
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile chemoenzymatic synthesis of a novel stable mimic of NAD+†‡
Zhefu
Dai§
a,
Xiao-Nan
Zhang§
a,
Fariborz
Nasertorabi
b,
Qinqin
Cheng
a,
Hua
Pei
c,
Stan G.
Louie
c,
Raymond C.
Stevens
*b and
Yong
Zhang
 *adef
*adef
aDepartment of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, 1985 Zonal Ave, Los Angeles, CA 90089, USA. E-mail: yongz@usc.edu
bDepartments of Biological Sciences and Chemistry, Bridge Institute, Michelson Center for Convergent Bioscience, University of Southern California, Los Angeles, CA 90089, USA. E-mail: stevens@usc.edu
cTitus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, 1985 Zonal Ave, Los Angeles, CA 90089, USA
dDepartment of Chemistry, Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
eNorris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA 90089, USA
fResearch Center for Liver Diseases, University of Southern California, Los Angeles, CA 90089, USA
First published on 15th October 2018
Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor participating in a variety of important enzyme-catalyzed physiological and pathophysiological processes. Analogues of NAD+ provide key and valuable agents for investigating NAD+-dependent enzymes. In this study, we report the preparation of a novel stable NAD+ mimic, 4′-thioribose NAD+ (S-NAD+), using a facile and efficient chemoenzymatic approach. Substrate activity assays indicated the resulting S-NAD+ is chemically inert to human CD38 and sirtuin 2 enzymes, but capable of participating in redox reactions in a manner similar to NAD+. X-ray crystallographic analysis revealed binding of S-NAD+ to the active site of human CD38 and critical residues involved in leaving group activation and catalysis. By more closely mimicking NAD+ in geometry and electrostatics, the generated S-NAD+ offers a unique and important tool that can be extended to study enzymes utilizing NAD+.
Introduction
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor involved in numerous biological processes, such as energy metabolism and homeostasis, chromatin stability, gene expression, protein homeostasis, and signaling transduction.1–4 As a cosubstrate, NAD+ participates in chemical reactions catalyzed by various types of enzymes that are responsible for controlling those important processes (Fig. 1).2,3 NAD+ together with its reduced form NADH are utilized by oxidoreductases for electron transfer reactions. In the presence of NAD+, sirtuins catalyze protein deacetylation, forming acetyl-ADP-ribose (acetyl-ADPR). In addition, NAD+-dependent enzymes covalently add the ADP-ribose moiety of NAD+ to side chains of target proteins, resulting in protein ADP-ribosylation. This is catalyzed by a superfamily of ADP-ribosyltransferases (ARTs), including intracellular poly-ADP-ribose polymerases (PARPs) and sirtuins and ecto-ARTs. The last category of NAD+-dependent enzymes includes CD38 and CD157 catalyzing formation of ADP-ribose (ADPR) and cyclic ADPR (cADPR) by consuming NAD+ (Fig. 1). As membrane glycoproteins, CD38 and CD157 are multifunctional enzymes. In addition to catalyzing formation of cADPR from NAD+, they possess NAD+ glycohydrolase and base exchange activities. Compared with CD157, CD38 exhibits significantly higher enzymatic activity and catalytic efficiency in cleaving circulating NAD+, suggesting a major role in controlling tissue and cellular NAD+ homeostasis.5,6 Recent studies indicated that CD38 deficiency in mouse leads to ameliorated pathology of Alzheimer's disease.7 Pharmacological inhibitions of CD38 can suppress glioma progression in mice, improve physiological and metabolic parameters, and provide beneficial effects on metabolic syndrome.8–11 CD38 was also shown to regulate immune responses in physiological and pathological conditions.12,13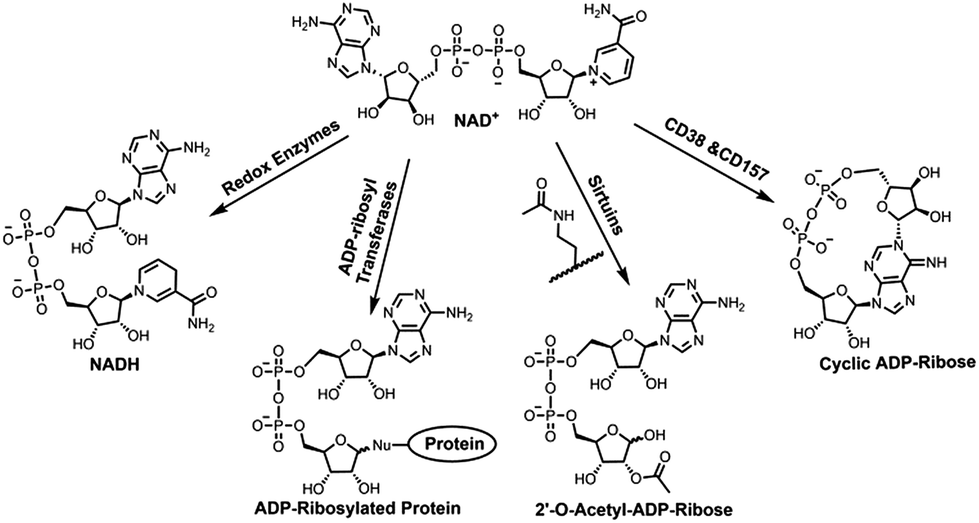 | ||
| Fig. 1 NAD+ participates in chemical reactions catalyzed by distinct classes of enzymes. | ||
Given the extensive involvements of NAD+-dependent enzymes in human diseases, a substantial amount of studies have been performed to determine their mechanisms of action. For this purpose, X-ray crystal structures of reactive complexes can provide direct and valuable information at the molecular levels.14 Since the complexes of NAD+-bound catalytically active enzymes are short-lived, catalytically inactive enzymes or modified substrate proteins were used for solving crystal structures of such binary or ternary enzyme complexes, which often miss important information about functions and roles of key residues involved in enzyme catalysis.15–17 Moreover, currently available NAD+ analogues for this purpose are suboptimal in mimicking NAD+ due to the lack of nicotinamide leaving group for redox reactions or the altered ribosyl geometry by methylene substitution.18–23 Herein we report chemoenzymatic synthesis of a novel stable NAD+ mimic, 4′-thioribose NAD+ (S-NAD+), which was generated through two-step enzymatic reactions in a high yield following chemical synthesis of nicotinamide 4′-thioriboside (S-NR). Biochemical studies indicated that the resulting S-NAD+ is resistant to enzymatic cleavage by human CD38 and sirtuin 2 enzymes, but still able to participate in redox reactions. The determined high-resolution X-ray crystal structure of S-NAD+ with human CD38 revealed molecular interactions between S-NAD+ and key catalytic residues. These results demonstrate a facile chemoenzymatic approach for the preparation of S-NAD+. As a novel stable NAD+ mimic, the generated S-NAD+ may offer a unique and important tool that can be extended to investigate mechanisms of action for enzymes utilizing NAD+.
Results and discussion
Considering the chemical structure of NAD+, we envisioned that replacing the endocyclic oxygen with sulfur can possibly generate a stable analogue more closely mimicking NAD+ in geometry and electrostatics. Furthermore, inspired by nicotinamide riboside (NR) kinase (NRK)- and nicotinamide mononucleotide adenylyltransferase (NMNAT)-mediated biosynthesis of NAD+ from its NR precursor, we hypothesized that S-NR may be enzymatically converted to S-NAD+ in high efficiency by human NRK and NMNAT enzymes (Fig. 2A). The human genome encodes two NRK isoforms (NRK1 and 2) and three NMNAT isoforms (NMNAT1-3). Previous studies indicated that human NRK1 and NMNAT1 exhibit adequate catalytic activities for NR+adenosine triphosphate (ATP) and nicotinamide mononucleotide (NMN)+ATP, respectively, and promiscuity towards other substrate analogues.24–26 In contrast to total chemical synthesis of NAD+ analogues which has proven challenging due to synthetic complexity and low yields for the difficult pyrophosphate coupling, using recombinant NRK and NMNAT may afford a facile and efficient approach for the generation of stable NAD+ mimics.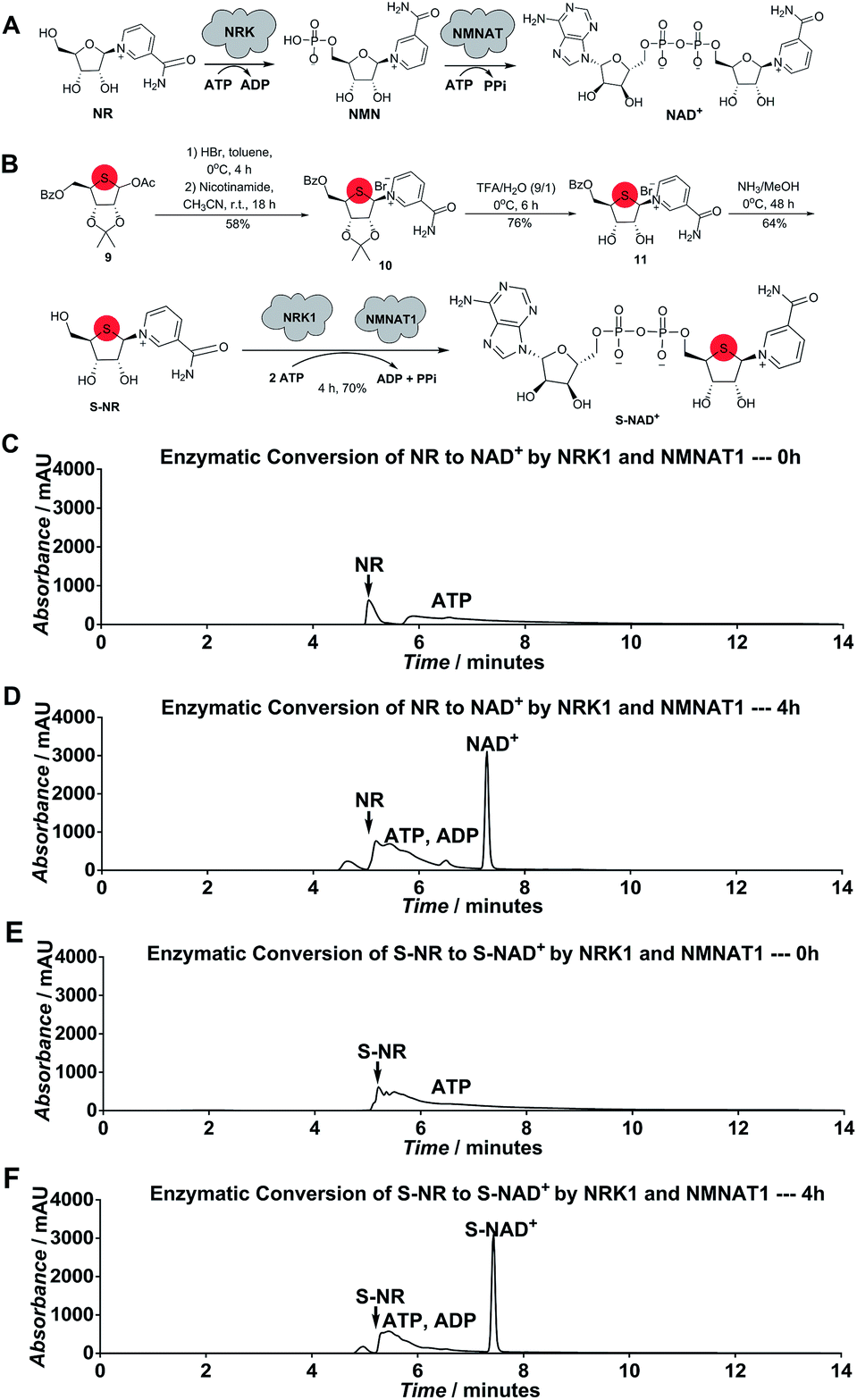 | ||
| Fig. 2 Enzymatic synthesis of NAD+ and chemoenzymatic synthesis of S-NAD+. (A) Scheme of enzymatic conversion of NR to NAD+. (B) Scheme of chemoenzymatic synthesis of S-NAD+. (C)–(F) HPLC analysis of enzymatic synthesis of NAD+ ((C) and (D)) and S-NAD+ ((E) and (F)) by human NRK1 and NMNAT1 as measured by UV absorbance at 260 nm. 2 mM NR or S-NR was incubated with 6 mM ATP and 5 μM NRK1 and 5 μM NMNAT1 at RT for 0 h and 4 h, followed by HPLC analysis. | ||
To test this notion, S-NR was first chemically synthesized. As shown in Scheme S1,‡ 1-acetoxy thioribose 9 was prepared from D-gulonic acid γ-lactone according to the reported procedures.27 Despite established methods for synthesis of NR via introduction of nicotinamide onto ribose, S-NR is yet to be developed.28–31 By testing the substitution of nicotinamide for OAc group of 9, it was found that treatment of 9 with sequential HBr (33% (wt) in acetic acid) in toluene at 0 °C for 4 hours and nicotinamide in CH3CN at room temperature for 18 hours successfully afforded 10 in a 58% yield (Fig. 2B). S-NR was then generated through subsequent deprotections of the 4′-thioribose ring.
Human NRK1 and NMNAT1 were then expressed and purified from Escherichia coli (Fig. S1‡). In vitro biosynthesis of NAD+ and S-NAD+ from NR and S-NR, respectively, were carried out using purified NRK1 and NMNAT1. In the presence of NRK1 and NMNAT1 and ATP, a large amount of NAD+ and S-NAD+ were rapidly formed within 4-hour incubation (Fig. 2B–F and S2–S4‡). The formation rate of S-NAD+ was comparable to that of NAD+. In contrast to the last two steps of chemical synthesis of NAD+ analogues that usually take two to four days and are characterized by tedious purification procedures and low yields, enzymatic conversion of S-NR to S-NAD+ by NRK1 and NMNAT1 could be completed within four hours with a final yield of 70% and requires significantly less efforts on purification (Fig. 2B and Scheme S1‡). These results demonstrate a facile and efficient chemoenzymatic approach for the generation of S-NAD+. The sulfur substitution seemed to have no effect on substrate activities of S-NR and 4′-thioribose nicotinamide mononucleotide (S-NMN) for human NRK and NMNAT, respectively. In addition, the UV absorption spectrum of the generated S-NAD+ is nearly identical to that of NAD+ (Fig. S5‡).
To evaluate chemical stability of the generated S-NAD+, recombinant extracellular domain of human CD38 was chosen as a model enzyme (Fig. S6‡), which catalyzes rapid formation of ADPR and cADPR from NAD+. HPLC analysis revealed that in contrast to NAD+ that was completely consumed by human CD38 after an overnight reaction, S-NAD+ still remained unchanged after overnight incubation with CD38 (Fig. 3). These results indicate that relative to NAD+, the synthesized S-NAD+ is more resistant to cleavage catalyzed by CD38 enzyme and support that 4′-thioribose substitution for NR ribosyl group results in an NAD+ analogue chemically inert to catalysis for N-glycosidic bond breakage.
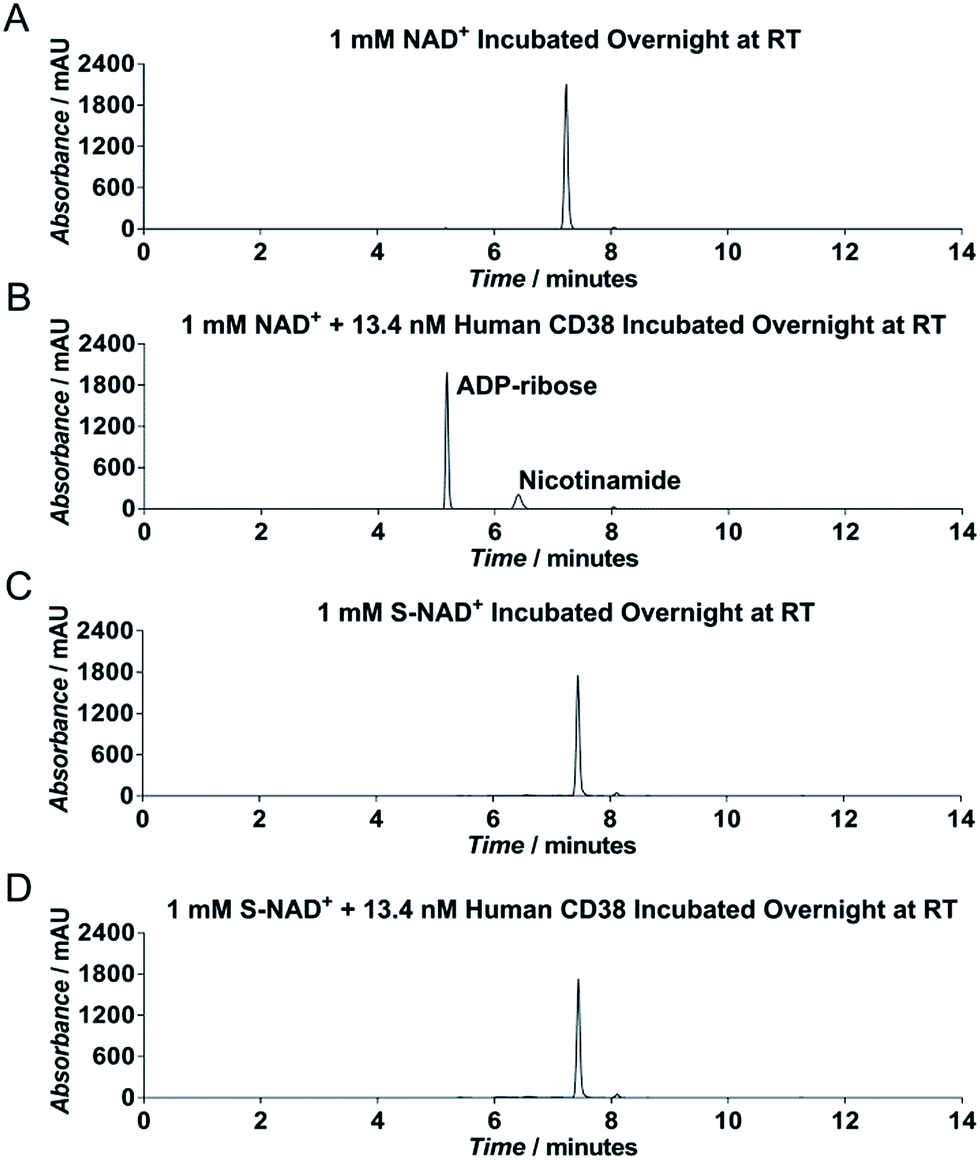 | ||
| Fig. 3 Activity of NAD+ and S-NAD+ for human CD38. HPLC analysis of substrate activities of NAD+ (A and B) and S-NAD+ (C and D) with human CD38 as measured by UV absorbance at 260 nm. 1 mM NAD+ or S-NAD+ was incubated without and with CD38 at RT overnight, followed by HPLC analysis. | ||
Next, the generated S-NAD+ was examined for its competitive inhibition activity for human CD38 enzyme by performing a continuous fluorescence-based activity assay using nicotinamide guanine dinucleotide (NGD+) as a substrate. The cyclase activity of CD38 catalyzes formation of fluorescent cyclic GDP-ribose (cGDPR) from NGD+.32 On the basis of cGDPR-derived fluorescence, kinetics for human CD38-catalyzed conversion of NGD+ to cGDPR was examined and the Km of NGD+ for human CD38 cyclase activity was determined to be 8.5 ± 0.6 μM (Fig. S7‡). Competitive inhibition assays showed that S-NAD+ exhibits a dose-dependent inhibition of cGDPR formation by CD38 with a Ki of 28.5 ± 3.1 μM (Fig. 4), comparable to previously determined Km values of NAD+ for CD38 enzyme.33,34 These results suggest that by mimicking NAD+, S-NAD+ could bind to the active site of human CD38 to inhibit its enzymatic activity and a single atomic substitution within the ribosyl ring seems to induce no adverse effects on its binding to the CD38 enzyme. Additionally, no slow-onset inhibition was observed for S-NAD+ in direct competition assays with extended incubation or in studies of preincubating CD38 with S-NAD+.
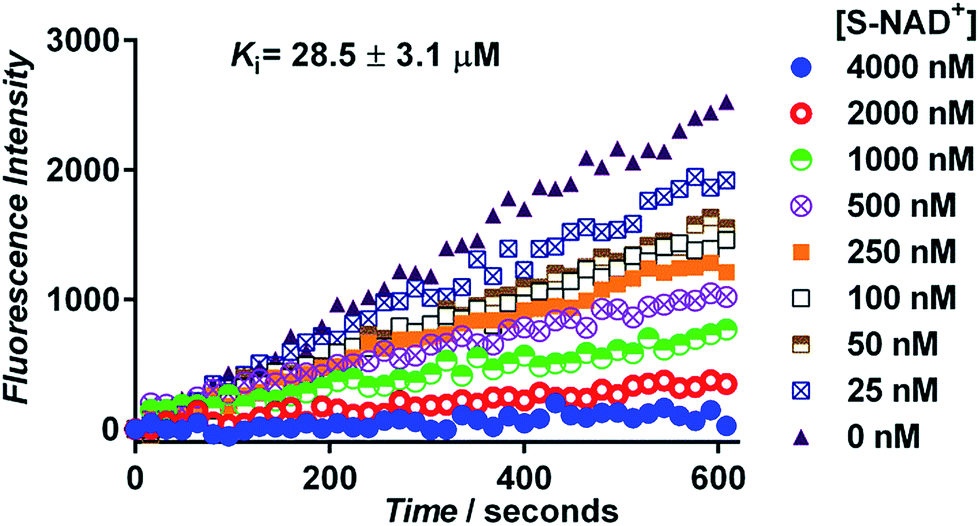 | ||
| Fig. 4 Inhibition activity of S-NAD+ for human CD38. Recombinant human CD38 (8 nM) was incubated with 50 μM NGD+ in the presence of varied concentrations of S-NAD+. CD38 cyclase activities were monitored on the basis of the formation of fluorescent cGDPR as measured at 410 nm. | ||
In addition to human CD38, the chemical stability of S-NAD+ was evaluated for human sirtuin 2 (SIRT2), an NAD+-dependent protein deacetylase. Trypsin-coupled fluorescence-based activity assays indicated that in contrast to NAD+ that could participate in SIRT2-catalyzed protein deacetylation, S-NAD+ alone revealed no substrate activity for SIRT2 after overnight incubation with SIRT2 and a fluorogenic, acetylated peptide substrate and co-incubation of S-NAD+ with NAD+ significantly inhibited protein deacetylation catalyzed by SIRT2 (Fig. S8‡). These results suggest that as a stable mimic of NAD+, S-NAD+ could bind to the SIRT2 active site and inhibit its catalytic activity. It is expected that the replacement of the endocyclic oxygen with less electronegative sulfur would reduce chemical reactivity of the N-glycosidic bond, making it more resistant to enzymatic cleavage. Moreover, sulfur substitution may induce minimal changes in ribosyl geometry, causing little effects on enzyme binding. By altering bond stability and retaining molecular geometry, the single atomic substitution results in the S-NAD+ as a stable mimic of NAD+.
In addition to enhancing stability of the N-glycosidic linkage between nicotinamide and ribose ring, replacing the endocyclic oxygen with sulfur is expected to have no impact on reduction of nicotinamide ring in oxidation–reduction reactions. To determine whether S-NAD+ could participate in electron transfer reactions, bovine glutamate dehydrogenase (GDH) and Leuconostoc mesenteroides glucose-6-phosphate dehydrogenase (G6PDH) were utilized, which are known to catalyze oxidation of glutamate and glucose-6-phosphate by NAD+, respectively.35,36 Enzymatic activity assays revealed that incubation of S-NAD+ with bovine GDH and L. mesenteroides G6PDH resulted in characteristic increases of UV absorbance at 340 nm in a manner similar to those of NAD+-containing reactions (Fig. S9‡). HPLC analyses of the reduction of NAD+ and S-NAD+ by bovine GDH and L. mesenteroides G6PDH showed enzyme-dependent formation of NADH and S-NADH peaks, which were confirmed by mass spectrometry (Fig. S10 and S11‡). These results suggest that the single atomic substitution within NR ribosyl ring has little effects on accepting electrons and S-NAD+ can participate in redox reactions.
To explore the binding mode of S-NAD+ with human CD38, X-ray crystallographic analysis was carried out. The extracellular domain of wild-type human CD38 except four mutated glycosylation sites was recombinantly expressed in mammalian cells (Fig. S6‡). Enzymatic activities of purified CD38 were verified by fluorescence- and HPLC-based activity assays using NGD+ and NAD+ as substrates. Recombinant human CD38 in high purity was successfully cocrystallized with S-NAD+ and its X-ray crystal structure was solved at a resolution of 2.4 Å (Fig. 5 and Table S2‡). The determined crystal structure clearly revealed the binding of S-NAD+ to the active site of human CD38 and multiple interactions formed between S-NAD+ and active site residues. The 4′-thioribose ring of S-NAD+ adopted a C3′-endo-C2′-exo conformation. It was seen that the distances between S-NR 2′-OH of S-NAD+ and side chains of S193 and E226 are 2.8 Å and 3.6 Å, respectively. The S-NR 3′-OH of S-NAD+ is 2.7 Å away from the side chain of E146 (Fig. 5). These observations support that these three residues play important roles in catalysis of human CD38 and are consistent with previous reports, in which it was shown that E226 and S193 are catalytic residues involved in stabilization of formed oxacarbenium ion intermediate and E146 controls cyclizing and hydrolyzing activities.6,15,37 The most striking finding for the determined X-ray structure is that the nicotinamide ring of S-NAD+ is fully stacked on top of the indole ring of W189 (Fig. 5). The planes of S-NAD+ nicotinamide and W189 indole are nearly parallel with an interplanar spacing of approximately 3.4 Å. On the basis of the solved X-ray structure, the relative position, orientation, and distance of the nicotinamide moiety of S-NAD+ at the active site of human CD38 suggest that W189 plays an important role in activation of the leaving group upon binding of substrate. This finding is consistent with previous studies which showed that mutations of W189 cause significantly decreased enzymatic activities of human CD38.
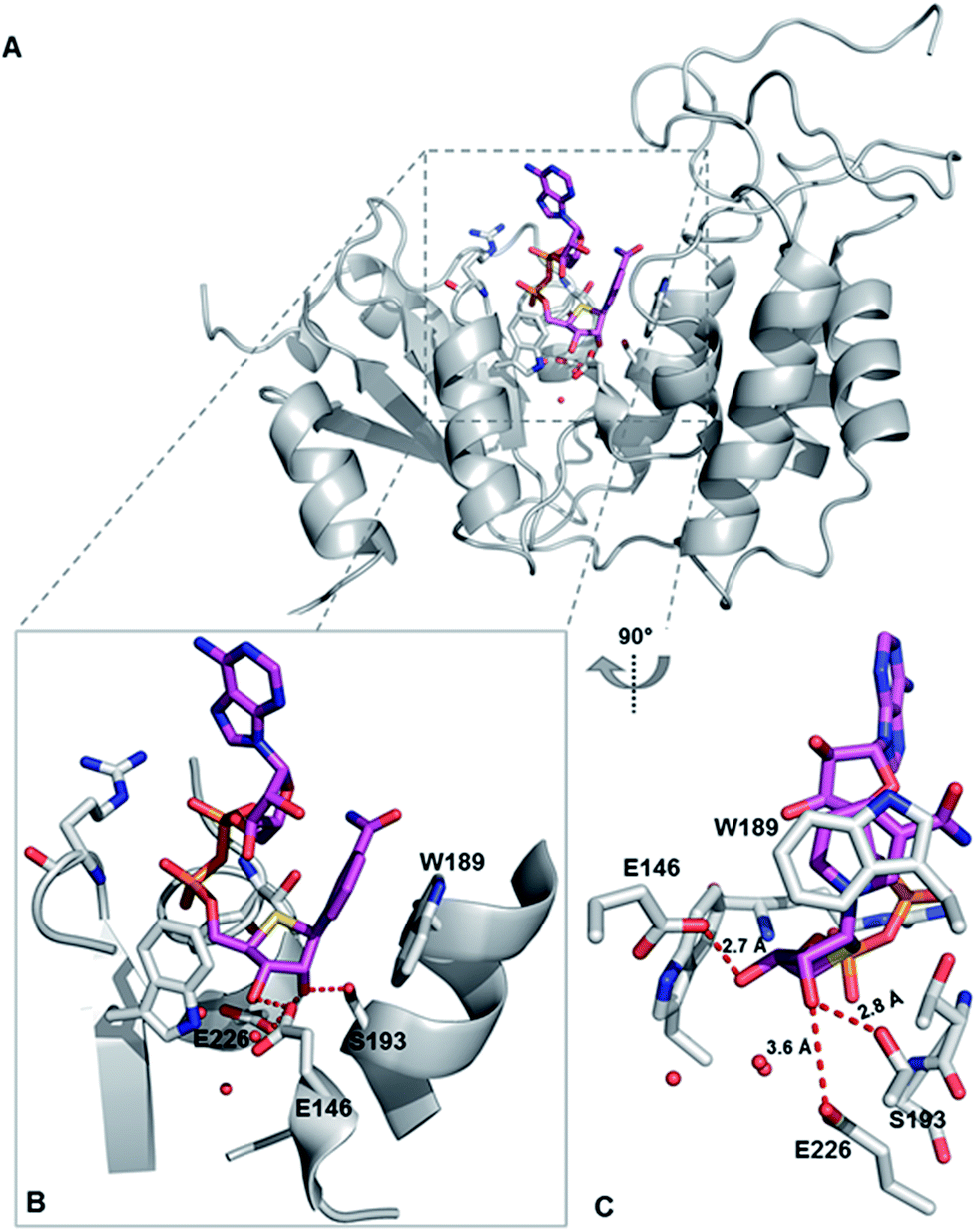 | ||
| Fig. 5 X-ray crystal structure of catalytically active human CD38 in complex with S-NAD+. CD38 and S-NAD+ are shown in grey and magenta, respectively. (A) Overall crystal complex structure of human CD38-S-NAD+. (B) and (C) Bound S-NAD+ at the active site of human CD38 with indicated catalytic residues and interacting water molecules. PDB ID: 6EDR. | ||
Comparative analysis of S-NAD+-bound CD38 with apo-CD38 (PDB ID: 1YH3) and NAD+-bound CD38 (E226Q) (PDB ID: 2I65) showed no significant changes in overall structure (Fig. S12‡).15,38 Relative to S-NR of S-NAD+, the NR moiety of NAD+ at the binding site of catalytically inactive CD38 (E226Q) is closer to the bottom of the binding pocket. As a result, the nicotinamide ring of NAD+ forms very minor stacking interaction with the indole ring of W189 (Fig. S12‡). The nicotinamide rings of NAD+ and S-NAD+ at the CD38 active sites reveal distinct orientations with C2-N1-C1′-C2′ dihedral angles of 174.3° and 34.7°, respectively, which enable the amide group of NAD+ to be in hydrogen bond distance to the side chain of E146. In addition, the ribose ring of NAD+ adopted a C2′-endo-C3′-exo conformation. These structural differences between NAD+ and S-NAD+ at the active sites of CD38 suggest that the sulfur substitution results in an NAD+ analogue more resistant to the N-glycosidic bond breakage. Taken together, the X-ray structure of S-NAD+ with catalytically active human CD38 is consistent with the substrate and inhibition activities studies and demonstrates that as a stable mimic of NAD+, the generated S-NAD+ allows X-ray crystallographic characterization of reactive complexes of CD38 to elucidate its catalytic mechanism.
Conclusions
As a novel stable NAD+ mimic, S-NAD+ was successfully generated. By exploiting human NRK1 and NMNAT1, S-NAD+ could be efficiently prepared from its chemically synthesized S-NR precursor through a two-step enzymatic process in a high yield. The generated S-NAD+ is chemically inert to cleavage by human CD38 and sirtuin 2, while functioning as an electron acceptor in redox reactions. The X-ray structure of S-NAD+ with human CD38 demonstrates its binding to enzyme active site and revealed residues important for catalysis along the reaction coordinate. This work provides a facile and efficient chemoenzymatic approach for the generation of S-NAD+ and a unique and important tool that can be extended to investigate NAD+-dependent enzymes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the staff at Synchrotron Radiation Light Source (SSRL) (beamline 12-2) for their excellent support during data collections and Dr Gye Won Han for her advice on the refinement and checking the quality of the final structure. This work was supported by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, Sharon L. Cockrell Cancer Research Fund, The V Foundation for Cancer Research V Scholar Grant V2016-021 (to Y. Z.), and University of Southern California Research Center for Liver Diseases Pilot Grant P30DK048522 (to Y. Z.).References
- K. W. Ryu, D. S. Kim and W. L. Kraus, New Facets in the Regulation of Gene Expression by ADP-Ribosylation and Poly(ADP-ribose) Polymerases, Chem. Rev., 2015, 115, 2453 CrossRef CAS PubMed.
- H. Lin, Nicotinamide adenine dinucleotide: beyond a redox coenzyme, Org. Biomol. Chem., 2007, 5, 2541 RSC.
- S. Imai and L. Guarente, NAD+ and sirtuins in aging and disease, Trends Cell Biol., 2014, 24, 464 CrossRef CAS PubMed.
- E. Verdin, NAD(+) in aging, metabolism, and neurodegeneration, Science, 2015, 350, 1208 CrossRef CAS PubMed.
- S. Yamamoto-Katayama, A. Sato, M. Ariyoshi, M. Suyama, K. Ishihara, T. Hirano, H. Nakamura, K. Morikawa and H. Jingami, Site-directed removal of N-glycosylation sites in BST-1/CD157: effects on molecular and functional heterogeneity, Biochem. J., 2001, 357, 385 CrossRef CAS PubMed.
- R. Graeff, C. Munshi, R. Aarhus, M. Johns and H. C. Lee, A single residue at the active site of CD38 determines its NAD cyclizing and hydrolyzing activities, J. Biol. Chem., 2001, 276, 12169 CrossRef CAS PubMed.
- E. Blacher, T. Dadali, A. Bespalko, V. J. Haupenthal, M. O. Grimm, T. Hartmann, F. E. Lund, R. Stein and A. Levy, Alzheimer's disease pathology is attenuated in a CD38-deficient mouse model, Ann. Neurol., 2015, 78, 88 CrossRef CAS PubMed.
- C. Escande, V. Nin, N. L. Price, V. Capellini, A. P. Gomes, M. T. Barbosa, L. O'Neil, T. A. White, D. A. Sinclair and E. N. Chini, Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome, Diabetes, 2013, 62, 1084 CrossRef CAS PubMed.
- M. G. Tarrago, C. C. S. Chini, K. S. Kanamori, G. M. Warner, A. Caride, G. C. de Oliveira, M. Rud, A. Samani, K. Z. Hein, R. Huang, D. Jurk, D. S. Cho, J. J. Boslett, J. D. Miller, J. L. Zweier, J. F. Passos, J. D. Doles, D. J. Becherer and E. N. Chini, A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline, Cell Metab., 2018, 27, 1081 CrossRef CAS PubMed.
- J. Camacho-Pereira, M. G. Tarrago, C. C. S. Chini, V. Nin, C. Escande, G. M. Warner, A. S. Puranik, R. A. Schoon, J. M. Reid, A. Galina and E. N. Chini, CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism, Cell Metab., 2016, 23, 1127 CrossRef CAS PubMed.
- E. Blacher, B. Ben Baruch, A. Levy, N. Geva, K. D. Green, S. Garneau-Tsodikova, M. Fridman and R. Stein, Inhibition of glioma progression by a newly discovered CD38 inhibitor, Int. J. Cancer, 2015, 136, 1422 CrossRef CAS PubMed.
- F. Morandi, A. L. Horenstein, A. Chillemi, V. Quarona, S. Chiesa, A. Imperatori, S. Zanellato, L. Mortara, M. Gattorno, V. Pistoia and F. Malavasi, CD56brightCD16-NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation, J. Immunol., 2015, 195, 965 CrossRef CAS PubMed.
- S. Chatterjee, A. Daenthanasanmak, P. Chakraborty, M. W. Wyatt, P. Dhar, S. P. Selvam, J. Fu, J. Zhang, H. Nguyen, I. Kang, K. Toth, M. Al-Homrani, M. Husain, G. Beeson, L. Ball, K. Helke, S. Husain, E. Garrett-Mayer, G. Hardiman, M. Mehrotra, M. I. Nishimura, C. C. Beeson, M. G. Bupp, J. Wu, B. Ogretmen, C. M. Paulos, J. Rathmell, X. Z. Yu and S. Mehrotra, CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response, Cell Metab., 2018, 27, 85 CrossRef CAS PubMed.
- P. O. Hassa, S. S. Haenni, M. Elser and M. O. Hottiger, Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going?, Microbiol. Mol. Biol. Rev., 2006, 70, 789 CrossRef CAS PubMed.
- Q. Liu, I. A. Kriksunov, R. Graeff, C. Munshi, H. C. Lee and Q. Hao, Structural basis for the mechanistic understanding of human CD38-controlled multiple catalysis, J. Biol. Chem., 2006, 281, 32861 CrossRef CAS PubMed.
- Q. Liu, I. A. Kriksunov, R. Graeff, H. C. Lee and Q. Hao, Structural basis for formation and hydrolysis of the calcium messenger cyclic ADP-ribose by human CD38, J. Biol. Chem., 2007, 282, 5853 CrossRef CAS PubMed.
- K. G. Hoff, J. L. Avalos, K. Sens and C. Wolberger, Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide, Structure, 2006, 14, 1231 CrossRef CAS PubMed.
- A. Zatorski, K. A. Watanabe, S. F. Carr, B. M. Goldstein and K. W. Pankiewicz, Chemical synthesis of benzamide adenine dinucleotide: inhibition of inosine monophosphate dehydrogenase (types I and II), J. Med. Chem., 1996, 39, 2422 CrossRef CAS PubMed.
- M. F. Langelier, L. Zandarashvili, P. M. Aguiar, B. E. Black and J. M. Pascal, NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains, Nat. Commun., 2018, 9, 844 CrossRef PubMed.
- B. G. Szczepankiewicz, H. Dai, K. J. Koppetsch, D. Qian, F. Jiang, C. Mao and R. B. Perni, Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5, J. Org. Chem., 2012, 77, 7319 CrossRef CAS PubMed.
- A. A. Sauve and V. L. Schramm, Mechanism-based inhibitors of CD38: a mammalian cyclic ADP-ribose synthetase, Biochemistry, 2002, 41, 8455 CrossRef CAS PubMed.
- J. H. Shrimp, J. Hu, M. Dong, B. S. Wang, R. MacDonald, H. Jiang, Q. Hao, A. Yen and H. Lin, Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe, J. Am. Chem. Soc., 2014, 136, 5656 CrossRef CAS PubMed.
- H. Jiang, J. Congleton, Q. Liu, P. Merchant, F. Malavasi, H. C. Lee, Q. Hao, A. Yen and H. Lin, Mechanism-based small molecule probes for labeling CD38 on live cells, J. Am. Chem. Soc., 2009, 131, 1658 CrossRef CAS PubMed.
- L. Sorci, F. Cimadamore, S. Scotti, R. Petrelli, L. Cappellacci, P. Franchetti, G. Orsomando and G. Magni, Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis, Biochemistry, 2007, 46, 4912 CrossRef CAS PubMed.
- F. Berger, C. Lau, M. Dahlmann and M. Ziegler, Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms, J. Biol. Chem., 2005, 280, 36334 CrossRef CAS PubMed.
- W. Tempel, W. M. Rabeh, K. L. Bogan, P. Belenky, M. Wojcik, H. F. Seidle, L. Nedyalkova, T. Yang, A. A. Sauve, H. W. Park and C. Brenner, Nicotinamide riboside kinase structures reveal new pathways to NAD+, PLoS Biol., 2007, 5, e263 CrossRef PubMed.
- L. S. Jeong, H. W. Lee, K. A. Jacobson, H. O. Kim, D. H. Shin, J. A. Lee, Z.-G. Gao, C. Lu, H. T. Duong and P. Gunaga, Structure – Activity Relationships of 2-Chloro-N 6-substituted-4 ‘-thioadenosine-5 ‘-uronamides as Highly Potent and Selective Agonists at the Human A3 Adenosine Receptor, J. Med. Chem., 2006, 49, 273 CrossRef CAS PubMed.
- T. Yang, N. Y.-K. Chan and A. A. Sauve, Syntheses of nicotinamide riboside and derivatives: effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells, J. Med. Chem., 2007, 50, 6458 CrossRef CAS PubMed.
- J. Dowden, C. Moreau, R. S. Brown, G. Berridge, A. Galione and B. V. Potter, Chemical synthesis of the second messenger nicotinic acid adenine dinucleotide phosphate by total synthesis of nicotinamide adenine dinucleotide phosphate, Angew. Chem., 2004, 116, 4737 CrossRef.
- J. Dowden, R. S. Brown, C. Moreau, A. Galione and B. V. Potter, Chemical synthesis of the novel Ca2+ messenger NAADP, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 513 CrossRef CAS.
- Y. Cen and A. A. Sauve, Transition state of ADP-ribosylation of acetyllysine catalyzed by Archaeoglobus fulgidus Sir2 determined by kinetic isotope effects and computational approaches, J. Am. Chem. Soc., 2010, 132, 12286 CrossRef CAS PubMed.
- R. M. Graeff, T. F. Walseth, K. Fryxell, W. D. Branton and H. C. Lee, Enzymatic synthesis and characterizations of cyclic GDP-ribose. A procedure for distinguishing enzymes with ADP-ribosyl cyclase activity, J. Biol. Chem., 1994, 269, 30260 CAS.
- G. Orsomando, V. Polzonetti and P. Natalini, NAD(P)(+)-glycohydrolase from human spleen: a multicatalytic enzyme, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2000, 126, 89 CrossRef CAS.
- K. A. Wall, M. Klis, J. Kornet, D. Coyle, J. C. Ame, M. K. Jacobson and J. T. Slama, Inhibition of the intrinsic NAD+ glycohydrolase activity of CD38 by carbocyclic NAD analogues, Biochem. J., 1998, 335(3), 631 CrossRef CAS PubMed.
- M. Li, C. J. Smith, M. T. Walker and T. J. Smith, Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics, J. Biol. Chem., 2009, 284, 22988 CrossRef CAS PubMed.
- C. Olive, M. E. Geroch and H. R. Levy, Glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides. Kinetic studies, J. Biol. Chem., 1971, 246, 2047 CAS.
- C. Munshi, R. Aarhus, R. Graeff, T. F. Walseth, D. Levitt and H. C. Lee, Identification of the enzymatic active site of CD38 by site-directed mutagenesis, J. Biol. Chem., 2000, 275, 21566 CrossRef CAS PubMed.
- Q. Liu, I. A. Kriksunov, R. Graeff, C. Munshi, H. C. Lee and Q. Hao, Crystal structure of human CD38 extracellular domain, Structure, 2005, 13, 1331 CrossRef CAS PubMed.
Footnotes |
| † The X-ray structure of S-NAD+ with human CD38 is deposited in Protein Data Bank (https://www.rcsb.org) (PDB ID: 6EDR). |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, and supplementary figures, table, and data. See DOI: 10.1039/c8sc03899f |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |