Herein, we have developed polystyrene resin supported bi-functional Fe(III) complexes and studied their catalytic performance towards chemical fixation of CO2. The synthesized complex PFe-1 showed excellent catalytic activity towards aromatic/terminal aryloxy and aliphatic epoxides for achieving mono- and bis-cyclic carbonates in the absence of any co-catalysts. Higher conversion with excellent selectivity for cyclic carbonates was obtained with the catalyst PFe-1 with an added advantage of catalyst recyclability. This catalytic protocol was further extended for the synthesis of linear/cross polyhydroxyurethanes with good yield in a single step via addition polymerisation between bis-cyclic carbonate and different aliphatic diamines/triamines.
Introduction
The current climatic behaviour is undeniably associated with the increasing emission of anthropogenic carbon dioxide (CO2, >400 ppm) into the atmosphere, which raises several environmental and societal concerns.1 Carbon dioxide is the final product of every chemical combustion process;2–5 on the other hand, this ubiquitously available source can serve as a renewable C1 feedstock.6–8 Utilization of CO2 is a big challenge, presently receiving worldwide attention and a fascinating dream for the chemical community.8c Particularly, catalytic processes that can convert CO2 into chemicals have become a topic of significant interest. The sluggish reactivity of CO2 due to its thermodynamic stability can be surmounted by designing suitable catalytic systems to enhance the reactivity and sustainability profile.9,10 To date, various CO2 utilization processes have been developed. In line with the principle of green chemistry, cycloaddition of epoxide to CO2 is one of the 100% atom-economical approaches and frequently accompanied by wider applications in fine chemicals and pharmaceuticals.11–13 Although the consumption of CO2 during this process is very meagre, it eliminates the use of toxic reagents such as phosgene.14 In the past few decades, a series of homogeneous and heterogeneous catalytic systems have been investigated for the formation of cyclic carbonates.15
In this context, numerous catalysts including quaternary ammonium salts, organocatalysts, ionic liquids and various transition metal complexes were explored.16–20 Among the transition metal complexes, ironically, only a few attempts were made to use iron based catalysts for the cycloaddition of epoxide. Iron is an attractive metal due to its high availability, and non-toxic and environmentally benign nature. In this direction, William's group has developed a series of dinuclear iron complexes for the synthesis of cyclic and polycarbonates.21 Besides, an iron(II)-Schiff base complex was also used as a catalyst, and it gave good conversion of epoxides into the corresponding cyclic carbonates.22
Kleij et al. reported the catalytic activity of an aminotris(phenolates) iron(III) chelated complex for the coupling of CO2 and epoxides; the catalyst gave cyclic carbonates with higher conversion and selectivity in the presence of TBAB as the co-catalyst.23 However, due to the higher solubility of these catalysts in the reaction medium, separation and recycling of the catalysts is not that easy. Therefore, heterogenization of homogeneous systems is highly desirable and makes the process economically viable even in the absence of a co-catalyst. The excellent catalytic activity, thermal stability and recyclability are the necessary pre-requisites for a catalyst to scale-up reactions during process development. Therefore, with these considerations, we set out to develop heterogeneous bi-functional monometallic iron(III) complexes immobilized on polystyrene resin (Scheme 2) and explored their use for the formation of different aliphatic and aromatic substituted cyclic carbonates under moderate catalytic reaction conditions. Cyclic carbonates with higher conversion up to 97% and excellent selectivity were obtained under co-catalyst free conditions.
The catalyst can be recycled four times with retention of its activity and selectivity. This catalytic protocol worked successfully for the synthesis of linear/cross polyhydroxyurethanes in a single step (Scheme 1).
 |
| | Scheme 1 Catalytic transformation of CO2 into valuable cyclic carbonates and different polyurethanes. | |
Results and discussion
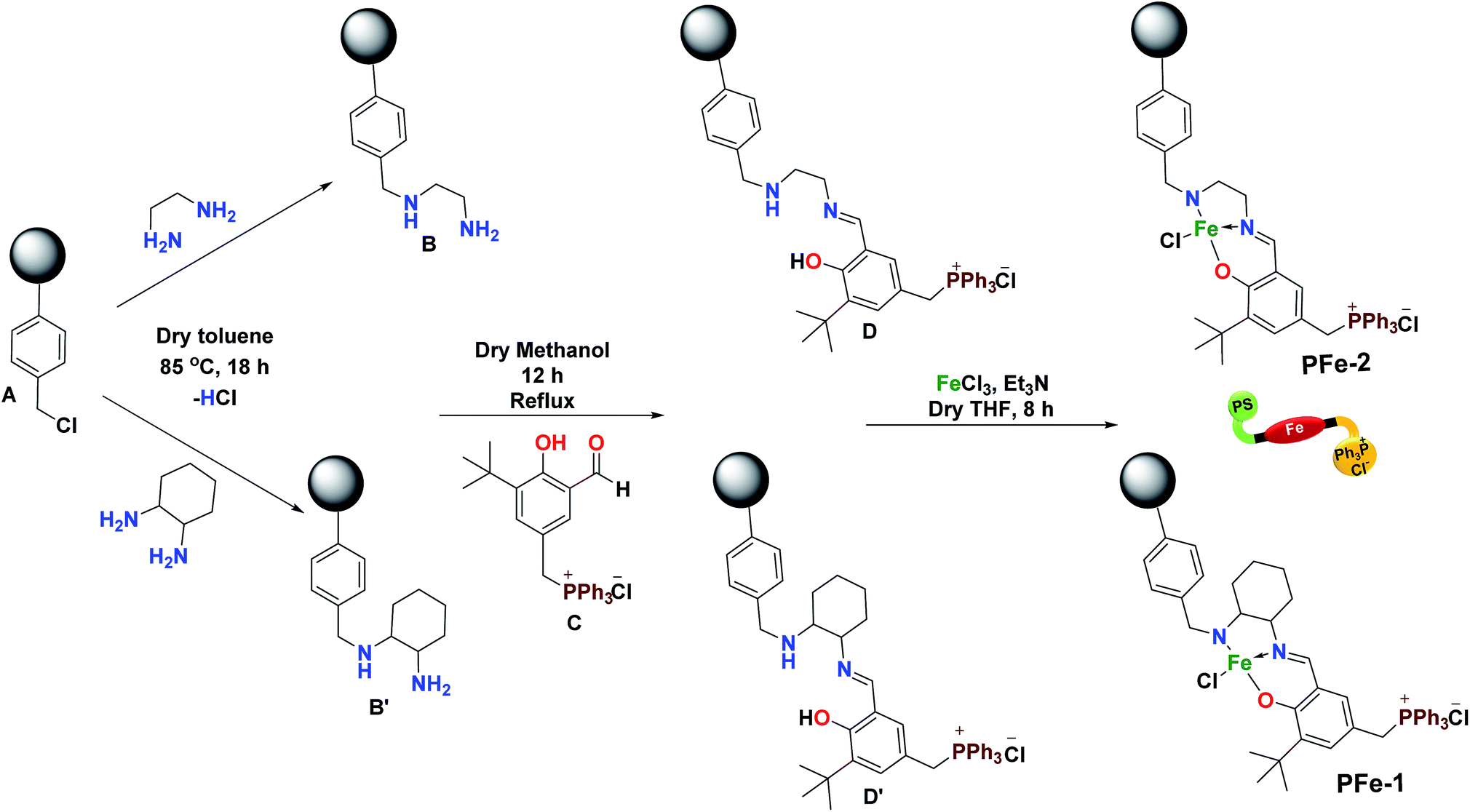
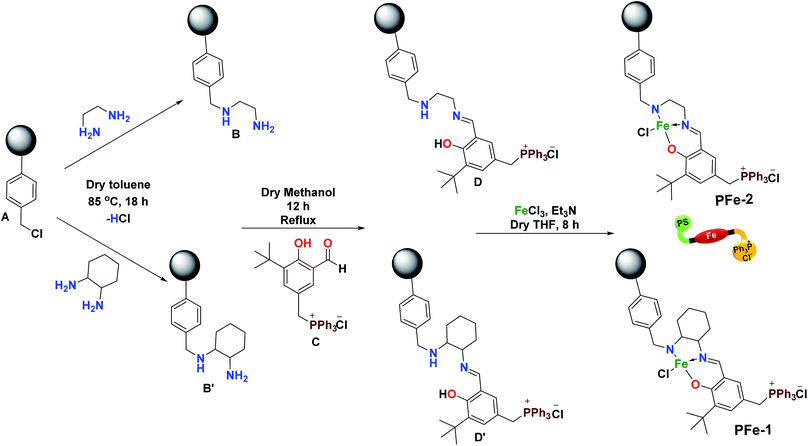
In general, several catalysts were explored for mediating the cycloaddition reaction of epoxide to CO2 but most of them require a nucleophilic co-catalyst that can help in improving the conversion of cyclic carbonates.24 In this investigation, our prime efforts were aimed at developing a highly sustainable bi-functional catalyst, which can simultaneously activate both the epoxide and CO2, without the need for any external co-catalyst. Accordingly, polymeric resin supported catalysts PFe-1 and PFe-2 were synthesized in a stepwise manner. Initially, the chloromethyl polystyrene resin A was treated with diamines (ethylene diamine and cyclohexane-1,2-diamine) in dry toluene at 85 °C to give polystyrene-supported amines (B and B′). The supported amines were further treated with aldehyde C in dry methanol, yielding tridentate ligands D and D′. Subsequently, complexation of ligands D and D′ with FeCl3 in the presence of triethylamine gave complexes PFe-1 and PFe-2, respectively (Scheme 2). Characterization of the precursor ligands D and D′ as well as the complexes PFe-1 and PFe-2 was accomplished by thermogravimetric analysis, FTIR, SEM and EDX.
 |
| | Scheme 2 Synthesis of bifunctional heterogeneous catalysts PFe-1 & PFe-2. | |
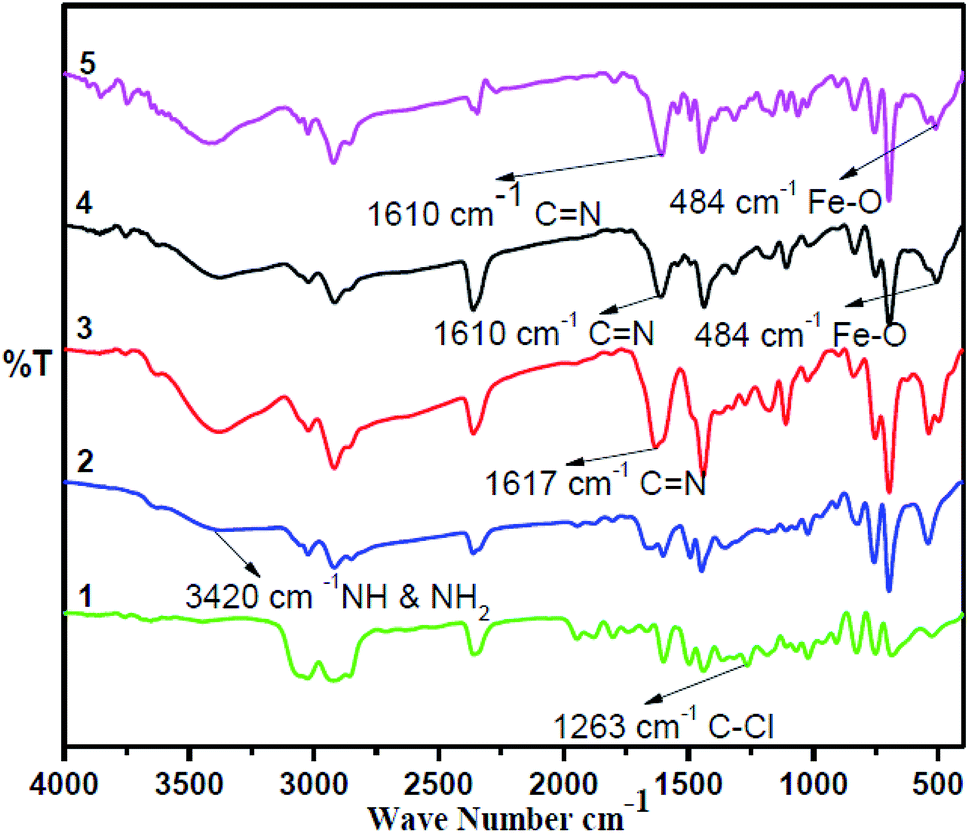
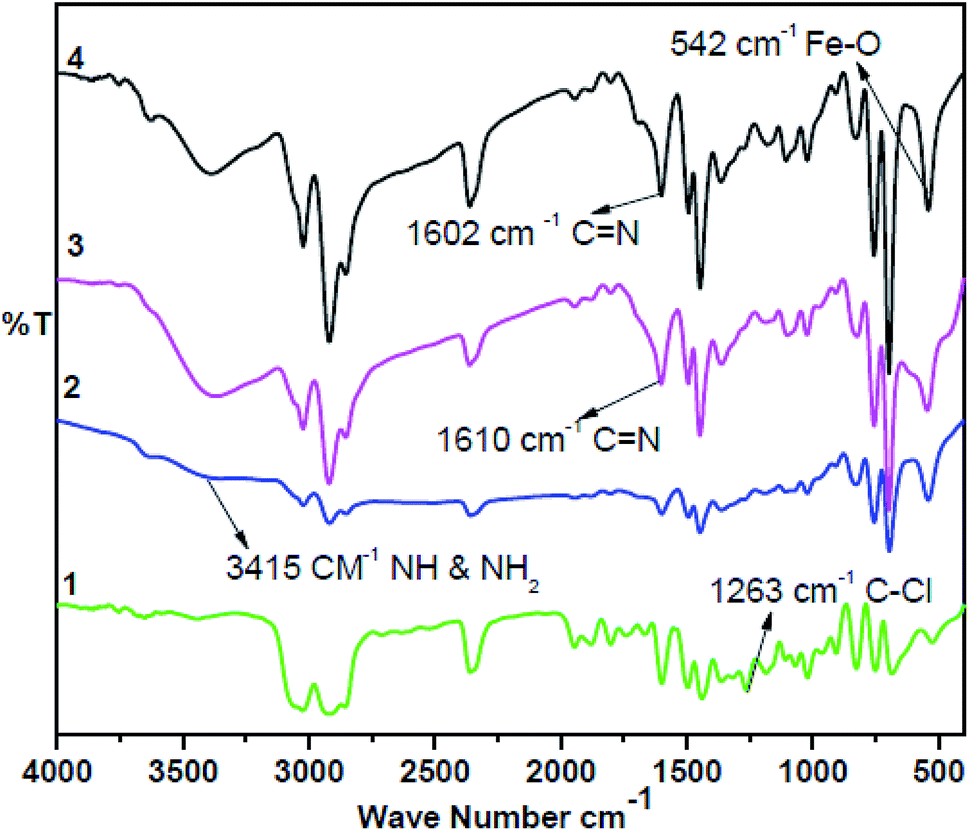
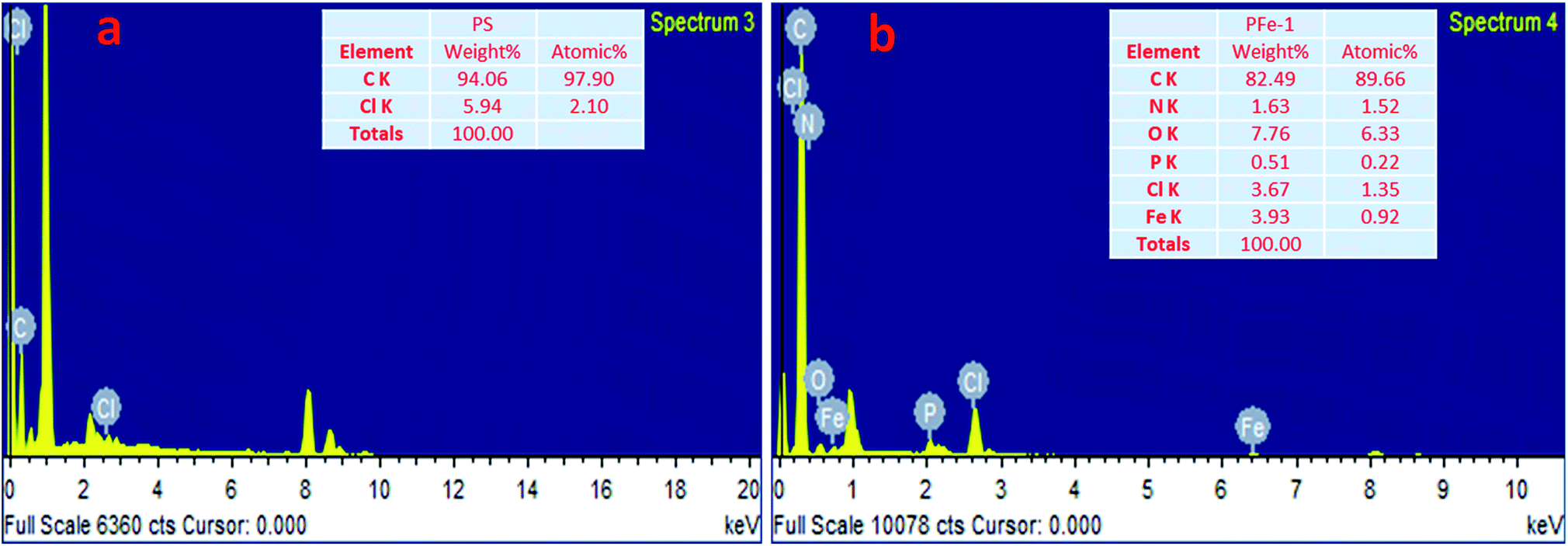
The synthesis of the catalysts was monitored by analyzing the samples after each step by FT-IR spectroscopy (Fig. 1 & 2). In the FT-IR spectrum, an amine functionalized polystyrene gave a broad peak at 3375 cm−1, which was assigned to NH and NH2 groups of the polystyrene resin supported amines B and B′, which confirms the replacement of Cl present in A with an amine group.24 On further reaction of B or B′ with C, there was a merging of bands due to NH, NH2 and OH groups, which gave a new band at 3386 cm−1 with an additional band of the azomethine group at 1617 cm−1. This confirms the formation of ligands D and D′.25 After complexation the shift in the azomethine band to a lower wavenumber (1610 cm−1) and the decrease in the intensity of the band corresponding to merged OH and amine functionalities confirm the formation of the metal complex. Also, the presence of the Fe–O stretching vibration at 536 cm−1 further confirms the metalation of D and D′ (Fig. 1 & 2). A morphological study was carried out by examining the field-emission scanning electron microscopy (FESEM) images of A and PFe-1 & PFe-2 catalysts, which clearly show that a morphological change has occurred on the surface of A after loading of iron metal on ligands D and D′ (Fig. 3a–c). Energy dispersive X-ray spectroscopy (EDX) investigation of PFe-1 & PFe-2 also confirmed the formation of immobilised iron(III) Schiff base complexes with the ligands D and D′ (Fig. 4). The iron content in the catalysts PFe-1 and PFe-2 was determined by ICP-EOS; it was found to be 9.11% (1.60 mmol g−1) and 8.55% (1.53 mmol g−1), respectively.26b The thermal stability of the samples after each step was also investigated by thermogravimetric analysis (TGA). The analysis shows that the chloromethyl polystyrene resin A was stable up to 260 °C, and beyond this range 0.88 mg (10.40%) weight loss was observed due to the removal of the C–Cl bond (Fig. 5A). The diamine supported moieties (B & B′) initially showed negligible weight loss due to the physically adsorbed solvent molecules while decomposition of the diamines with 2.35 (79.78%) & 3.65 mg (79.71%) weight loss was observed at 220 & 240 °C. The supported ligands (D & D′) also showed negligible weight loss due to solvent molecules while decomposition of ligands occurred with 0.70 mg (21.66%) and 0.62 mg (19.11%) weight loss at 170 and 140 °C, respectively. The complexes (PFe-1 & PFe-2) were stable up to 220 and 200 °C, respectively. However, negligible weight loss was also observed below 200 °C due to the solvent molecules present in the complexes.
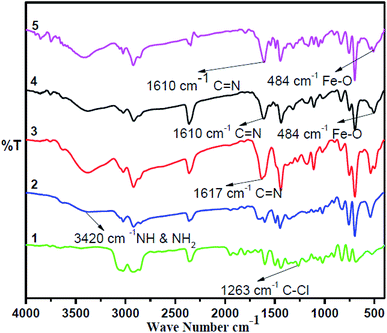 |
| | Fig. 1 FTIR spectra of the chloromethyl polystyrene resin (1), polystyrene-supported amine (2), supported Schiff base (3), supported Fe(III) Schiff base complex (PFe-1) (4) and recycled supported Fe(III) Schiff base complex (PFe-1) (5). | |
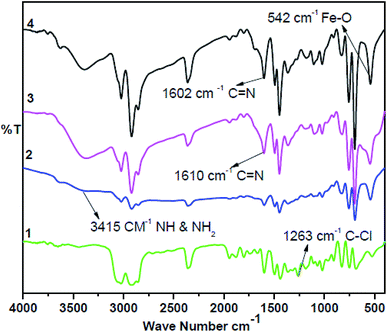 |
| | Fig. 2 FTIR spectra of the chloromethyl polystyrene resin (1), polystyrene-supported amine (2), supported Schiff base (3) and supported Fe(III) Schiff base complex (PFe-2) (4). | |
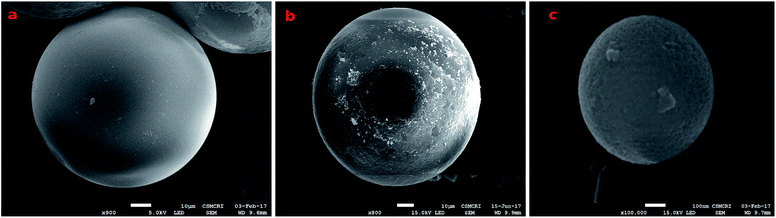 |
| | Fig. 3 FESEM image of the chloromethyl polystyrene resin (a) at 10 μm, PFe-1 (b) at 10 μm and PFe-2 (c) at 100 nm. | |
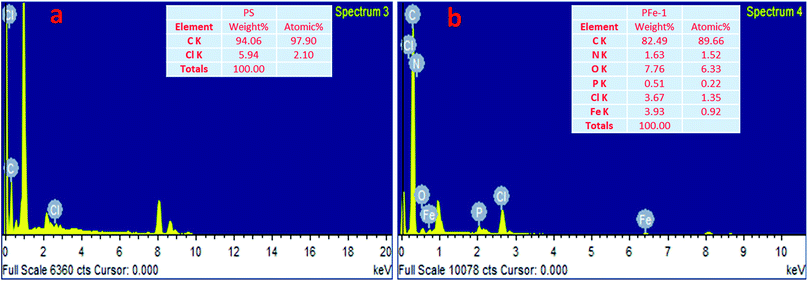 |
| | Fig. 4 EDX analysis of the chloromethyl polystyrene resin (a) and supported Fe(III) Schiff base complexes PFe-1 (b) and PFe-2 (present in the ESI†). | |
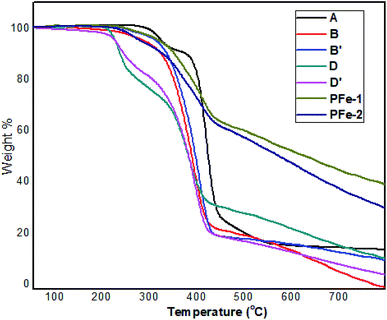 |
| | Fig. 5 The TG curves of the chloromethyl polystyrene resin (A), supported diamines (B and B′), supported ligands (D and D′) and supported iron(III) Schiff base complexes PFe-1 & PFe-2. | |
The well-characterized polystyrene supported catalysts PFe-1 & PFe-2 were first screened for their performance in the cycloaddition reaction of CO2 with phenyl glycidyl ether 1a as the model substrate. In order to establish the optimization of reaction conditions we begin with a cycloaddition reaction of CO2 to phenyl glycidyl ether 1a in the absence of any catalyst, where no product formation was observed (Table 1, entry 1).
Table 1 Optimisation of reaction conditions for the formation of cyclic carbonate using phenyl glycidyl ether 1a as the model substratea
|

|
| Entry |
Substrate |
Catalyst mg(mmol) |
Solvent |
Conversionb (%) |
|
Reaction conditions: substrate, phenyl glycidyl ether (1.5 g, 10 mmol); catalysts, PFe-1 & PFe-2.
Conversion was obtained by 1H/13C NMR.
|
| 1 |
1a
|
— |
Toluene |
— |
| 2 |
1a
|
25(0.04), PFe-1 |
Toluene |
35 |
| 3 |
1a
|
27(0.04), PFe-2 |
Toluene |
27 |
| 4 |
1a
|
50(0.08), PFe-1 |
Toluene |
53 |
| 5 |
1a
|
75(0.12), PFe-1 |
Toluene |
87 |
| 6 |
1a
|
100(0.16), PFe-1 |
Toluene |
96 |
| 7 |
1a
|
125(0.20), PFe-1 |
Toluene |
97 |
| 8 |
1a
|
100(0.16), PFe-1 |
DCM |
93 |
| 9 |
1a
|
100(0.16), PFe-1 |
THF |
89 |
| 10 |
1a
|
100(0.16), PFe-1 |
EtAc |
85 |
| 11 |
1a
|
100(0.16), PFe-1 |
Dioxane |
91 |
In contrast, in the presence of PFe-1 & PFe-2 (25 and 27 mg, 0.04 mmol of iron) as a catalyst, the conversion of phenyl glycidyl carbonate 1a′ was 35 and 27%, respectively (Table 1, entries 2 & 3). This shows that the presence of a catalyst is necessary for the cycloaddition reaction. Among the two catalysts used, PFe-1 performed better in terms of conversion of cyclic carbonate (Table 1, entry 2) and was used for further optimization of the reaction conditions. To improve the conversion of the product, the catalyst amount was varied from 50 mg (0.08 mmol) to 100 mg (0.16 mmol) where a substantial rise in the conversion of phenyl glycidyl carbonate 1a′ (53–96%) was observed (Table 1, entries 4–6). On further, increasing the amount of catalyst to 125 mg (0.2 mmol), the conversion of the product remained unchanged (Table 1, entry 7). Hence, a catalyst loading of 100 mg (0.16 mmol) was found to be optimum. It is well known that the solvent plays a crucial role in catalytic reactions. Therefore, several solvents, namely, toluene, dichloromethane, tetrahydrofuran, ethylacetate and dioxane, were screened (Table 1, entries 6 and 8–11), and the best result in terms of the conversion to the product was obtained for the cycloaddition reaction in toluene (entry 7).
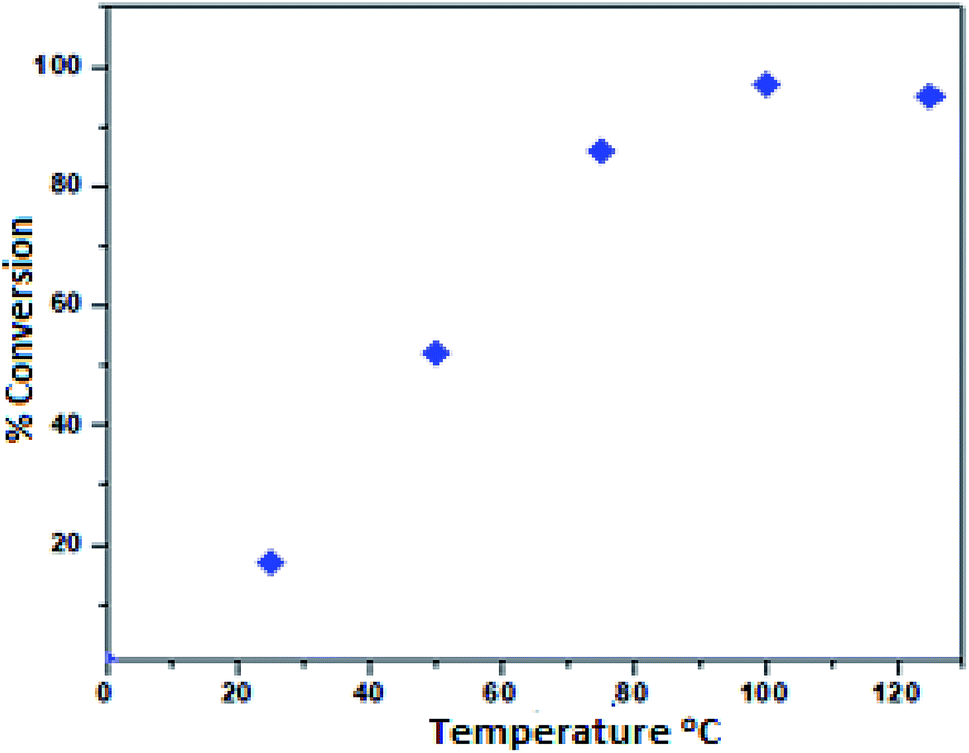
To investigate the effect of temperature on the formation of the phenyl glycidyl carbonate product 1a′, we varied the temperature from 25 to 125 °C using toluene as the solvent (Fig. 6). A high conversion (97%) of phenyl glycidyl carbonate was achieved at 100 °C. Furthermore, increasing the temperature from 100 to 125 °C showed a detrimental effect, possibly due to the formation of a small amount of polymerized PGC as a by-product at a higher temperature.27
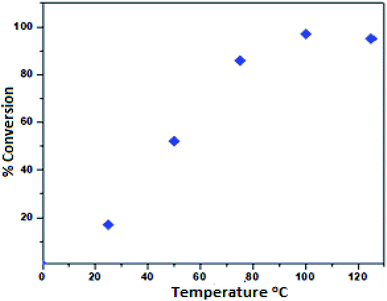 |
| | Fig. 6 Dependence of the phenyl glycidyl carbonate 1a′ conversion on temperature. Reaction conditions: phenyl glycidyl ether 1a (10 mmol), catalyst PFe-1: 100 mg (0.16 mmol), CO2 pressure (2 MPa), time 10 h. | |
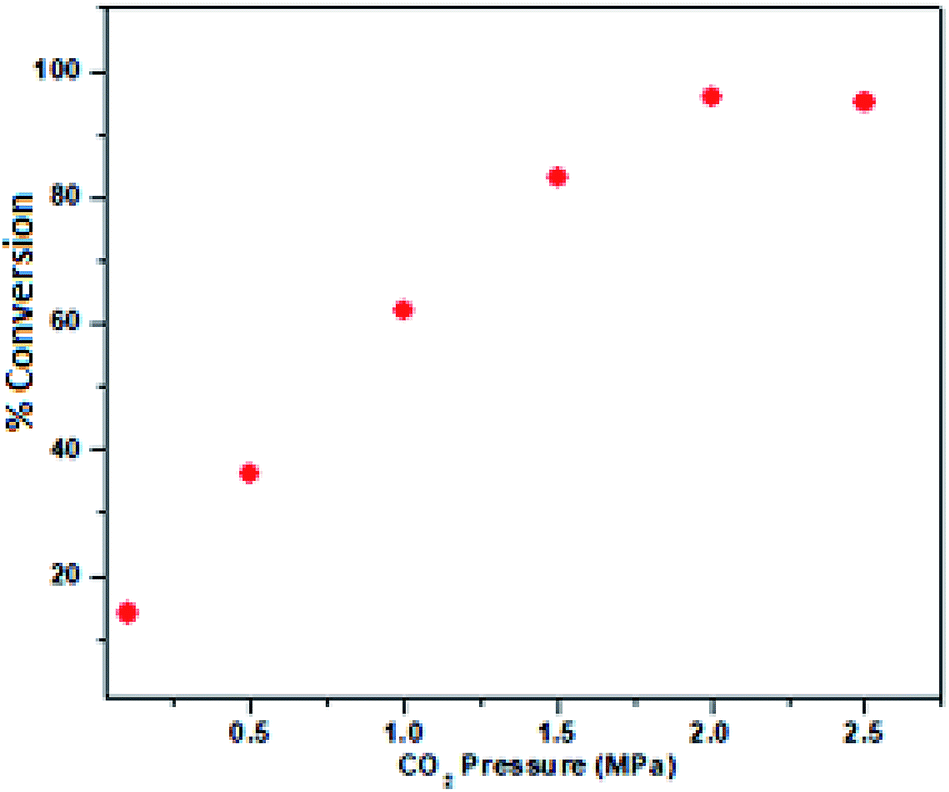
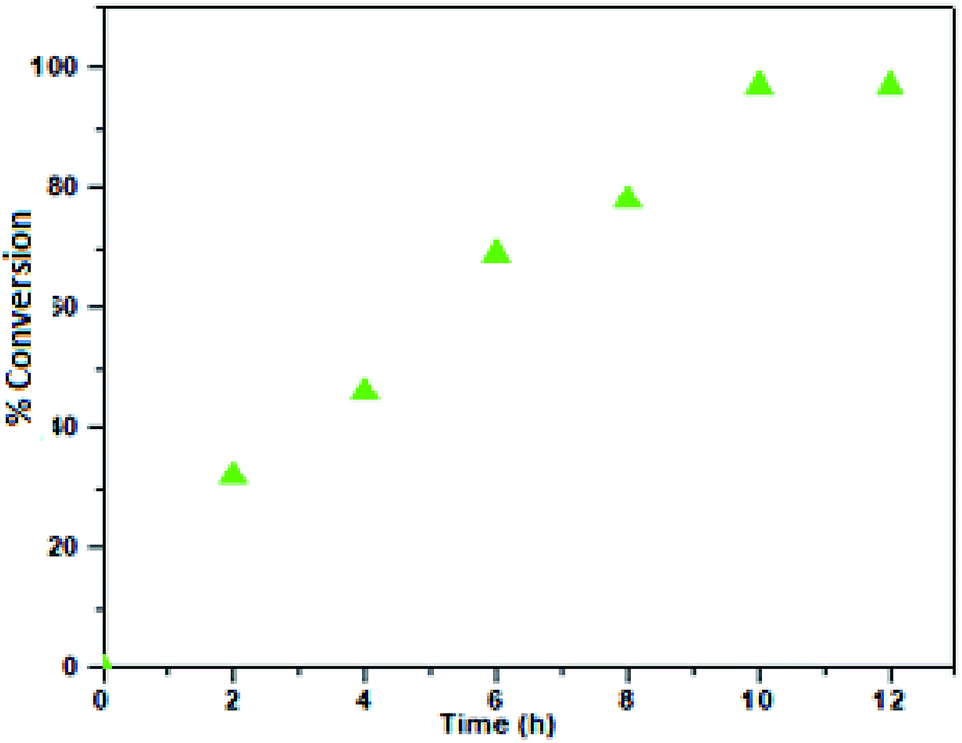
Therefore, 100 °C was found to be optimum for the synthesis of cyclic carbonate (Fig. 6). Subsequently, we also studied the effect of CO2 pressure on the cycloaddition reaction at 100 °C. As shown in Fig. 7, the CO2 pressure in the cycloaddition reaction had a great influence on the conversion of cyclic carbonate (1a′). Initially, the conversion of 1a′ was increased with an increase in pressure from 0.1 to 2.5 MPa, and a maximum conversion could be achieved at 2.0 MPa. However, a slight decrease in the conversion of 1a′ was observed upon further increase from 2.0 to 2.5 MPa. This kind of phenomenon has also been observed in other catalytic systems.27 The dependence of cyclic carbonate formation on reaction time was also investigated. As shown in Fig. 8, the reaction proceeded smoothly in the range of 2–10 h with the conversion of 1a′ up to 96% within 10 h. Furthermore, prolonging the reaction time had no significant effect on the conversion. Therefore, a reaction time of 10 h was considered as the optimum reaction time for the cycloaddition reaction.
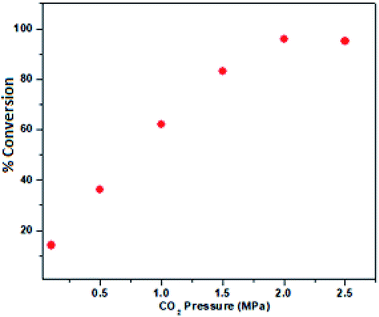 |
| | Fig. 7 Dependence of the phenyl glycidyl carbonate 1a′ conversion on the pressure of CO2. Reaction conditions: phenyl glycidyl ether 1a (10 mmol), catalyst PFe-1: 100 mg (0.16 mmol), time 10 h. | |
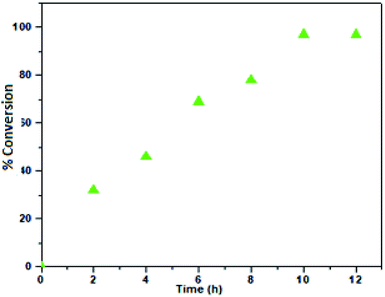 |
| | Fig. 8 Dependence of the phenyl glycidyl carbonate 1a′ conversion on time. Reaction conditions: phenyl glycidyl ether 1a (10 mmol), catalyst PFe-1: 100 mg (0.16 mmol), CO2 pressure 2 (MPa), temperature 100 °C. | |
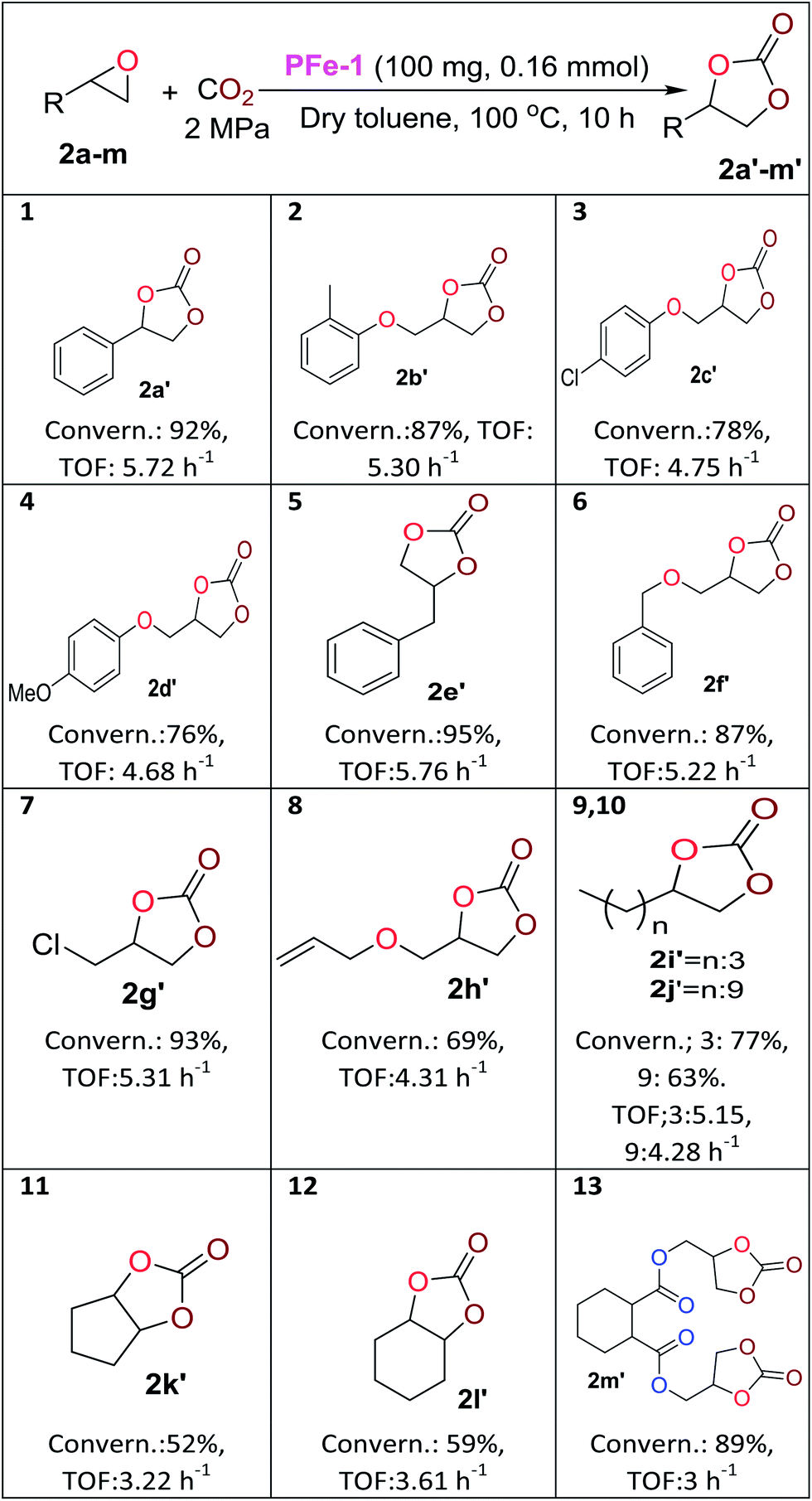
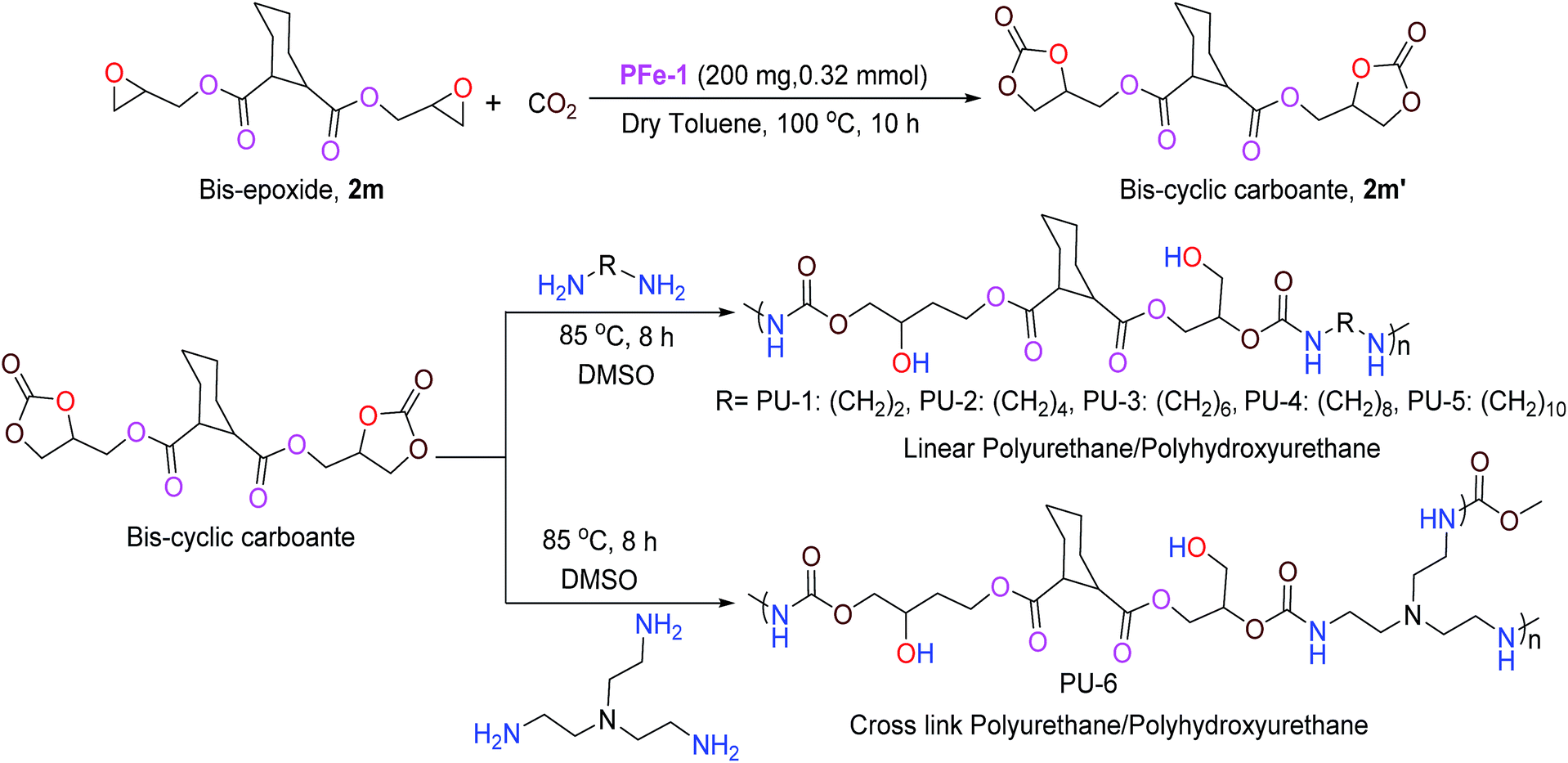
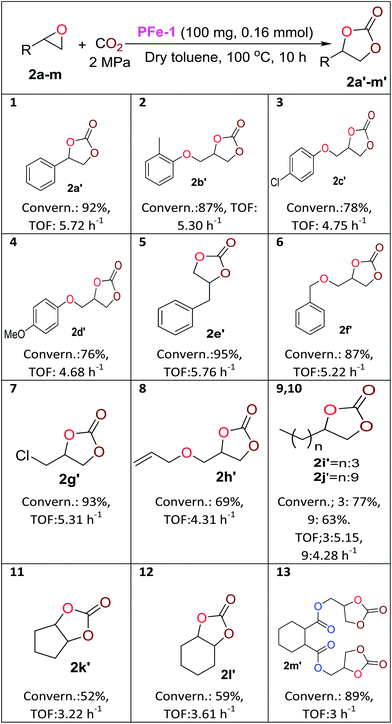
With the optimized reaction conditions in hand, we further investigated the cycloaddition reaction for a range of commercially available terminal and non-terminal epoxides and the results are summarized in Table 2. The iron(III) Schiff base complex PFe-1 worked efficiently for aromatic terminal/aryloxy epoxides with both electron-withdrawing and donating substituents resulting in the conversion (76–95%) of the corresponding cyclic carbonate with high selectivity (Table 2, entries 1–6). In case of aliphatic terminal epoxides, epichlorohydrin showed better results (93%, entry 7) compared to other aliphatic substrates with longer alkyl chain lengths (63–77%) (Table 2, entries 8–9). Besides that, internal epoxides were also explored for the cycloaddition reaction using PFe-1 as the catalyst. A relatively lower conversion (52–59%) was obtained in the case of cyclic epoxides, viz., cyclopentene oxide and cyclohexene oxide (Table 2, entries 11–12), which may be due to the ring steric effect. Bis-epoxide (diglycidyl 1,2-cyclohexanedicarboxylate 2m) was also investigated for the cycloaddition reaction with CO2 in the presence of PFe-1 as the catalyst and the bis-cyclic carbonate (diglycidyl carbonate 1,2-cyclohexanedicarboxylate 2m′) product was obtained with good conversion (89%) (entry 13). Bis-cyclic carbonates are important precursors for the synthesis of non-isocyanate polyurethanes.
Table 2 Cycloaddition reaction of aromatic/terminal aryloxy and aliphatic terminal/meso epoxides with CO2a
|
Reaction conditions: substrates (10 mmol); catalyst PFe-1; the conversion was determined by 1H/13C NMR.
|
|
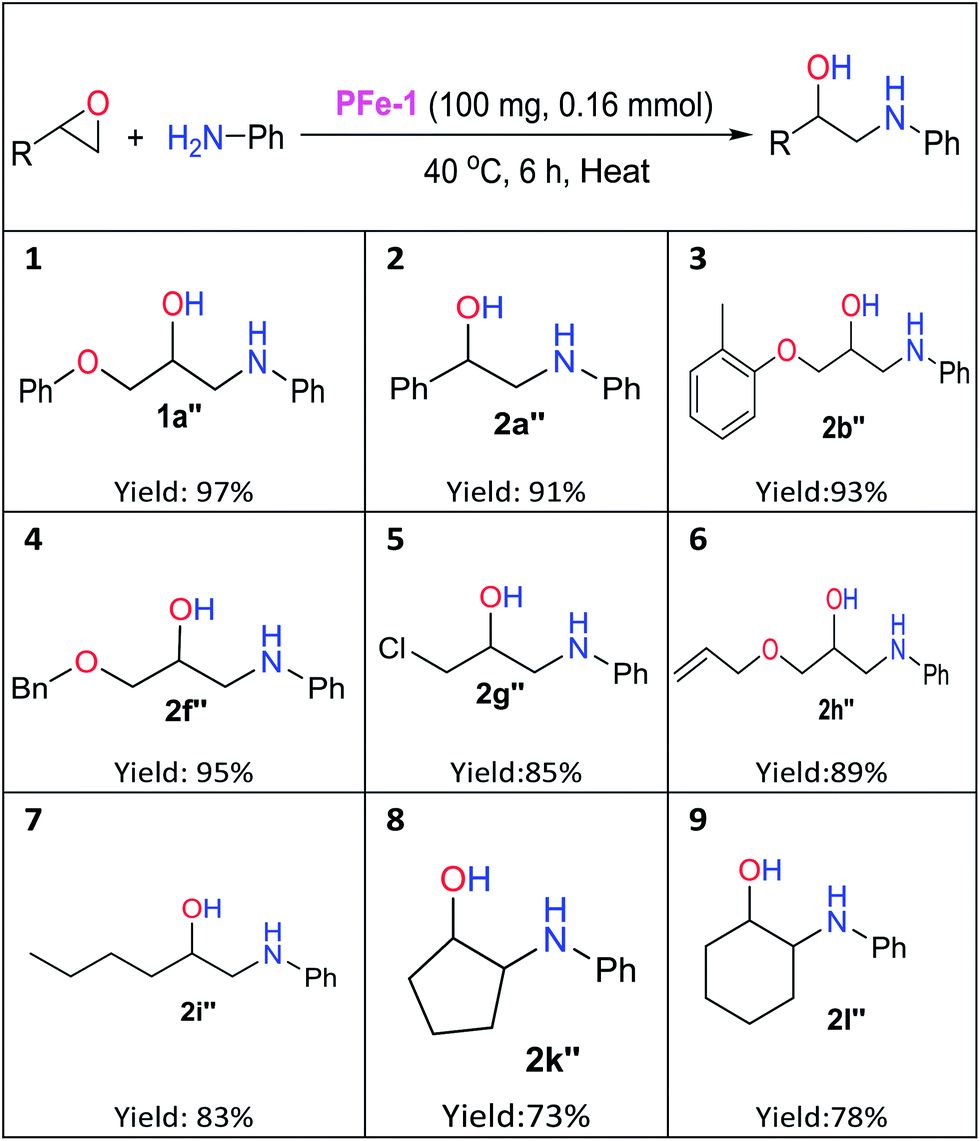
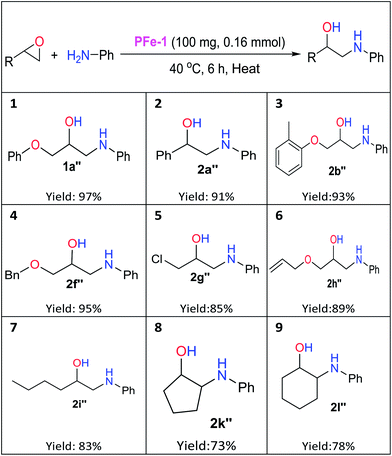
The catalytic activity of the polystyrene resin iron(III) Schiff base complex PFe-1 towards the cycloaddition reaction has encouraged us to expand the scope of PFe-1 in aminolysis of various epoxides viz., glycidyl phenyl ether 1a, styrene oxide 2a, glycidyl 2-methylphenyl ether 2b, (2,3-epoxypropyl)benzene 2e, benzyl glycidyl ether 2f, epichlorohydrin 2g, allyl glycidyl ether 2h, cyclopentane oxide 2j and cyclohexane oxide 2k to form their respective β-aminoalcohols (well-known building blocks for pharmaceuticals) under solvent-free conditions. As depicted in Table 3, PFe-1 gave high yields (91–97%, Table 3, entries 1–4) for β-amino alcohols (1a′′, 2a′′, 2b′′, and 2f′′) derived from the aromatic epoxides with aniline in the presence of 0.16 mmol of catalyst PFe-1 under solvent-free conditions. In the case of aliphatic epoxides, 83–89% yields (Table 3, entries 5–7) of the corresponding β-amino alcohols (2g′′, 2h′′, and 2i′′) were observed. On the other hand, aminolysis of cyclopentane/hexane oxides resulted in moderate yields (73–78%) of the corresponding β-amino alcohols (2k′′ and 2l′′) (Table 3, entries 8 and 9), which may be due to their steric hindrance and affects the substrate–catalyst interactions.
Table 3 Ring-opening reaction of terminal/meso epoxides with anilines in the presence of iron(III) Schiff base complex PFe-1a
|
Reaction conditions: substrates, epoxides 1a and 2a–l/amines (10 mmol); catalyst PFe-1. Isolated yield was characterised by 1H/13C NMR.
|
|
Mechanism for PFe-1 catalyzed cycloaddition of CO2 with various epoxides
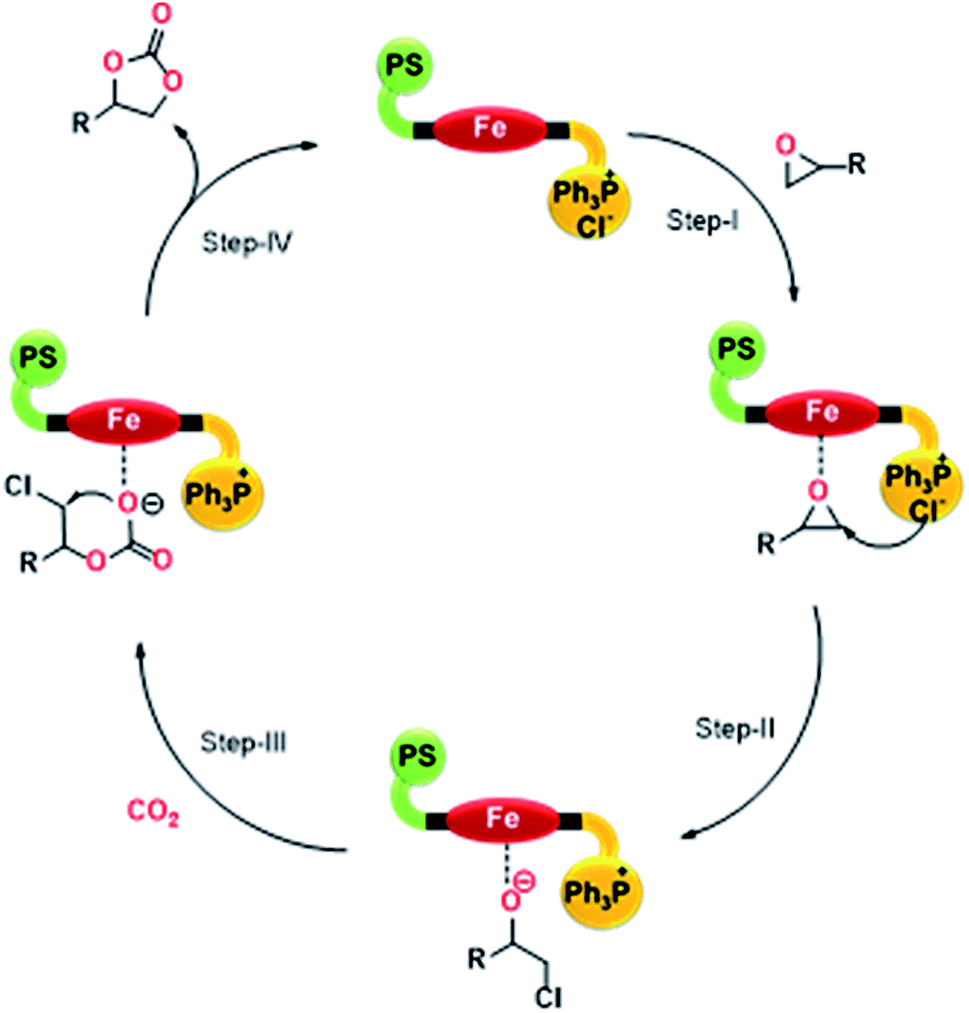
Based on the results achieved for the cycloaddition of CO2 with epoxides, a probable mechanism (Scheme 3) is proposed for the bi-functional PFe-1 catalyst, which possesses both Lewis acid and base centres. First, the epoxide was activated by the Lewis acid site Fe[III] through the interaction of Fe and the oxygen atom of the epoxide (Step I). Then, the Cl− ion from the phosphonium centre attacked the less-hindered i.e. terminal carbon atom of the activated epoxide and opened up the ring to form an active oxygen anion (Step II). Subsequently, CO2 was inserted between the oxygen anion and the metal (Fe) to generate an alkylcarbonate anion (Step III). Finally, ring-closure occurred by the alkylcarbonate anion attack on the terminal carbon (C–Cl) to produce the corresponding cyclic carbonates and the catalyst PFe-1 was regenerated (Step IV). Furthermore, it should be realized that the catalyst itself activates both the epoxide and CO2 without the need for any external nucleophilic co-catalysts.
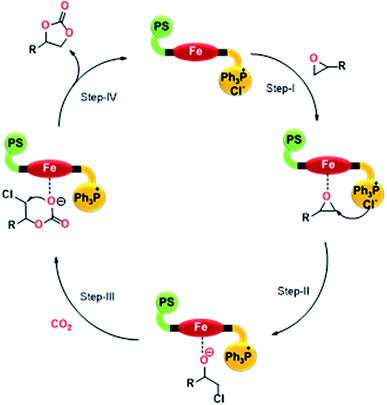 |
| | Scheme 3 The possible reaction mechanism for the cycloaddition of CO2 with epoxides catalyzed by PFe-1. | |
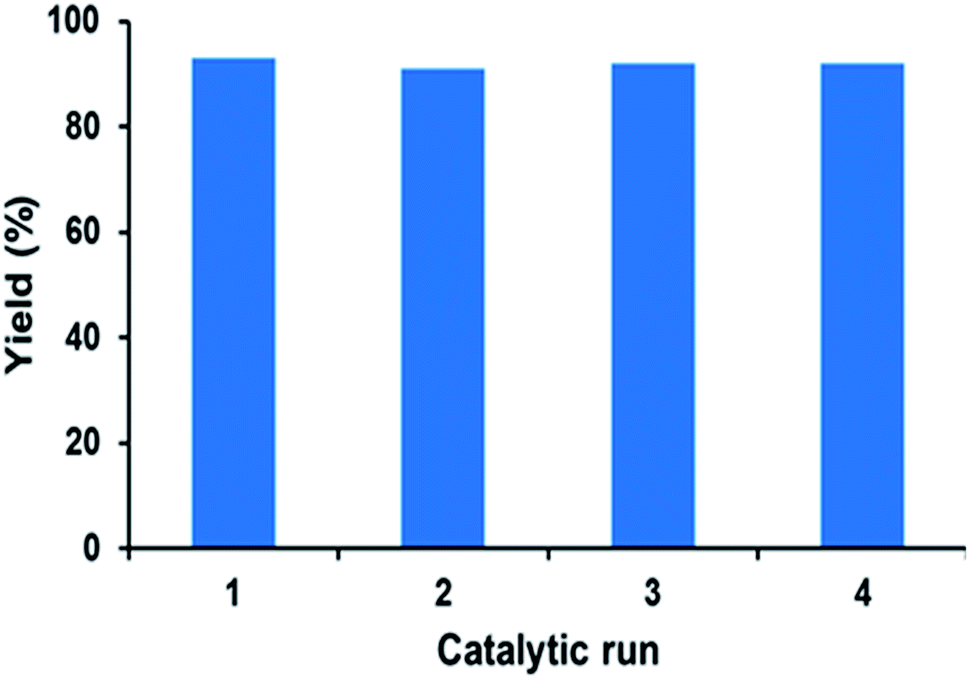
The reusability of catalyst PFe-1 was studied under the optimized reaction conditions on a 10 mmol scale using styrene oxide 2a as the representative substrate (Fig. 9). After the catalytic run, the catalyst was filtered, washed with toluene and dried in a vacuum for use in subsequent catalytic runs. The recovered catalyst worked well for the cycloaddition reaction of CO2 in the same manner as the fresh catalyst. Fig. 9 suggests that the recovered catalyst is stable and worked well for four cycles with retention of its catalytic activity, although a slight decrease in conversion was observed in the fifth cycle. The reused PFe-1 catalyst (after four cycles) was recovered and characterized by FT-IR (Fig. 1) and ICP. Although the FT-IR spectra of the fresh and recovered PFe-1 catalyst were found to be similar, indicating that there was no structural change in the reused catalyst, ICP analysis of the crude reaction mass after removal of the catalyst revealed there was a very small amount of leaching of the metal (Fe) (<1 ppm) from the PFe-1 catalyst, which seems to be responsible for hampering the catalytic activity for further re-use.
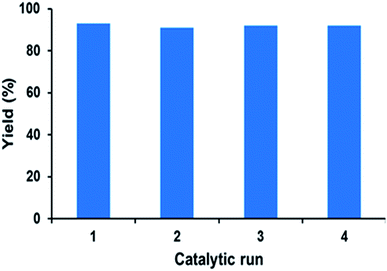 |
| | Fig. 9 The recyclability study of catalyst PFe-1 for the formation of cyclic carbonate from styrene oxide and CO2. | |
Application of the product bis-cyclic carbonate (2m′) in the synthesis of linear/cross polyhydroxyurethanes
Among the various classes of polymers, polyurethanes/polyhydroxyurethanes (PUs) occupy the sixth position with an annual worldwide production of up to 12 million tons.26a Conventionally, PUs are produced from moisture sensitive polyols and isocyanates, which are hazardous to human health and the environment. Hence, much attention has been devoted in the past to the development of a non-isocyanate route for the synthesis of polyurethanes. Generally, linear polyurethanes are obtained via addition copolymerization between bis-cyclic carbonate with aliphatic/aromatic diamines in the absence of any catalysts under aerobic conditions. In this work, a series of linear and cross-linked polyurethanes/polyhydroxyurethanes were synthesized in good yield via the reaction of bis(cyclic carbonate) 2m′ (Table 2, entry 13) with five different diamines viz., 1,2-diaminoethane, 1,4-diaminobutane, 1,6-diaminohexane, 1,8-diaminooctane, 1,10-diaminedecane and tris(2-aminoethyl)amine. The linear/cross-linked polyurethanes were characterized by various physico-chemical techniques such as 1H/13C NMR, FT-IR, GPC and MALDI TOF.26b
Experimental section
Method and materials
Chloromethyl polystyrene resin (CMPSR), ethylene diamine, cyclohexane 1,2-diamine and the epoxides viz., phenyl glycidyl ether 1a, styrene oxide 2a, glycidyl 2-methylphenyl ether 2b, 4-chlorophenyl glycidyl ether 2c, 4-methoxyphenyl glycidyl ether 2d, benzyl glycidyl ether 2e, benzyl glycidyl ether 2f, epichlorohydrin 2g, allyl glycidyl ether 2h, 1,2-epoxyhexane 2i, 1,2-epoxydodecane 2j, cyclopentane oxide 2k, cyclohexane oxide 2l and diglycidyl 1,2-cyclohexanedicarboxylate 2m were purchased from Aldrich and TCI Chemicals. Elemental analysis of the catalysts and products was done on a Perkin Elmer 2400 CHNS analyser. 1H and 13C NMR spectra were recorded on a Bruker 500/600 MHz spectrometer at ambient temperature using CDCl3 as the solvent and TMS as an internal standard. The FT-IR spectra were recorded on a Perkin-Elmer Spectrum GX spectrophotometer in a KBr/Nujol mull. High-resolution mass spectra were recorded using an LCMS (Q-TOFF) LC (Waters) and an MS (Micromass) instrument. The surface morphology of supported complexes PFe-1 and PFe-2 was scanned using a Field Emission Scanning Electron Microscope (FESEM; JEOL, JSM 7100F) after applying a thin sputter coating of conducting Au–Pd alloy. Flash chromatography was carried out for product purification, using silica gel (60–200 mesh size) purchased from SD Fine-Chemicals Limited, India. The purity of the solvents, diamines and epoxide was measured by gas chromatography (GC) on a Shimadzu GC 1020 instrument using a fused silica capillary column (30 m × 0.25 mm × 0.12 μm film thickness).
The synthesis of polymer supported N-diamines B and B′
Chloromethyl polystyrene resin A (1 g, 2.4 mmol) was added into a 250 mL round-bottom flask containing dry toluene (100 mL) and equipped with a magnetic bead under a nitrogen atmosphere and stirred for 1 h. Then, 1,2-diamines (ethylene diamine 12 mmol, 0.6 g and cyclohexane 1,2-diamine 12 mmol, 1.37 g) were added to the solution, which was then heated at 85 °C for 18 h. The B and B′ polymer supported N-diamine particles were filtered, washed with toluene, MeOH, and MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O and then washed several times with aqueous 5% NaHCO3 and water for the removal of excess diamines. Further supported diamines were extracted overnight with acetone in a Soxhlet extractor and dried overnight in a vacuum (Scheme 2).24
:H2O and then washed several times with aqueous 5% NaHCO3 and water for the removal of excess diamines. Further supported diamines were extracted overnight with acetone in a Soxhlet extractor and dried overnight in a vacuum (Scheme 2).24
Preparation of polymer-supported Schiff base ligands D and D′
The cationic aldehyde C was prepared by our previously reported procedure.28 In the next step, the cationic aldehyde C (1.2 g, 2.4 mmol) was added into a 100 mL round-bottom flask containing dry methanol and equipped with a magnetic bead, and stirred for 30 minutes. The supported diamines B and B′ (1 g, 2.4 mmol) were then added to this solution and the resulting solution was allowed to reflux for 12 h. The yellow colour polymer supported Schiff base ligands D and D′ were obtained after filtration and washed with MeOH and acetone for the removal of excess cationic aldehyde C (Scheme 2). The solid residue obtained was dried overnight in a vacuum.
Preparation of polymer supported Fe(III) Schiff base complexes PFe-1 & PFe-2
The polymer supported ligands D and D′ (1 g, 2.4 mmol) were dispersed in 60 mL of dry tetrahydrofuran in a three-necked 100 mL round-bottom flask equipped with a magnetic bead. Then triethylamine (0.7 mL, 5 mmol) was added under a nitrogen atmosphere, and the reaction mixture was stirrer for 45 minutes at room temperature. Anhydrous FeCl3 (0.390 g, 2.4 mmol) dissolved in dry THF was added slowly into the reaction mixture, which was then refluxed for 15 h. The progress of the reaction was checked by TLC. On completion of the reaction, the resulting solution was cooled to room temperature, filtered, washed thoroughly with MeOH (3 × 30 mL) and acetone and finally dried in a vacuum for 24 h (Scheme 2). The solid immobilized residue thus obtained was repeatedly extracted with THF in a Soxhlet extractor until the washing solution became colourless. The characterization of the supported Fe(III) Schiff base complexes PFe-1 & PFe-2 was done by various physico-chemical techniques (Scheme 2).
Typical cycloaddition reaction of epoxides with CO2 in the presence of supported Fe(III) Schiff base complexes PFe-1 & PFe-2 as the catalyst
The cycloaddition reactions were performed in a 25 mL SS autoclave, which was charged with 8 mL of solvent, an appropriate epoxide (10 mmol, 1.5 g) and the catalyst (0.1 g, 0.16 mmol) and flushed and pressurized with CO2 to the required pressure. The reaction solution was then allowed to stir (900 rpm) and heated to the specified temperature for 10 h. The catalyst was separated via simple filtration, and the conversion and selectivity of cyclic carbonates were investigated by 1H NMR by taking an aliquot from the reaction mass at regular intervals. Purification of the product was done by flash column chromatography using a silica gel column (eluted with 20% hexane/ethyl acetate) and characterized by 1H/13C NMR, CHNS and FTIR (given in the ESI†).
General procedure for the synthesis of β-aminoalcohols in the presence catalyst PFe-1
The epoxides (10 mmol) and aniline (10 mmol) were introduced into a 10 mL reaction vial to which polystyrene resin supported Fe(III) Schiff base complex PFe-1 was added and the resulting mixture was allowed to stir with the help of a magnetic bar at 40 °C for 6 h. The reaction was monitored by TLC and after completion of the reaction the catalyst was separated by simple filtration. For the determination of the isolated yield of β-amino alcohols, flash column chromatography on a silica gel column was carried out. The characterization of the product was accomplished by 1H/13C NMR, CHNS and FT-IR.
Addition polymerization of diglycidyl carbonate 1,2-cyclohexanedicarboxylate with diamines for the formation of polyurethanes/polyhydroxyurethanes
To a dry test tube containing a magnetic stirring bar diglycidyl carbonate 1,2-cyclohexanedicarboxylate 2m′ (0.7 g, 1.90 mmol) and various diamines (1,2-diaminoethane: 0.110 g, 1.9 mmol; 1,4-diaminobutane: 0.170 g, 1.9 mmol, 1,6-diaminohexane: 0.220 g, 1.9 mmol, 1,8-diaminooctane: 0.270 g, 1.9 mmol, 1,10-diaminedecane: 0.330 g, 1.9 mmol and tris(2-aminoethyl)amine: 0.130 g, 0.85 mmol) were added. To the resulting solution, 2 mL of DMSO was added and the resulting solution was allowed to stir at 85 °C for 8 h under aerobic conditions. The monitoring of the reaction was done by TLC until completion, and the resulting mixture was then poured into ether giving ether-insoluble additives. It was then dissolved in DMSO and precipitated with water. The precipitate was filtered and dried under vacuum to give the corresponding polyhydroxyurethane (Scheme 4). The characterization of the polymeric products was accomplished by 1H/13C NMR, GPC and MALDI-MS.29
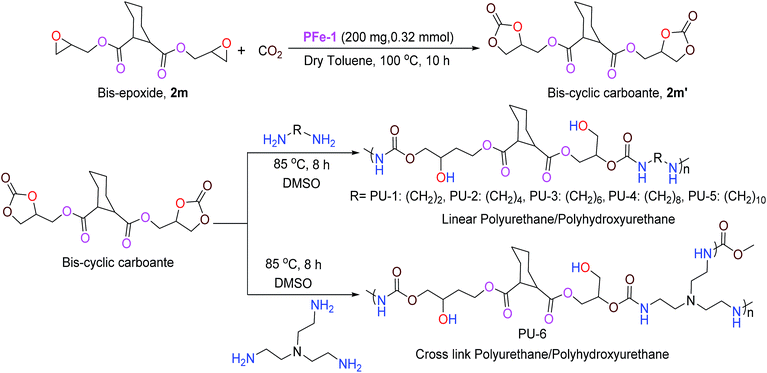 |
| | Scheme 4 Formation of bis-cyclic carbonate and linear/cross-linked polyhydroxyurethanes. | |
Conclusions
Monometallic bi-functional heterogeneous Fe(III) Schiff base complexes (PFe-1 & PFe-2) were synthesized and characterized using various physico-chemical technique and used for the cycloaddition reaction of epoxide with CO2 in the absence of any co-catalyst under moderate conditions. PFe-1 exhibited excellent catalytic activity with a wide range of terminal/non-terminal epoxides and yielded good to high conversion (up to 97%) with an excellent selectivity of cyclic carbonates. The supported Fe(III) catalyst was recycled four times with retention of its activity and selectivity. This catalytic protocol was further used for the formation of linear/cross-linked polyurethanes/polyhydroxyurethanes with good yield in a single step.
Characterization data of the products15–20
4-Phenyl-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 4.33 (1H, t, J = 8 Hz), 4.80 (1H, t, J = 8.4 Hz), 5.68 (1H; t, J = 8 Hz), 7.35–7.37 (2H, m), 7.41–7.45 (3H, m) and 13C NMR (125 MHz, CDCl3): 71.09, 77.92, 125.80, 129.11, 129.63, 135.70, 154.81. Elemental analysis for C9H8O3: C 65.85, H 4.91. Found C 65.92, H 5.12, ESI-MS (M + Na)+: m/z 186.38.
4-(Phenoxymethyl)-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 4.14 (1H, dd, J = 3.5 Hz, 3.5 Hz), 4.21 (1H, dd, J = 4 Hz, 4 Hz), 4.52 (1H, q), 4.60 (1H, t, J = 8.5 Hz), 5.02 (1H, m), 6.91 (2H, d, J = 8 Hz), 7.01 (1H, t, J = 7.5 Hz), 7.30 (2H, t, J = 8.5 Hz) and 13C NMR (125 MHz, CDCl3): 66.14, 66.79, 74.14, 114.53, 121.88, 129.61, 154.70, 157.69. Elemental analysis for C10H10O4: C 61.85, H 5.19. Found C 61.97, H 5.06, ESI-MS (M + H)−: m/z 195.00.
4-((4-Chlorophenoxy)methyl)-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 4.12 (1H, dd, J = 3.5 Hz, 3.5 Hz), 4.21 (1H, dd, J = 4 Hz, 3.5 Hz), 4.52 (1H, q), 4.62 (1H, t, J = 8.5 Hz), 5.02 (1H, m), 6.85 (2H, d, J = 9 Hz), 7.25 (2H, d, J = 8.5 Hz) and 13C NMR (125 MHz, CDCl3): 66.05, 67.23, 73.93, 115.90, 126.98, 129.98, 154.51, 156.32. Elemental analysis for C10H9ClO4: C 52.53, H 3.97. Found C 65.92, H 5.12, ESI-MS (M + K + H2O)+: m/z 283.31.
4-((o-Tolyloxy)methyl)-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 2.22 (3H, s), 3.70–3.74 (1H, m), 376–380 (1H, m), 4.02 (2H, d, J = 5 Hz), 4.61 (1H, t, J = 8 Hz), 4.22 (1H, m), 6.79 (1H, d, J = 3 Hz), 6.88 (1H, t, J = 9 Hz), 7.14 (1H, t, J = 8.4 Hz) and 13C NMR (125 MHz, CDCl3): 16.16, 68.88, 69.29, 72.74, 111.10, 120.80, 126.64, 126.83, 130.69, 155.45. Elemental analysis for C9H8O3: C 65.85, H 4.91. Found C 65.92, H 5.12, ESI-MS (M + Na+ + H2O)+: m/z 263.11.
4-((Benzyloxy)methyl)-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 3.58 (1H, dd, J = 3.5 Hz, 3.5 Hz), 3.68 (1H, dd, J = 4 Hz, 4 Hz), 4.36 (1H, q), 4.45 (1H, t, J = 8 Hz), 4.89 (1H, m), 4.56 (2H, q), 4.80 (1H, m), 7.20–7.31 (5H, m) and 13C NMR (125 MHz, CDCl3): δ 66.31, 68.87, 73.67, 75.10, 127.76, 128.59, 137.15, 155.05. Elemental analysis for C9H8O3: C 65.85, H 4.91. Found C 65.92, H 5.12, ESI-MS (M + Na)+: m/z 131.39.
4-Decyl-1,3-dioxolan-2-one
1H NMR (500 MHz, CDCl3): δ 0.88 (3H, s), 1.27 (16H, m), 1.68 (1H, m), 1.78 (1H, m), 4.07 (1H, t, J = 7.4 Hz), 4.53 (1H, t, J = 7.8 Hz), 4.71 (1H, m) and 13C NMR (125 MHz, CDCl3): 14.06, 22.63, 24.34, 29.12, 29.25, 29.32, 29.42, 29.50, 29.65, 31.84, 33.82, 69.41, 155.13. Elemental analysis for C13H24O3: C 68.38, H 10.59. Found C 68.87, H 10.79, ESI-MS (M + H2O)+: m/z 246.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The CSMCRI communication number is 127/2017. The authors are thankful for the CSD HSP 009 project for financial assistance. Shailesh Verma would like to thank the Council of Scientific & Industrial Research (CSIR) for a SRF. Shailesh Verma is also thankful to the Academy of Scientific and Innovative Research (AcSIR) for enrolment into the PhD program and the “Analytical Discipline and Centralized Instrumental Facilities” for providing instrumentation facilities.
Notes and references
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- T. M. Lenton, Clim. Change, 2006, 76, 7–29 CrossRef CAS.
- P. P. Pescarmona and M. Taherimehra, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- N. V. D. Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 Search PubMed.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
-
(a) T. Sakakura and K. Kohnoa, Chem. Commun., 2009, 1312–1330 RSC;
(b)
P. Styring, D. Jansen, H. de Coninck, H. Reith and K. Armstrong, Carbon Capture and Utilisation in the Green Economy, CO2 Chem Media and Publishing, UK, 2011, ISBN: 978-0-9572588-1-5 Search PubMed;
(c) F. Chen, D. Chen, L. Shi, N. Liu and B. Dai, J. CO2 Util., 2016, 16, 391–398 CrossRef CAS.
-
(a) M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS;
(b) M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
-
(a) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC;
(b) K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876–1880 RSC;
(c) B. H. Xu, J. Q. Wang, J. Sun, Y. Huang, J. P. Zhang, X. P. Zhang and S. J. Zhang, Green Chem., 2015, 17, 108–122 RSC.
-
(a) K. M. K. Yu, I. Curcic, J. Gabriel and S. C. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed;
(b)
Carbon Dioxide as a Chemical Feedstock, ed. M. Aresta, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed;
(c) P. Styring and K. Armstrong, Chim. Oggi, 2011, 1, 34–37 Search PubMed.
-
(a) A. L. Marshall and P. J. Alaimo, Chem.–Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed;
(b) P. N. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–105029 CrossRef PubMed;
(c) P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2012, 2, 1487–1499 CrossRef CAS;
(d) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
-
(a) A. A. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed;
(b) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS;
(c) J. P. Parrish, R. N. Salvatore and K. W. Jung, Tetrahedron, 2000, 56, 8207–8237 CrossRef CAS;
(d) Q. Sun, Y. Jin, B. Aguila, X. Meng, S. Ma and F. S. Xiao, ChemSusChem, 2017, 10, 1160–1165 CrossRef CAS PubMed.
- J. Bayardon, J. Holz, B. Schaeffner, V. Andrushko, S. Verevkin, A. Preetz and A. Boerner, Angew. Chem., Int. Ed., 2007, 46, 5971–5974 CrossRef CAS PubMed.
-
(a) C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed;
(b) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed;
(c) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed;
(d) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
-
(a) R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef CAS PubMed;
(b) S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC;
(c) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
-
(a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC;
(b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
-
(a) G. Rokicki, W. Kuran and B. Pogorzelska-Marciniak, Monatsh. Chem., 1984, 115, 205–214 CrossRef CAS;
(b) J. W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705–6709 CrossRef CAS PubMed;
(c) K. Kossev, N. Koseva and K. Troev, J. Mol. Catal. A: Chem., 2003, 194, 29–37 CrossRef CAS;
(d) F. Li, C. Xia, L. Xu, W. Sun and G. Chen, Chem. Commun., 2003, 2042–2043 RSC.
-
(a) W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS;
(b) W. L. Wong, K. C. Cheung, P. H. Chan, Z. Y. Zhou, K. H. Lee and K. Y. Wong, Chem. Commun., 2007, 2175–2177 RSC;
(c) J. Wang, J. Wu and N. Tang, Inorg. Chem. Commun., 2007, 10, 1493–1495 CrossRef CAS;
(d) X. B. Lu, Y. J. Zhang, K. Jin, L. M. Luo and H. Wang, J. Catal., 2004, 227, 537–541 CrossRef CAS.
-
(a) R. A. Shiels and C. W. Jones, J. Mol. Catal. A: Chem., 2007, 261, 160–166 CrossRef CAS;
(b) K. K. Yu, I. Curcic, J. Gabriel, H. Morganstewart and S. C. Tsang, J. Phys. Chem. A, 2009, 114, 3863–3872 CrossRef PubMed.
- X. Zhang, Y. B. Jia, X. B. Lu, B. Li, H. Wang and L. C. Sun, Tetrahedron Lett., 2008, 49, 6589–6592 CrossRef CAS.
-
(a) S. Zhang, Y. Chen, F. Li, X. Lu, W. Dai and R. Mori, Catal. Today, 2006, 115, 61–69 CrossRef CAS;
(b) J. Sun, S. I. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS;
(c) J. Peng and Y. Deng, New J. Chem., 2001, 1, 639–641 RSC;
(d) H. S. Kim, J. J. Kim, H. Kim and H. G. Jang, J. Catal., 2003, 220, 44–46 CrossRef CAS.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- J. E. Dengler, M. W. Lehenmeier, S. Klaus, C. E. Anderson, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2011, 1, 336–343 CrossRef.
- C. J. Whiteoak, B. Gjoka, E. Martin, M. M. Belmonte, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef CAS PubMed.
-
(a) P. C. Perumgani, S. Keesara, S. Parvathaneni and M. R. Mandapati, New J. Chem., 2016, 40, 5113–5120 RSC;
(b) M. Bakherad, A. Keivanloo, B. Bahramian and T. A. Kamali, J. Braz. Chem. Soc., 2009, 20, 907–912 CrossRef CAS.
-
(a) T. P. Devi and R. K. H. Singh, Rasayan J. Chem., 2010, 3, 266–270 CAS;
(b) A. R. Patil, K. J. Donde, S. S. Raut, V. R. Patil and R. S. Lokhande, J. Chem. Pharm. Res., 2012, 4, 1413–1425 CAS.
-
(a) J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnama and A. R. Yuvarajc, RSC Adv., 2016, 6, 114453–114482 RSC;
(b) S. M. Islam, S. Mondal, P. Mondal, A. S. Roy, K. Tuhina and M. Mobarok, Inorg. Chem. Commun., 2011, 14, 1352–1357 CrossRef CAS.
- W. Chen, L.-X. Zhong, X.-W. Peng, R.-C. Sun and F.-C. Lu, ACS Sustainable Chem. Eng., 2015, 3, 147–152 CrossRef CAS.
- R. I. Kureshy, K. J. Prathap, T. Roy, N. C. Maity, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, Adv. Synth. Catal., 2010, 352, 3053–3060 CrossRef CAS.
-
(a) L. Zhang, X. Luo, Y. Qin and Y. Li, RSC Adv., 2017, 7, 37–46 RSC;
(b) S. Verma, G. Kumar, A. Ansari, R. I. Kureshy and N. H. Khan, Sustainable Energy Fuels, 2017, 1, 1620–1629 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00016f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Noor-ul H.
Khan
ab
*ab and
Noor-ul H.
Khan
ab