
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence enhancement of a ligand-activated fluorescent protein induced by collective noncovalent interactions†
Euihyun
Lee
 ab,
Sang-Hee
Shim
*ab and
Minhaeng
Cho
*ab
ab,
Sang-Hee
Shim
*ab and
Minhaeng
Cho
*ab
aCenter for Molecular Spectroscopy and Dynamics, Institute for Basic Science (IBS), Korea University, Seoul 02841, Republic of Korea. E-mail: mcho@korea.ac.kr; sangheeshim@korea.ac.kr
bDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea
First published on 8th October 2018
Abstract
Fluorescent proteins contain an internal chromophore constituted of amino acids or an external chromophore covalently bonded to the protein. To increase their fluorescence intensities, many research groups have attempted to mutate amino acids within or near the chromophore. Recently, a new type of fluorescent protein, called UnaG, in which the ligand binds to the protein through many noncovalent interactions was discovered. Later, a series of mutants of the UnaG protein were introduced, which include eUnaG with valine 2 mutated to leucine emitting significantly stronger fluorescence than the wild type and V2T mutant, in which valine 2 is mutated to threonine, emitting weaker fluorescence than the wild type. Interestingly, the single mutation sites of both eUnaG and V2T mutants are distant from the fluorophore, bilirubin, which renders the mechanism of such fluorescence enhancement or reduction unclear. To elucidate the origin of fluorescence intensity changes induced by the single mutations, we carried out extensive analyses on MD simulations for the original UnaG, eUnaG and V2T, and found that the bilirubin ligand bound to eUnaG is conformationally more rigid than the wild-type, particularly in the skeletal dihedral angles, possibly resulting in the increase of quantum yield through a reduction of non-radiative decay. On the other hand, the bilirubin bound to the V2T appears to be flexible than that in the UnaG. Furthermore, examining the structural correlations between the ligand and proteins, we found evidence that the bilirubin ligand is encapsulated in different environments composed of protein residues and water molecules that increase or decrease the stability of the ligand. The changed protein stability affects the mobility and confinement of water molecules captured between bilirubin and the protein. Since the flexible ligand contains multiple hydrogen bond (H-bond) donors and acceptors, the H-bonding structure and dynamics of bound water molecules are highly correlated with the rigidity of the bound ligand. Our results suggest that, to understand the fluorescence properties of protein mutants, especially the ones with noncovalently bound fluorophores with internal rotations, the interaction network among protein residues, ligand, and water molecules within the binding cavity should be investigated rather than focusing on the local structure near the fluorescing moiety. Our in-depth simulation study may offer a foundation for the design principles for engineering this new class of fluorescent proteins.
1. Introduction
Green fluorescent protein (GFP) discovered in the early 1960s has enabled investigators to monitor cellular processes in living systems using a light microscope when GFP is fused to various proteins of interest. This approach has revolutionized modern biology because it becomes possible to study functional cellular signals such as membrane potential, calcium concentration, reactive oxygen species, to name a few.1 Fluorescent proteins have either constitutive fluorophores or covalently bonded fluorogens. Jellyfish/coral-derived GFP and GFP-like proteins exhibit characteristic fluorescence emitted from the chromophores produced by a series of chemical reactions of their amino acids. For instance, the fluorophore of GFP is formed by chemical reactions of the three residues, Thr65, Tyr66, and Gly67 (Fig. 1a).2 There exist other types of FPs that acquire fluorescence by forming a covalent bond with a small molecule that serves as the fluorophore of external origin (Fig. 1b). Infrared FPs derived from photoreceptors consist of a host protein and a highly conjugated compound, usually a linear tetrapyrrole such as biliverdin.3–6 Upon binding, the external chromophore forms a thioether bond with a Cys residue located in the binding pocket autocatalytically or by a separate enzyme, lyase (Fig. 1b).7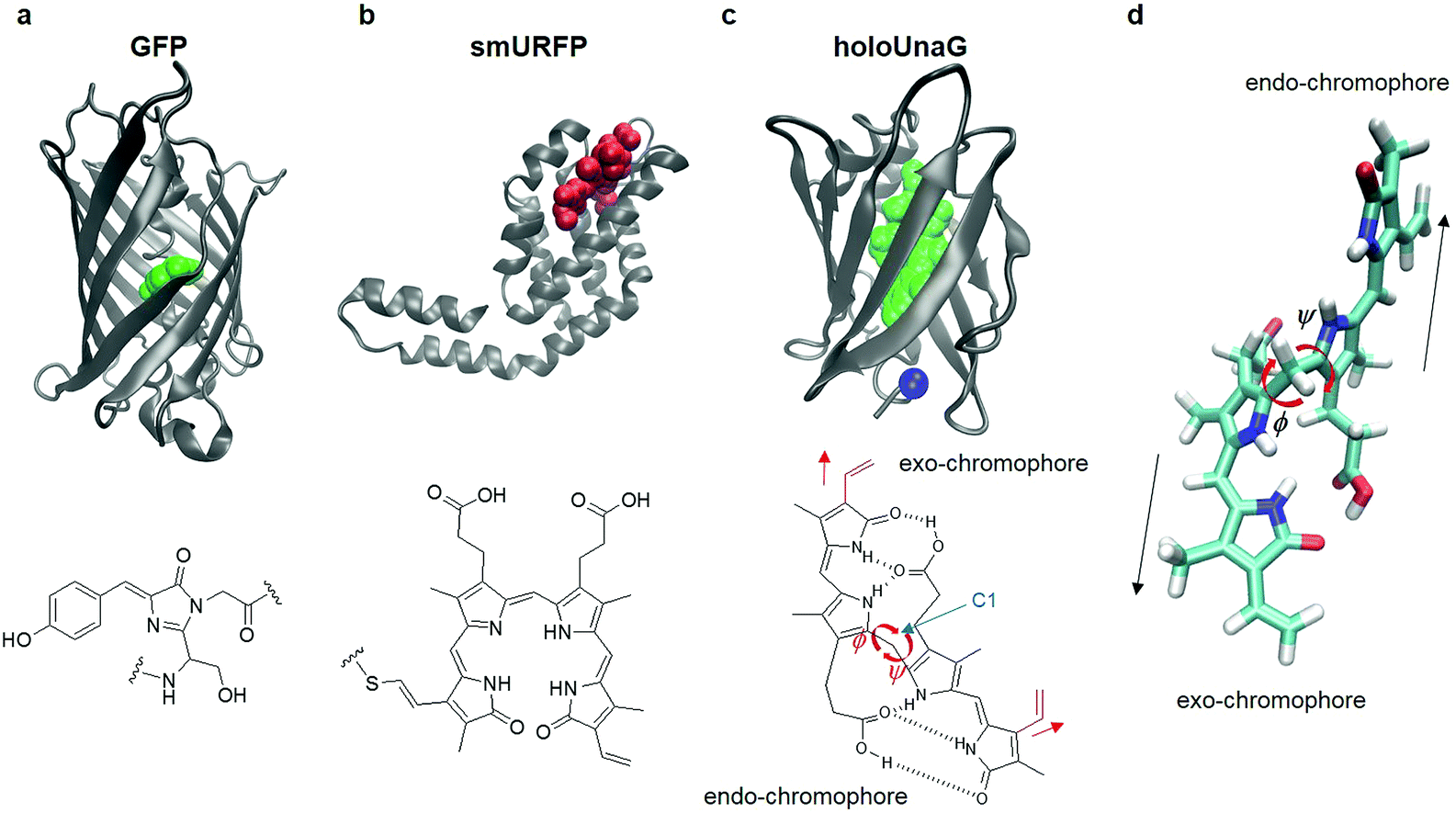 | ||
| Fig. 1 Fluorescent proteins (FP) and their chromophores. (a) Green fluorescent protein (GFP, PDB ID: 1EMA, top), a FP with a constitutive fluorophore, and its chromophore (green balls in the top panel and chemical structure in the bottom) composed of Ser65, Tyr66, and Gly67. (b) Small ultra-red FP (smURFP, PDB ID: 4PO5, top), a FP with covalently linked external chromophore, biliverdin (red balls in the top panel and chemical structure in the bottom). (c) HoloUnaG protein (PDB ID: 4I3B, top), a FP with noncovalently bound external chromophore, bilirubin (BR, green balls in the top panel and chemical structure in the bottom). Blue ball indicates the mutation site, Val 2. Note that the positions of the two vinyl groups (red) attached to the two terminal pyrrole-like rings are slightly different for the two constituent chromophores in BR. When the vinyl group stretches in the same direction as the chromophore, it is named an ‘exo’-chromophore, otherwise it is named an ‘endo’-chromophore. ϕ and ψ are the two main dihedral angles around the central carbon (C1), which essentially determine the relative orientations of the exo- and endo-chromophores, respectively. (d) In-line conformation of BR in UnaG proteins. The exo- and endo-chromophores are arranged along a line. The mutation site in the protein is close to the exo-chromophore rather than the endo-chromophore. | ||
Mechanistic insights of FPs offer design principles for engineering fluorescent proteins. For instance, mClover3 was engineered from avGFP by using key insights in the residues influencing the hydrogen-bonding network surrounding the chromophore, the stacking residues that shift the emission wavelength, and the residues associated with photostability and folding. As a result, mClover3 exhibits about 2.5 times stronger fluorescence intensity than mEGFP.8,9 These mechanistic insights come from combinatorial mutagenesis studies as well as structural and spectroscopic investigations.10 In addition, MD simulations were often found to be of critical use in elucidating the detailed mechanism of the observed fluorescence enhancement as well as other spectral and molecular characteristics.11–15 From these results, protein engineers often mutate amino acid residues within or near the chromophores, which is essentially to change the chemical structure of or the local environment around the chromophore.16–20 Constitutive engineering of FPs such as GFP usually involves mutations inducing direct changes of the molecular structure of the chromophore. Infrared FPs, originating from photoreceptors in bacteria and algae that bind to phytochromes nonexistent in higher animals, are derived from mutations resulting in incorporating a non-natural partner, biliverdin present in mammals. Since the incorporation of such chromophores proceeds through two steps (non-covalent binding to the protein and formation of a thioether bond between the chromophore and a Cys residue), a series of mutations are often needed to engineer those infrared FPs. In summary, fluorescent proteins have been engineered through a series of mutations on or near the chromophore to achieve noticeable enhancement in fluorescence intensity.
In 2013, a new class of fluorescent proteins whose fluorescence is induced by a noncovalently bound ligand was discovered from Japanese freshwater eel and named UnaG (Fig. 1c).21 The fluorescent biliprotein emits fluorescence upon noncovalent binding of a diffusible fluorogenic ligand to the protein and differs from other fluorescent biliproteins with covalently linked fluorogens (Fig. 1a–c).22–26 The ligand binding to the UnaG protein is bilirubin (BR) that is produced from the degradation of heme.21,27 BR has two chromophore units with the same chemical formula but slightly different structures that were called exo- and endo-chromophores (vinyl dipyrrinone) by Kumagai et al (Fig. 1c, bottom).21 These two chromophore units are covalently bonded to the central carbon atom (C1 in the chemical structure of BR in Fig. 1c) so that the two dihedral angles denoted as ϕ and ψ determine the relative orientations of and distances between the exo- and endo-chromophores (Fig. 1c). The free dihedral rotations and conformational flexibility of BR make non-radiative decay of the photo-excited BR in solution very efficient, which results in very weak fluorescence of BR. However, when it binds to UnaG protein, BR is tightly bound in a way that the two chromophores are stretched in line (Fig. 1d), which then increases its conformational rigidity, reduces the non-radiative decay rate, and consequently results in emission of strong fluorescence whose quantum yield is on par with enhanced GFPs.
Recently, Yeh et al.28 discovered a fluorescence-enhanced mutant of UnaG, named eUnaG, by randomly mutating the original UnaG protein and screening out those bright mutants. The BR-binding eUnaG exhibits almost twice stronger fluorescence intensity. In the eUnaG, residue 2 of the original UnaG, valine (Val), is replaced by a slightly larger hydrophobic residue, leucine (Leu). In contrast, another mutant named V2T, in which residue 2 of UnaG is replaced by threonine (Thr), resulted in reduced fluorescence intensity by about 2-fold compared with the original UnaG. It is surprising that these single subtle mutations resulted in such a significant increase in both the fluorescence intensity and thermal stability of eUnaG or decrease in the fluorescence intensity of V2T. Unlike the covalently linked chromophores in the other FPs, the entirely noncovalent interactions of the chromophore and the amino acid residues in UnaG may play different roles in the fluorescence enhancement of the eUnaG from those of conventional FPs. Therefore, it should be of great interest to investigate the mechanism of the fluorescence enhancement/diminishment in UnaG mutants in new perspectives, not limited to the ones investigated for other fluorescent proteins.
Here, we present MD simulation results with detailed comparisons of UnaG, eUnaG, and V2T structures as well as analysis results on the conformational rigidity of BR (radius of gyration, root-mean-square deviation/atomic fluctuation, distributions of dihedral angles, and global structure) to address the underlying mechanism of single-mutation-induced changes in the fluorescence and thermal stability of UnaG and its mutants. It turns out that, even though the single mutation site is remote from the BR fluorophore, the protein undergoes extensive, overall structural changes that significantly affect the dynamics and H-bonding characters of water molecules captured between the protein and the ligand, which influence the conformational rigidity of the ligand.
2. Methods
A. Preparation of simulation systems
To examine and compare the conformational rigidity of the BR molecule in water and aqueous protein solutions, we considered four systems: BR in water, BR in aqueous UnaG solution, BR in aqueous eUnaG solution, and BR in aqueous V2T solution. The initial structure of UnaG was obtained from the Protein Data Bank (https://www.rcsb.org, PDB ID: 4I3B).21 Among the 6 chains (monomers) of UnaG in PDB, we chose chain A (first monomer). For eUnaG and V2T, there are no crystal structures available so that we used the PDB structure of UnaG with a single substitution of Val with Leu and of Val with Thr, respectively (Fig. 1c). All systems composed of protein, BR, water molecules and counter ions (for charge neutrality) were generated using the Packmol program.29B. MD simulation methods
MD simulations were carried out using the AMBER 16 program package30 employing the parm99 force field parameters for the proteins.31 Each periodic box contains 5500 TIP3P water molecules and free BR, BR–UnaG complex, BR–eUnaG complex or BR–V2T complex. Additionally, in the box containing the BR–protein complex, 14 Na+ and 15 Cl− were added to the box to neutralize the whole composite system. Dang's force field parameters32 were used for Na+ and the general AMBER parameters were for Cl−. The corresponding box sizes are 55.13 × 55.15 × 55.14 Å3 for the free BR in water, and 57.203 Å3 for the three BR–protein complex systems, respectively. A cut-off distance of 10 Å was applied to all the non-bonding interactions and the particle mesh Ewald (PME) method30,33 was used to approximately calculate long-range electrostatic interactions. SHAKE algorithm was used for the TIP3P water and the simulation time step was set to be 1 fs. Three composite systems were first energy-minimized for 6000 steps, by using the steepest descent method and the conjugate gradient method with position restraint with a force constant of 500 kcal mol−1 Å−2, applied to protein. After that, energy-minimizations of the systems were repeated without position restraints for 6000 steps. Then, a 4 ns NPT simulation was performed at 1 atm and 298 K using a Berendsen barostat and thermostat with a coupling constant of 2 ps. After confirming that the whole system reached its thermal equilibrium, 14 ns NVT simulation at 298 K (Berendsen thermostat) was carried out for further equilibration. Finally, the production MD runs were performed for 110 ns at 298 K under constant NVT conditions. Trajectories were saved every 1 ps for subsequent statistical analyses. In our 110 ns MD trajectories, BRs in three protein systems show no notable conformational changes, indicating that the whole composite system is in a thermal equilibrium state.C. Quantum chemistry calculations
To calculate electronic circular dichroism spectra and electronic structures of BR bound to proteins, we performed quantum chemistry calculations.34 The conformation of each BR in the protein was set by using the average angles derived from the entire MD trajectories (the average ϕ and ψ angles are −58.37° and −63.69° for BR in UnaG, −59.31° and −61.41° in eUnaG, and −55.68° and −57.18° in V2T, respectively). First, a partial optimization of isolated BR was performed with the fixed dihedral angles to avoid conformational change. After the optimization, rotational strengths (in cgs unit) were obtained from the time-dependent DFT calculations with the B3LYP/6-31+G(d) basis set. To further carry out time-dependent DFT calculations for BR with H-bonded water molecules, the positions of the two water molecules were obtained from the MD trajectory of the eUnaG system.3. Results and discussion
A. Conformational rigidity of BR
Fluorescence intensity is often related to the rigidity of the chromophore structure. Here, using the MD simulation results for BR in water and in proteins, we calculated various measures of molecular rigidity of BR. In Fig. 2a–d, the entire 110 ns trajectories of radius of gyration (RG), root-mean-square deviation (RMSD), and the two dihedral angles (ϕ and ψ) are plotted. As expected, these measures of conformational rigidity of BR dissolved in water show very large fluctuations over time (black line in Fig. 2a–d). In contrast, the standard deviations of fluctuating RG values of BR bound to the three proteins, UnaG, eUnaG, and V2T (red, green and blue lines in Fig. 2a, respectively), are very small, i.e., σRG(BR–UnaG) < 0.20 Å, σRG(BR–eUnaG) < 0.12 Å and σRG(BR–V2T) < 0.28 Å (Table S1†). These clearly indicate that restraints by the protein environment strongly affect the BR structure. The reduced or increased standard deviation of the fluctuating RG of BR in eUnaG or V2T, respectively, compared to that of BR in UnaG, already shows that the conformational flexibility of BR is significantly reduced or increased by the site mutation in eUnaG or V2T, respectively.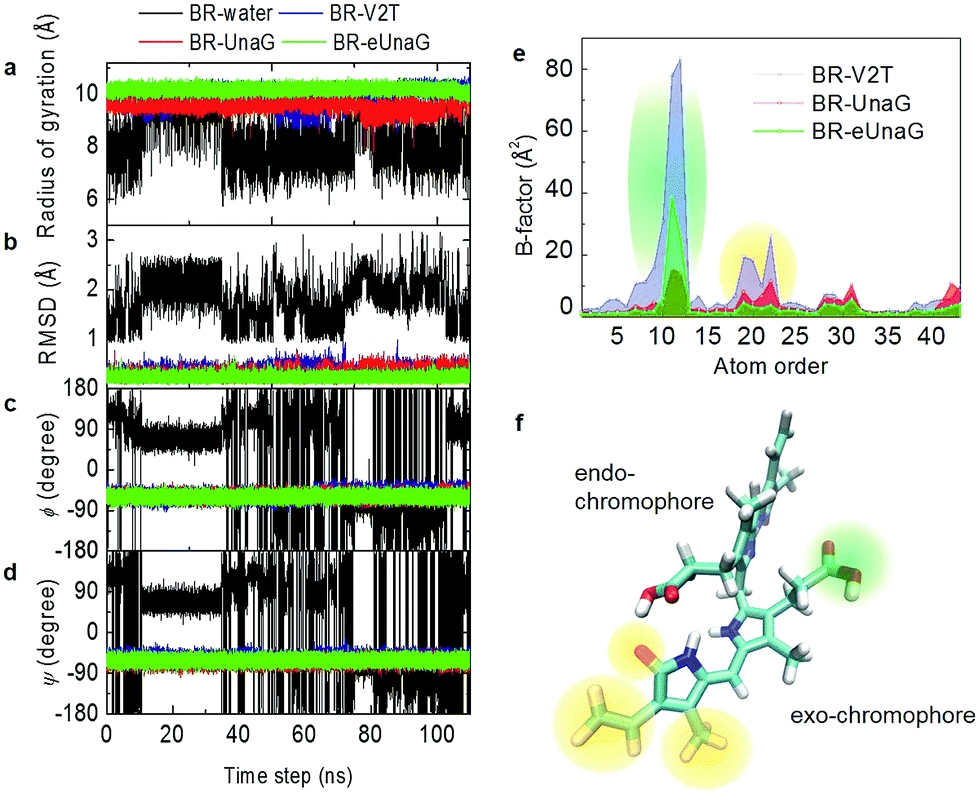 | ||
| Fig. 2 Trajectories of the radius of gyration (RG), RMSD, and ϕ and ψ angles taken from MD simulations. (a) Fluctuating radius of gyration of BR in water (black), in UnaG (red), in eUnaG (green), and in V2T (blue). Unlike the BR in water, the fluctuation amplitudes of the radius of gyration of BR bound to proteins are small and their average radii of gyration suggest that the BR adopts an extended linear structure. (b) RMSDs of BR in water, UnaG, eUnaG, and V2T. Here, only the heavy atoms of the conjugated backbone of BR are taken into consideration. (c and d) Trajectories of ϕ (c) and ψ (d) angles of BR in water, UnaG, eUnaG, and V2T. (e) Distribution of B-factors of the heavy atoms of BR in UnaG, eUnaG, and V2T. The atom order is identical to the order given in the PDB file. (f) BR structure with the highly fluctuating atoms highlighted with a green region for atom numbers of 10–12 and an orange region for atom numbers of 18–22 in (e). The green region is the carboxyl group of the exo-chromophore and the orange regions are the ends of the exo-chromophore. | ||
The conformation of BR bound to the three proteins is close to a linear structure (Fig. 1d) due to the constraints imposed by the neighboring amino acid residues around the BR in the proteins. This is in contrast with the BR molecule in water, where the conformational heterogeneity of BR in water originates from free-rotations of exo- and endo-chromophores along the two dihedral angles ϕ and ψ (Fig. 1c). Previous studies indicate that the intramolecular hydrogen bonding interactions make the ridge-tile conformation the most stable conformer of free bilirubin in water (Fig. 1c, bottom) whereas the porphyrin-like conformation is the most stable conformer of free biliverdin (Fig. 1b, bottom).35–38 In our RG trajectory shown in Fig. 2a, there appear to be two stable states of BR in water, which in fact contain many different states in terms of RMSD and dihedral angles. BR in water with RG ∼ 7.5 Å (low values) has intramolecular hydrogen bonds, but still the dihedral angles fluctuate widely and undergo transitions among different conformation states frequently. BR in water with RG ∼ 9 Å (high values) correspond to two different sets of dihedral angles, one of which is similar to that of BR in proteins, but again it undergoes conformational changes. Overall, BR in water is highly fluctuating, while BR in the three proteins remains in one stable conformation as expected.
To examine the detailed origins of the conformational flexibility of BR bound to the three proteins, we plot the two main dihedral angle trajectories in Fig. 2c and d. The ϕ angle is related to the dihedral rotation of the exo-chromophore and the ψ angle to the dihedral rotation of the endo-chromophore (Fig. 1c–d). Unlike the case of BR in water, BRs in the three proteins show constant ϕ and ψ angles of about −60° with small fluctuations for both (Table S1†). The standard deviations of ϕ and ψ angles of eUnaG (σϕ = 7.11°, σψ = 7.12°) smaller than those in UnaG (σϕ = 7.55°, σψ = 7.65°) and V2T (σϕ = 9.12°, σψ = 7.90°) also support the increased rigidity of BR in eUnaG compared to those in UnaG and V2T. Interestingly, even though the difference of standard deviations of ψ angles of BR in the three protein systems is less than that of ϕ angles, this difference indicates that the change of stability of the protein structures results from small but collective conformational changes throughout the whole protein, not just a few residues in the vicinity of the mutation site. Especially, it should be noted that the mutation site (Leu2 in eUnaG and Thr2 in V2T) is rather close to the exo-chromophore, the orientation of which is determined by the ϕ angle, instead of the endo-chromophore. Thus, the more (less) flexible ϕ and ψ angle rotations of BR in V2T (eUnaG) provide a clue as to the reduced (increased) fluorescence intensity of V2T (eUnaG). In general, the restricted rotational motion of BR in eUnaG makes the rotation-related nonradiative decay pathway less efficient, hence enhancing the fluorescence quantum yield and intensity. Especially, the fluorescence quantum yield is found to be sensitive upon the change of ϕ angle associated with the rotation of the exo-chromophore that is close to the mutation site. Not only the RG, but also the RMSDs of BR in the three proteins show consistent results. The average RMSD of BR in eUnaG is less than 0.22 Å, which is smaller than that in UnaG (0.29 Å) and V2T (0.35 Å). This trend in RMSD also reveals that the BR in eUnaG is in a more restricted environment than in UnaG or V2T.
To compare the conformational rigidities of bound BRs, we calculated the so-called B-factors of heavy atoms of BR (Fig. 2e), which are related to the thermal motions of those atoms.39–41 In the present case, it is proportional to the mean-squared atomic fluctuation of individual heavy atoms of BR, which is defined as
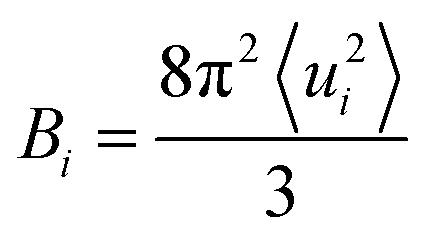 | (1) |
To investigate the energetics of the restricted conformations of BR, two-dimensional (2D) potentials of mean force (PMFs) with respect to ϕ and ψ angles were obtained from the MD trajectories. Here, the 2D PMF is defined as
ΔA(ϕ,ψ) = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P(ϕ,ψ)) ln(P(ϕ,ψ)) | (2) |
The PMF of BR in water (Fig. 3a) supports multiple possible conformers due to the rotational degrees of freedom along the two dihedral angles, ϕ and ψ. In contrast, there exists only one conformer of BR for each case of UnaG (Fig. 3c), eUnaG (Fig. 3d), and V2T (Fig. 3b), where the ψ and ϕ angles are about −60° for all. Quite interestingly, the curvatures of ψ- and ϕ-dependent PMFs of BR in eUnaG (Fig. 3d) are stiffer than those in UnaG and V2T (Fig. 3b and c), which is clear evidence showing that the BR in eUnaG is conformationally in a more constricted environment. Although the minimum energy ϕ and ψ angles do not change much upon the mutation from UnaG to eUnaG or V2T, the clear trends in force gradients suggest that the local environment around the BR is altered by the single mutation, even though these changes are not sufficient to make noticeable differences in electronic circular dichroism spectra (ECD, in Fig. S1†) obtained from the DFT calculations.34
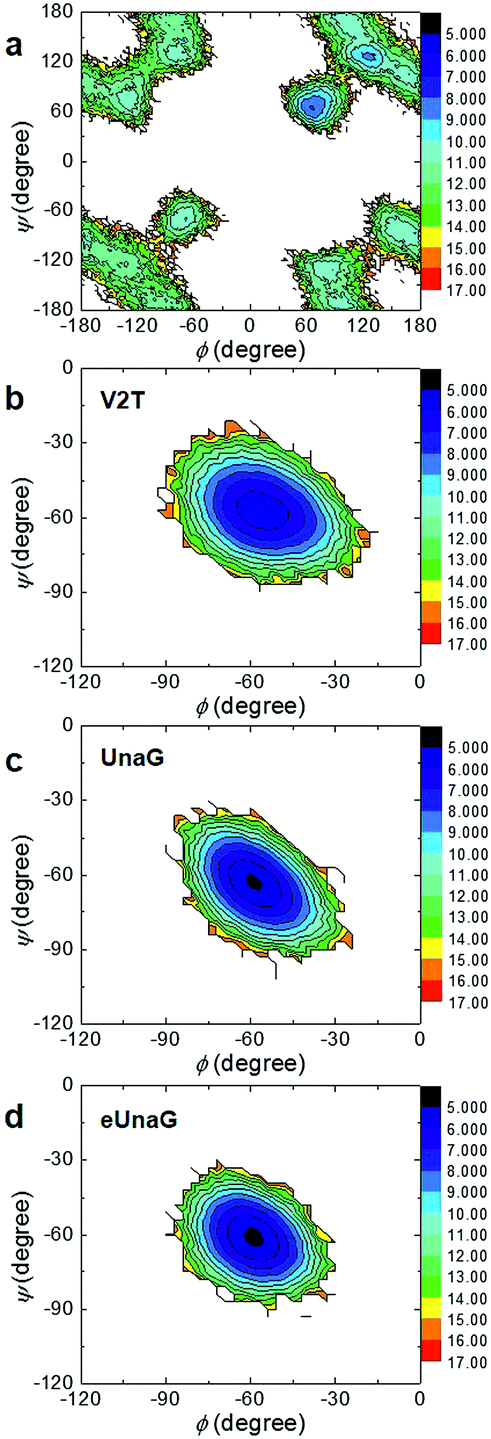 | ||
| Fig. 3 Two-dimensional potential of mean force (PMF) with respect to the two angles (ϕ and ψ angles) in water (a), V2T (b), UnaG (c), and eUnaG (d). Note the x- and y-axis scales in (a) differ from those in (b), (c), and (d). The unit of color scale (z-axis) is kcal mol−1. | ||
From the simulation and analysis results above, it is clear that the BR conformation is more rigid in eUnaG than in the other two proteins and the enhanced fluorescence stems from the increased dihedral rotational rigidity. However, it still remains unclear how the mutant proteins make the substrate BR structure more/less rigid and why the orientation of the endo-chromophore distant from the mutation site undergoes a change in orientation. To address these questions, we analyzed the environment of BR in the three proteins more in detail.
B. Local protein structure around BR
Many scattered protein residues (Asn57, Thr61, Glu77, Ser80, Asp81, Arg112, Arg132 and Tyr134) interact with various sites of the ligand in the reported PDB structure of UnaG.21 To examine how the amino acid residues adjacent to the bound BR restrict the motion of BR, we obtained the distance distribution of the protein atoms from each heavy atom of BR (Fig. S2†). For a given jth heavy atom of BR, we could find the closest protein atom and obtained the minimum distance value for that jth BR atom,42| RjBR–protein = min({|rjBR − rkprotein|}) for all k's | (3) |
A BR–protein interaction population map (B–P map) can be obtained by counting the encounter events of each BR atom to each protein atom from the MD trajectories (Fig. 4a–c). We counted the number of frames in which a particular pair of atoms becomes the closest neighbors among all BR–protein atom pairs. Then, it is normalized with respect to the total number of MD frames, which will be referred to as encounter probability. When the encounter probability between a chosen pair of atoms is 1, the two are always the nearest neighbors to each other. In Fig. 4a–c, the B–P encounter probability maps for the three protein systems are plotted along with the BR atom order in PDB as the x-axis and the protein atom order in PDB as the y-axis. Here, we do not consider the low (<0.2) encounter probability cases.
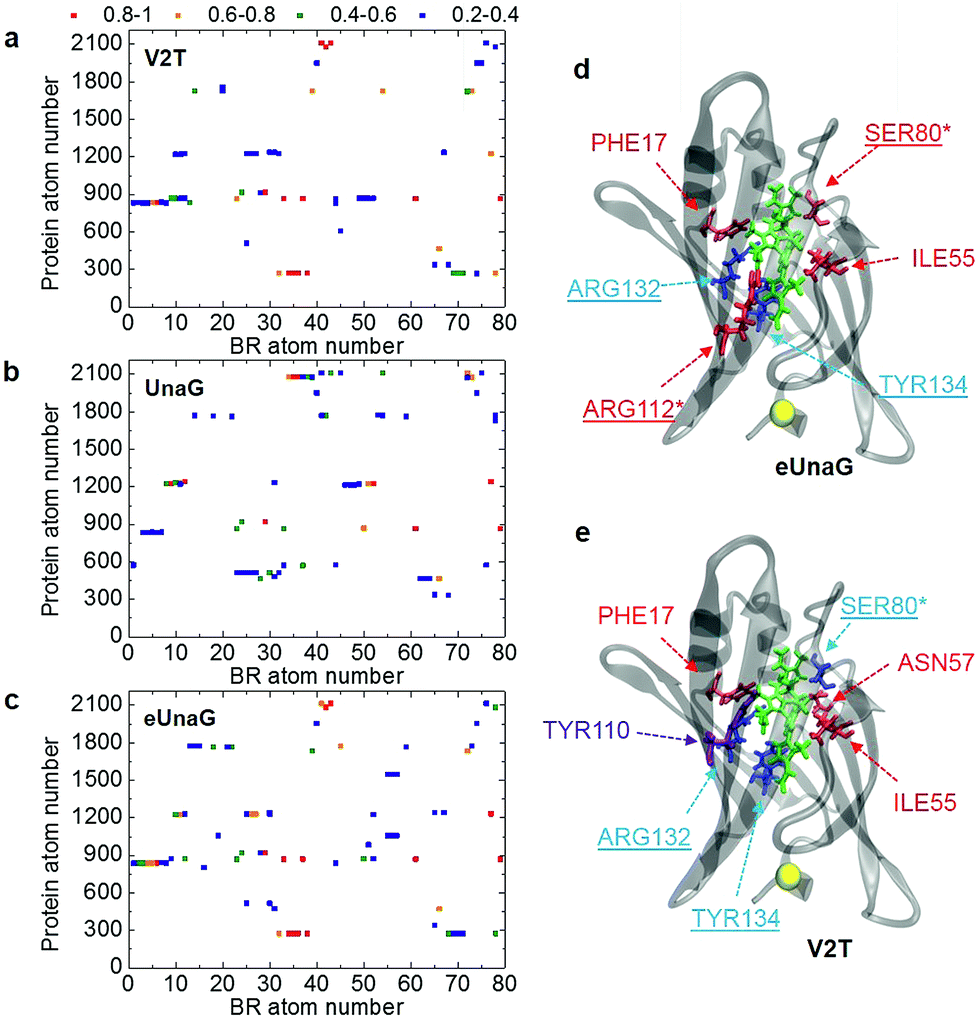 | ||
| Fig. 4 Probability of finding a chosen protein atom to be the closest atom to one of the BR atoms. This is referred to as encounter probability and the corresponding distribution with respect to protein and BR atom numbers is named the BR–protein encounter map. (a–c) The BR–protein encounter maps (B–P maps) for V2T, UnaG, and eUnaG are shown in these figures. The encounter probability from 0.8 to 1.0 is in red, from 0.6 to 0.8 is in orange, from 0.4 to 0.6 is in green, and from 0.2 to 0.4 is in blue. Those pairs of protein atom and BR atom, whose encounter probabilities are lower than 0.2, are left blank. (d and e) Protein residues binding to BR obtained from (a–c). In eUnaG (d) and V2T (e), the red-colored residues are more frequently found in the immediate vicinity of BR than those of UnaG, whereas the blue residues are less frequently found. A yellow ball in (d) and (e) indicates the mutation site. A violet colored residue, TYR110, is found to be a new residue close to BR only in the case of V2T (e). Underlined residues form H-bonds with BR and starred residues form water-mediated H-bonds. | ||
The B–P maps in Fig. 4 reveal that the BR-binding patterns of residues in eUnaG and V2T are different from those in UnaG, indicating that BRs in the three proteins are surrounded by different environments. Fig. 4d and e show residues in eUnaG and V2T with increased (red atoms) or decreased (blue atoms) encounter (interaction) probability in comparison to those in UnaG. In eUnaG, Phe17, Ser80, Ile55, and Arg112 strongly interact with BR, whereas those of Arg132 and Tyr134 with BR are weaker than those in UnaG. In V2T, Phe17, Ile55, and Asn57 interact stronger, and Ser80, Arg132, and Tyr134 interact weaker than those in UnaG. In addition, Tyr110 is found to be an additional interacting residue in V2T, though it does not participate in any interaction with BR in the case of UnaG. These changed interactions between BR and protein residues are found in various, widespread sites in the protein, not only in the vicinity of the mutation site, indicating the overall change of structure or stability of the mutated proteins.
Since many of the residues with increased/decreased interactions (underlined residues in Fig. 4d and e) are known to participate in H-bonding interactions with BR,21 we calculated the average H-bond number between BR and each protein (Table S2†). First, the average H-bond number between all atoms of BR and the protein is in excellent correlation with the conformational rigidity of BR, as eUnaG(35.18) > UnaG(34.09) > V2T(32.01). This means that the stronger H-bonding interaction between BR and eUnaG restricts the flexible motion of BR more than the other two proteins. Thus, the H-bonding environment is one of the dominant factors determining mutation-induced fluorescence enhancement of eUnaG. To correlate that with our B–P map, we also calculated the average H-bond number between all atoms of BR and each changed binding residue obtained from the B–P map (Fig. 4). Despite some exceptions (e.g., Ser80 of V2T, and Arg112 of eUnaG and V2T), the average number of H-bonds between BR and the highly binding residues in the B–P map (Table S2†) followed the trend of eUnaG > UnaG > V2T. These results support the mutation-induced changes in the environment near the BR as well as the representativeness of the B–P map.
C. Captured water molecules around BR
Among Glu77, Ser80, and Arg112 that notably interact with the ligand via hydrogen-bonding interactions with water molecules,21 the encounter probabilities of Ser80 and Arg112 with BR were increased in eUnaG and that of Ser80 was decreased in V2T (starred residues in Fig. 4d and e). To elucidate atomic details about H-bonding interactions between BR and water molecules, we obtained BR–water interaction population maps (B–W map) in the same way used to construct the B–P map. Fig. 5a–c display the B–W maps for the three proteins, where the color bar represents the BR–water H-bonding interaction probability. The B–W map reveals two interesting features: vertical blue lines and horizontal, color-varying lines. The two vertical blue lines correspond to water molecules that infrequently interact with the BR atoms near the end of the endo-chromophore (atom numbers of 28–30 and 62–63, Fig. 5d) in all the three proteins. These two vertical lines with low H-bonding interaction probability stem from bulk water molecules making contacts with the end atoms of the BR molecule that is exposed to solvent water (see Movie S1†). This region corresponds to the open top of the protein (Fig. S3a†), and therefore two vertical lines show the water exposure sites of BR (Fig. 5d). Since these water exposure sites exist in all three proteins, the H-bonding interactions of water molecules with those BR groups that are close to the open top of the proteins do not strongly affect the rigidity of BR. On the other hand, the horizontal high-population lines in Fig. 5a–c are mainly produced by a couple of water molecules frequently interacting with specific atoms of BR, which are mainly composed of the conjugate backbone of the exo-chromophore and the carboxyl group of the endo-chromophore, located in the closed bottom of the protein near the mutation site (Fig. 5d and S3b†). The horizontal lines with high encounter probabilities (red lines) of eUnaG indicate strong H-bonding interactions among the three protein systems, while the horizontal lines with low encounter probabilities of V2T clearly indicate weak H-bonding interactions with water molecules (Fig. 5a–c). These results indicate that two critical water molecules that are captured between the exo side of BR and neighboring protein residues play important roles in stabilizing BR–protein complexes (Fig. 5e and S4a and b†). In summary, while the water molecules near the endo-chromophore of BR pass through the water exposure site at the open top of the protein, the water molecules captured in the water-binding pocket mediate interactions between the exo-chromophore and the protein (Fig. 5d and e).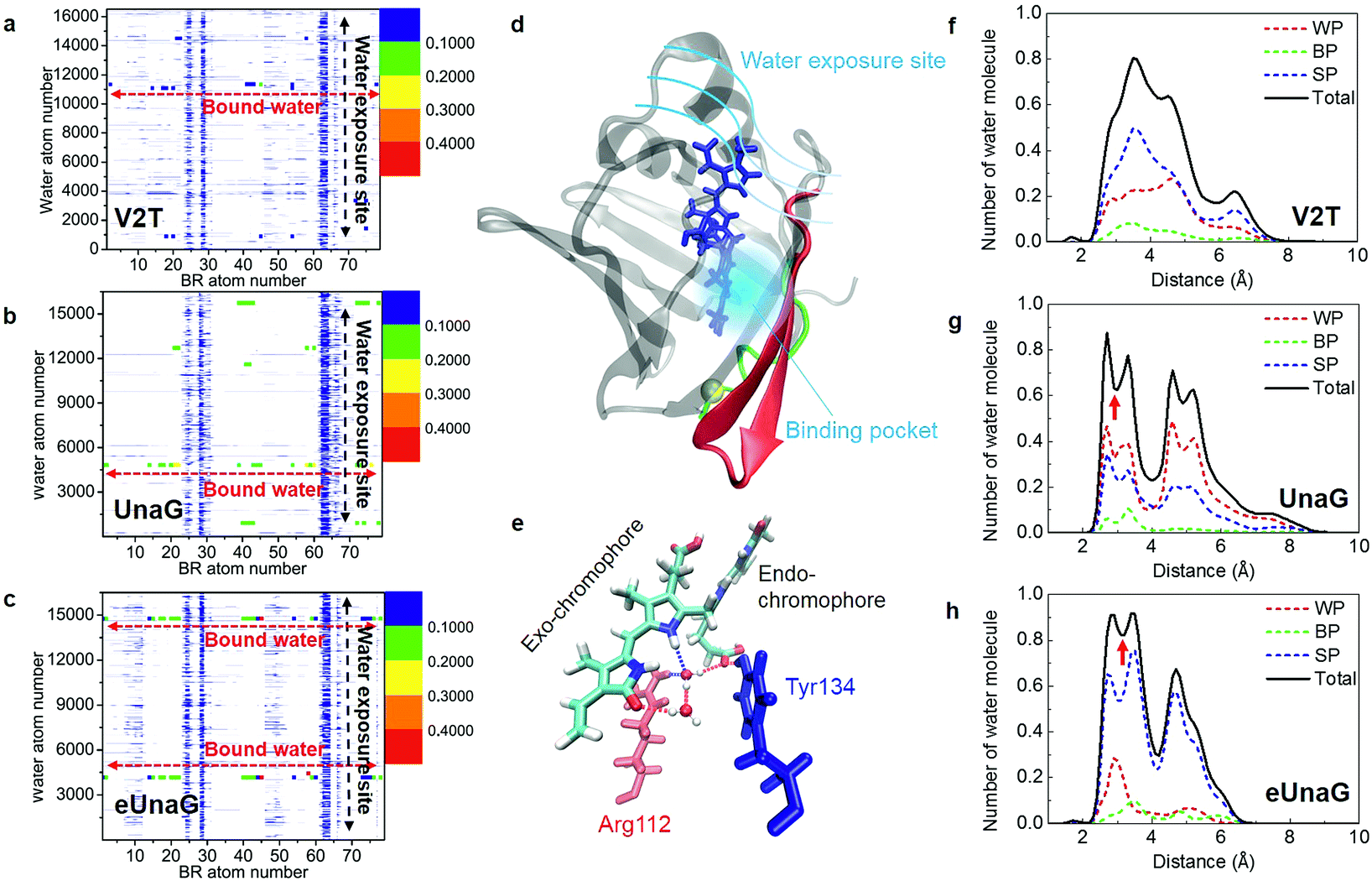 | ||
| Fig. 5 Probability of finding a specific water molecule interacting with BR. (a–c) The contour plots of BR–water interaction population distributions (B–W map) of V2T (a), UnaG (b), and eUnaG (c) are shown here. Strong horizontal lines appearing in the distribution map indicate that those water molecules are strongly bound to BR in the protein binding pocket. Noticeable vertical distributions mean that those BR atoms are interacting with all the water molecules in the periodic box because they are exposed to bulk-water. (d) Schematic of water-interacting loci of UnaG bound to BR (blue stick). The water binding pocket (light blue haze) encloses the end of the exo-chromophore and is formed by protein strands near the mutation site (yellow ball). The green-colored strand consists of protein residues 1–10 and the red colored β-strands correspond to protein residues 40–58. (e) Water-mediated H-bonding network between BR, two bound water molecules, Arg112 (red stick) and Tyr134 (blue stick) in eUnaG. H-bonds are plotted as dashed lines. (f–h) The number of oxygen atoms of the water molecules, obtained with eqn (3), in each shell between r Å and r +0.1 Å from each specific atom of BR, which are selected from the B–W maps in Fig. 5a–c for V2T (f), UnaG (g) and eUnaG (h). Black solid lines are counted from all the water molecules and dashed lines are from different H-bonding water configurations. Water-pointing water (WP, red dotted line) is a H-bond acceptor so that its O-atom forms H-bonds with BR, BR-pointing water (BP, green dotted line) is a double H-bond donor forming two H-bonds with BR, and BR surface-parallel water (SP, blue dotted line) straddles BR and protein. Unlike V2T, water distributions in UnaG (g) and eUnaG (h) clearly show minimum positions at about 3 Å (red arrow), indicating strongly interacting, stationary water layer. | ||
To examine how many water molecules are present in the vicinity of BR, we calculated the minimum distances between the oxygen atom of water and interacting BR atoms with eqn (3) and obtained BR–water distance distributions (Fig. 5f–h). In fact, water molecules in the binding pocket in V2T do not show any specific binding patterns, but are distributed broadly, indicating very weak BR–water interactions (Fig. 5f, black line). In contrast, the distributions of water molecules in UnaG and eUnaG show distinctive peaks, reflecting stable BR–water interactions (Fig. 5g and h, black lines). The first minimum position at 3 Å (red arrows in Fig. 5g and h) was chosen as the H-bond length threshold. By integrating the area <3 Å of the BR–water distance distributions, we counted the number of H-bonding water molecules as 3.78 water molecules in eUnaG, 3.50 in UnaG and 2.06 in V2T. Thus, BR in eUnaG is more stabilized by the increased number of H-bonding water molecules than those in UnaG and V2T. Especially those water molecules close to the exo-chromophore frequently participate in H-bonding interactions in the B–W map (red dots in Fig. 5c). In contrast, the B–W map of V2T does not show such specific H-bonding interaction patterns (all dots are blue in Fig. 5a). Indeed, the broad distribution of water–BR distances of V2T (Fig. 5f) indicates large amplitude spatial fluctuations of water molecules, which then affects the BR flexibility.43
To characterize the roles of the H-bonding water molecules, we categorized the H-bonding type of water molecules such as H-bond donor and acceptor for BR or protein. In our previous work,42 water configurations interacting with a protein were classified into three groups that are water-pointing water (H-bond accepting water from the protein), protein-pointing water (H-bond donating water to the protein) and protein surface-parallel water (one proton is H-bond donating to the protein and another proton is H-bond donating to bulk water). Applying this classification to the bound water molecules interacting with BR and protein, we classified three groups: (1) water-pointing water (WP), in which water oxygen points to the H-bond donor atoms of BR; (2) BR-pointing water (BP), in which two water H-atoms point to the H-bond acceptor atoms of BR; (3) BR surface-parallel water (SP), in which one water H-atom interacts with an H-bond acceptor atom of BR and another water H-atom interacts with another water molecule or H-bond acceptor atom of protein. In Fig. 5f–h, the number distributions of WP, BP, and SP are plotted as red-, green-, and blue-dotted lines, respectively. In all cases, because the BR in the cavity has 8 H-bond donor atoms and 3 H-bond acceptor atoms, BP does not populate much. In V2T, the SP population is almost twice higher than that of WP (Fig. 5f). All three distributions of WP, BP, and SP water molecules in V2T suggest that those water molecules are weakly interacting with BR. In contrast, those distributions of UnaG and eUnaG show clearly different patterns from those of V2T. First, in UnaG, the WP population is higher than the SP population, indicating that the bound water molecules are mainly H-bond acceptors (Fig. 5g). In eUnaG, the SP population is significantly higher than WP (Fig. 5h), reflecting the strong H-bonding interactions of water molecules bridging the carbonyl group of the exo-chromophore and the carboxyl group of the endo-chromophore with the neighboring protein residues (Fig. 5e). These two bound water molecules in eUnaG are capable of forming stable H-bonds with the oxygen atoms of the carbonyl and carboxyl groups, which further inhibit the dihedral rotation of BR. The bound water molecules rarely escape from the cavity, resulting in the sharp and high population distribution in the distance range from 2 to 3.5 Å from the specific atoms of BR.
To estimate how long each H-bonding interaction lasts, we considered water molecules within 3 Å from all the interacting BR atoms (Fig. S4c–e†). H-bonded water molecules associated with the horizontal lines in the B–W maps (Fig. 5a–c) will be denoted as bound water 1 and 2. In eUnaG, the two bound water molecules are located in different regions in the BR binding pocket (Fig. S4a and Movie S2†). In contrast, the bound water molecules in UnaG are distributed in a wider region than those in eUnaG. Those water molecules in the UnaG pocket sometimes move out of the pocket or make jumps from one to the other regions around the BR (Fig. S4b†). Thereby, the water molecules in the UnaG pocket tend to agitate BR strongly than those in eUnaG. The two captured water molecules in eUnaG remain H-bonded for 90% of the MD simulation time (Fig. S4e†), but those of UnaG and V2T have significantly low probabilities of forming H-bonds, which are 0.5 and 0.2, respectively (Fig. S4c and d†), which indicates that water molecules in the binding pocket of UnaG and V2T make frequent escapes from the pocket during more than half of the simulation time.
To quantitatively compare the motions of the two bound water molecules, we obtained velocity distributions of the oxygen atoms of the two bound water molecules (Fig. 6a–c). Fig. 6c shows the velocity distributions of the bound water molecules 1 and 2 and two free water molecules randomly chosen from bulk water in the aqueous eUnaG solution. Both bound water molecules 1 and 2 in the eUnaG solution have similarly very low velocities with narrow distributions. In contrast, bound water molecules in UnaG have overall low velocities with differently broad distributions (Fig. 6b). It is interesting to note that the bound water molecules in the V2T system are not significantly different from bulk water, albeit slightly slower (Fig. 6a). The slowest bound water molecules in the three protein systems are compared in Fig. 6d with the maximum points at 54 m s−1 for eUnaG, 60 m s−1 for UnaG, and 86 m s−1 for V2T. This is expected from the previous interaction maps shown in Fig. 5a–c, because strong interaction restricts the motion of water molecules (Fig. 5h and S4a†) and weakly binding water molecules occasionally escape from the binding pocket (Fig. S4b†). The results reveal that the mobility of adjacent water molecules can be related to the rigidity of the ligand and subsequently the fluorescence intensity.
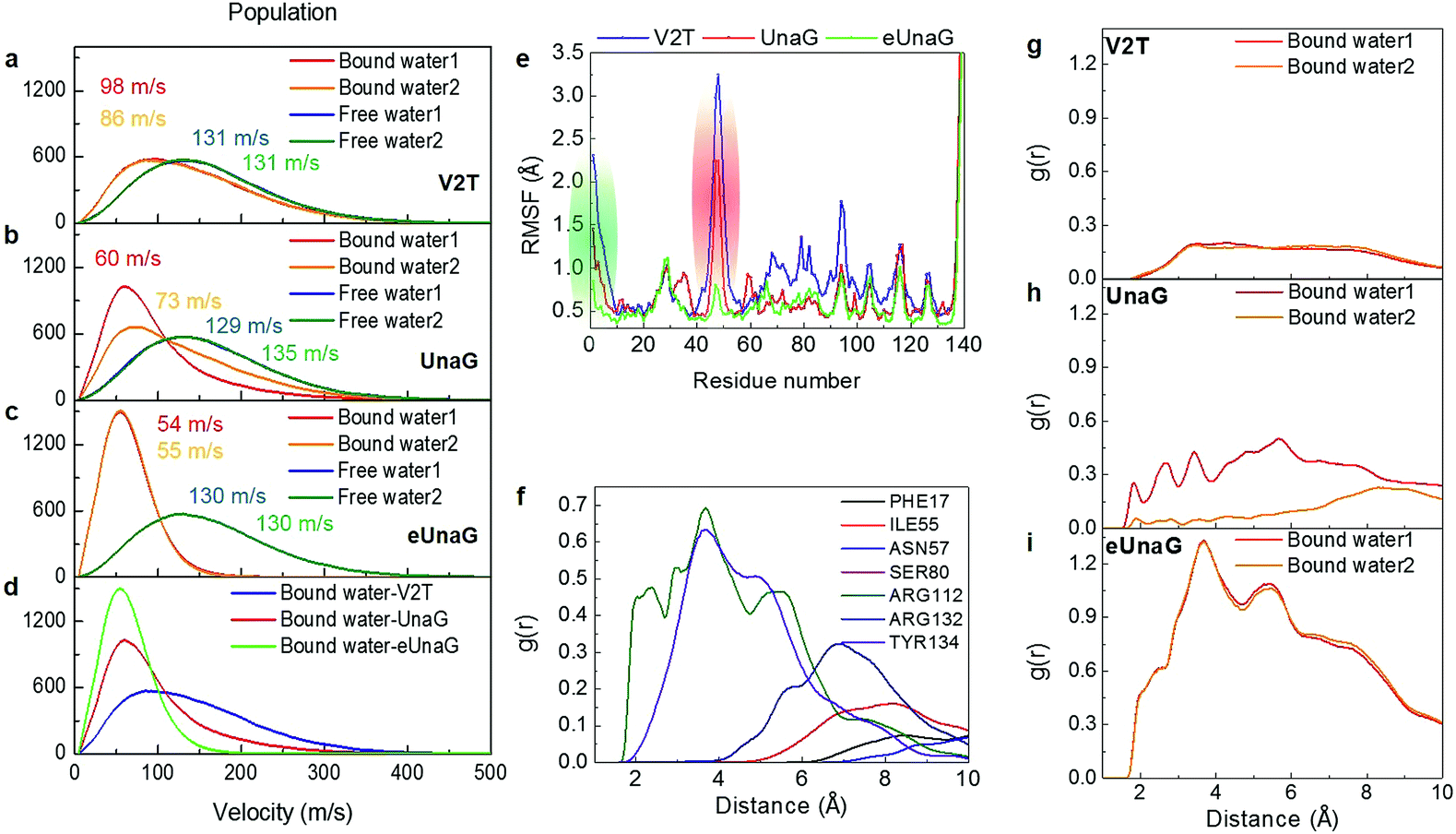 | ||
| Fig. 6 Velocity distributions and radial distributions of bound water molecules. (a–c) Velocity distributions of bound water molecules 1 and 2 inside the binding pocket. They are compared with those of free water molecules. Velocities (in m s−1) at the maximum of each velocity distribution are also given in each panel. (d) Comparison of the velocity distributions of the slowest bound water molecule in the three protein systems. (e) Root-mean-square fluctuation (RMSF) of protein residues. Green and red regions correspond to the green and red structures in Fig. 5d. (f) RDF between the bound water molecule 1 in eUnaG and all atoms of the interacting amino-acid residues that are chosen from the B–P map shown in Fig. 4. (g–i) Radial distribution function (RDF) between the bound water molecules and all the protein atoms for V2T (g), UnaG (h), and eUnaG (i), respectively. | ||
To correlate the water mobility and the protein stability, we calculated root-mean-square fluctuations (RMSFs) of protein residues (Fig. 6e). eUnaG is the most stable in all the protein residues, and V2T is the least stable. In particular, residue numbers of 1–10 and 40–58 show particularly large differences in RMSF. The residues 1–10 (green residues in Fig. 6e) constitute the N-terminus strand including the mutation site and the residues 40–58 (red residues in Fig. 6e) are on the β-sheets 2 and 3 next to the mutation site. This observation is consistent with the simulation result reported by Yeh et al.28 who showed the correlated distance fluctuations among the mutation site, Met51, and BR. Our results summarized in Fig. 5d further show how stabilization or destabilization of the BR structure occurs through the multifaceted changes involved in the protein secondary structures at the mutated N-terminus strand (green strand in Fig. 5d) and the neighboring two β-sheets (red ribbons in Fig. 5d) as well as bound water molecules in the binding pocket (blue region in Fig. 5d) near the exo-chromophore of BR. The increased fluctuation of the protein structure caused by the mutation from Val to Thr in the case of V2T can make the bound water molecules more mobile, which in turn induces an increased extent of fluctuation of the exo-chromophore. The enhanced flexibility of BR induces faster nonradiative relaxation of the excited BR, which reduces the fluorescence intensity in the V2T system. In contrast, the stabilized protein structure in eUnaG tightly entraps and slows down the captured water molecules that can stabilize the H-bonding network, resulting in the increase in the rigidity of BR, leading to the enhanced fluorescence intensity.
To further quantify the contribution of the bound water molecules to BR rigidity through H-bond chains that link BR, water, and protein,21 we obtained radial distribution functions (RDFs) between the oxygen atoms of the bound water molecule and protein atoms (Fig. 6g–i). Interestingly, the RDF of eUnaG exhibits strong radial correlation in the distance less than 10 Å (Fig. 6i), while the other two proteins show no notable correlation peaks in the same distance range (Fig. 6g and h). We then performed residue-specific RDF analyses by separately calculating RDFs between the bound water molecule and specific water-interacting protein residues for the eUnaG system (Fig. 6f). Since the velocity distribution and water-protein RDFs of both the bound water molecules are almost indistinguishable (Fig. 6c and i), we plot the residue-specific RDF only for the bound water 1 (Fig. 6f). Among 7 BR-interacting residues, Arg112 and Tyr134 make strong interactions with the bound water molecules, reflecting that these two residues form a H-bond network of BR–water–protein (Table S2†). The additional water-mediated H-bond of Arg112 effectively strengthens the interactions between the conjugated backbone of BR and Arg112 of eUnaG (Fig. 5e). The carboxylic group of the endo-chromophore is affixed by two H-bonds with Tyr134 and a bound water (Fig. 5e). The bound water molecules either mediate H-bond chains of ligand–water–protein or supplement additional H-bondings, thereby further stabilizing the dihedral rotation of BR.
To study the electronic structures of both ground and excited states and the effect of the H-bonded water molecules on the electronic structures of BR in three protein systems, we carried out time-dependent DFT calculations for BR conformations taken from MD trajectories. We first obtained molecular orbitals of quantum states involved in the absorption and fluorescence for BR without and with two H-bonded water molecules (Tables S3–S4 and Fig. S5–S6†). The quantum transition probability determined by the associated oscillator strength from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) is found to be about two orders of magnitude smaller than that associated with the transition to the second lowest excited state (see Tables S3 and S4†). Since the dominant transition in all cases was the electronic transition 2 which has the highest oscillator strength and the transition wavelength close to the experimental absorption wavelength, we focused on this transition to the excited state 2, which is given by a linear combination of three transition configurations. Two of them involve electron redistributions within either the exo- or endo-chromophore, but the remaining configuration resulting from the transition from HOMO−1 to LUMO+1 involves charge transfer from the endo-chromophore to the exo-chromophore (Fig. S5 and S6†), which suggests non-negligible electronic couplings between the two constituent chromophores.
In both cases of BR's with and without water, there are no noticeable effects coming from the ϕ and ψ angle differences in the three BR–protein complexes, indicating that the small ϕ and ψ angle changes do not induce any notable change in the electronic structure of BR. But, the effect of bound water molecules on the electronic structure of BR is not negligible. For all three protein systems, BR with the two bound water molecules shows slightly lower transition energies and slightly higher oscillator strengths (Tables S3 and S4†). However, the differences in electronic transitions due to BR's H-bonding interaction with water molecules are too subtle to explain the large differences in fluorescence quantum yield, which are better explained in terms of the interplay of the H-bonding structure and dynamics of bound water molecules with enhanced or decreased rigidity of the bound ligand, BR, (Fig. 5 and 6).
4. A few remarks and summary
In the present work, we report a variety of structural analysis results of three BR-binding proteins in water, in order to ultimately elucidate the mechanism of the enhanced/reduced fluorescence intensity of the non-covalently bound ligand, which is induced by a single mutation at the N-terminus of the UnaG protein. First, we estimated the conformational rigidity of BR and found an enhanced rigidity of BR in eUnaG, especially in terms of stiffness of the potential energy surfaces associated with the main dihedral angles of BR. The chromophore rigidity can be the direct cause of the reduced non-radiative decay rate, which consequently increases the fluorescence quantum yield of BR in eUnaG. The rigidity increase of BR was found to be notably large on the exo-chromophore part the orientation of which is determined by the ϕ angle, even though it is distant from the mutation site. While the smaller, but noticeable, increase of rigidity of the endo-chromophore indicates stronger contact between the mutated residue and BR, the larger increase of rigidity of the exo-chromophore indicates additional weak restraints that stabilize the protein in a wide range. The stability changes were found not only in the vicinity of the mutated residue but also at various residues spread out throughout the protein cavity in varying degrees. Not only the protein environment surrounding the ligand varied in the three proteins, but also the water molecules captured near the mutation site that mediate the H-bonding between the protein and the ligand exhibit different mobilities in the three proteins. Overall, the global changes of the multiple players (protein residues and water molecules) in the environment surrounding BR caused the rigidity change of BR.Recently, Shitashima et al.44 reported a possible existence of two distinct fluorescence states of holoUnaG (BR–UnaG). They claimed that the two different holoUnaG states with different fluorescence intensities share the same CD and absorption spectra. From the spectroscopic data, they concluded that the chemical structure and conformation of the encapsulated BR and the secondary structures of the two different holoUnaGs remain very similar. Although they didn't consider eUnaG, their experimental results appear to be similar to those of our study on UnaG, eUnaG, and V2T. The CD spectra of BR in the three proteins calculated by the quantum chemical calculation method are not different from each other (Fig. S1†), even though the average ψ angles differ from each other by about 6.5° (Table S1†). Thus, the average angle differences between BRs in the three proteins are negligible. Our interaction population mapping suggests that the changed environment around BR (e.g., the binding degree of protein interaction residues and the mobilities and H-bonding structure of bound water molecules), rather than the conformational change of protein and BR, is the main cause of the change of rigidity of BR, consistent with the findings of Shitashima et al.44
Biliverdin-induced fluorescent proteins share similarity in the chromophore structure with UnaG (Fig. 1b and c). The only difference between biliverdin and bilirubin is the link between the central C1 and C2 atoms being double and single bond, respectively. But, this small difference makes the mechanism of fluorescence enhancement quite different. BV can undergo cis–trans transition upon light absorption, but the endo/exo chromophores in BR can freely rotate without light exposure. Also, BV is linked to the fluorescent protein via a covalent thioether bond at the end of the BV molecule, while UnaG holds the BR through multiple H-bonding interactions all over the BR chromophore, without any covalent linkage. Therefore, engineering BV-binding proteins for fluorescence enhancement has often involved preventing the photoisomerization of BV by holding the other end of the molecule or blocking the free space required for the rotational motion. In contrast, UnaG engineering would require tuning of the entire barrel arrangement of the protein and the dynamics of H-bonding water molecules in the cavity.
Water molecules play a crucial role in mediating ligand–protein interactions. High-resolution crystallographic studies have revealed that the interactions of water molecules in ligand–protein complexes are energetically more favorable than bulk solvent interactions.45 A single water molecule can increase the ligand binding affinity when overcoming the entropic penalty from restricting water molecules by the enthalpic gain from the formation of hydrogen bonds.46 Therefore, for a water molecule to increase the binding free energy, the binding cavity should provide more H-bonding partners at the right proximities and orientations. eUnaG offers such a binding cavity with Arg112 and Tyr134 precisely positioned to form stable H-bonds with water molecules and the ligand. Indeed, the two bound water molecules lower transition energies and enhance oscillator strengths associated with the electronic transitions (Tables S3 and S4†). Arg112 is the only charged residue in the cavity whose interaction is enhanced in eUnaG (Fig. 4d), which can potentially make a charge transfer interaction with BR. In fact, the molecular orbitals of HOMO and LUMO+1, which participate the most among the three transitions constituting the excited state from which fluorescence photon is emitted (Tables S3 and S4†), are mainly located at the conjugated backbone of the exo-chromophore (Fig. S5 and S6†) with which Arg112 can interact (Fig. 5e). Thus, Arg112 has two counteracting roles in fluorescence: charge transfer destabilizes the excited state largely localized on the exo-chromophore; H-bonds with two water molecules stabilize the excited state. In eUnaG, the distance and orientation of Arg112 may be suited for stabilizing the water-mediated H-bonding network while preventing charge transfer. Thus, we here suggest that the fluorescence quantum yield can be further increased from eUnaG by substituting Arg112 with a noncharged, H-bonding residue. In addition, many H-bond donating and accepting groups in the bilirubin ligand offer potential sites for increasing the enthalpic gain from H-bonds. Therefore, UnaG is a powerful system to study the roles of bound waters in ligand–protein interaction because the facile experimental readout of fluorescence emission can sense the free-energetic favor or disfavor.
In conclusion, the single mutation of UnaG to produce eUnaG or V2T affects the overall protein structure stability, mostly in the strands near the mutation site, which leads to an increase or decrease of the rigidity of the ligand through various influences of the surroundings (i.e., binding residues and captured water molecules). The present work shows that a seemingly negligible, indirect change of a single-residue mutation on a small fluorescent protein with an open barrel structure that noncovalently binds to a large elongated chromophore spanning the entire length of the binding cavity can strongly affect the structural and spectral properties, which clearly provides a new insight for engineering such noncovalent ligand-inducible fluorescent proteins.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by IBS-R023-D1. We thank Dr Jonggu Jeon for helpful comments.Notes and references
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- J. D. Pedelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 1170 CrossRef CAS.
- X. K. Shu, A. Royant, M. Z. Lin, T. A. Aguilera, V. Lev-Ram, P. A. Steinbach and R. Y. Tsien, Science, 2009, 324, 804–807 CrossRef PubMed.
- H. Dortay and B. Mueller-Roeber, Microb. Cell Fact., 2010, 9, 29 CrossRef PubMed.
- G. S. Filonov, K. D. Piatkevich, L. M. Ting, J. H. Zhang, K. Kim and V. V. Verkhusha, Nat. Biotechnol., 2011, 29, 757 CrossRef CAS PubMed –U133.
- A. Miyawaki, Nat. Methods, 2016, 13, 729–730 CrossRef CAS PubMed.
- K. G. Chernov, T. A. Redchuk, E. S. Omelina and V. V. Verkhushaa, Chem. Rev., 2017, 117, 6423–6446 CrossRef CAS PubMed.
- B. T. Bajar, E. S. Wang, A. J. Lam, B. B. Kim, C. L. Jacobs, E. S. Howe, M. W. Davidson, M. Z. Lin and J. Chu, Sci. Rep., 2016, 6, 20889 CrossRef CAS PubMed.
- A. J. Lam, F. St-Pierre, Y. Y. Gong, J. D. Marshall, P. J. Cranfill, M. A. Baird, M. R. McKeown, J. Wiedenmann, M. W. Davidson, M. J. Schnitzer, R. Y. Tsien and M. Z. Lin, Nat. Methods, 2012, 9, 1005–1012 CrossRef CAS PubMed.
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. K. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS PubMed.
- R. Nifosi and V. Tozzini, Proteins, 2003, 51, 378–389 CrossRef CAS PubMed.
- A. A. Samma, C. K. Johnson, S. A. Song, S. Alvarez and M. Zimmer, J. Phys. Chem. B, 2010, 114, 15362–15369 CrossRef CAS PubMed.
- L. Viani, C. Curutchet and B. Mennucci, J. Phys. Chem. Lett., 2013, 4, 372–377 CrossRef CAS PubMed.
- F. V. Escobar, T. Hildebrandt, T. Utesch, F. J. Schmitt, I. Seuffert, N. Michael, C. Schulz, M. A. Mroginski, T. Friedrich and P. Hildebrandt, Biochemistry, 2014, 53, 20–29 CrossRef PubMed.
- K. Anwer, R. Sonani, D. Madamwar, P. Singh, F. Khan, K. Bisetty, F. Ahmad and M. I. Hassan, J. Biomol. Struct. Dyn., 2015, 33, 121–133 CrossRef CAS PubMed.
- R. Heim and R. Y. Tsien, Curr. Biol., 1996, 6, 178–182 CrossRef CAS PubMed.
- A. W. Nguyen and P. S. Daugherty, Nat. Biotechnol., 2005, 23, 355–360 CrossRef CAS PubMed.
- H. W. Ai, J. N. Henderson, S. J. Remington and R. E. Campbell, Biochem. J., 2006, 400, 531–540 CrossRef CAS PubMed.
- M. E. Auldridge, K. A. Satyshur, D. M. Anstrom and K. T. Forest, J. Biol. Chem., 2012, 287, 7000–7009 CrossRef CAS PubMed.
- A. Baldridge, S. H. Feng, Y. T. Chang and L. M. Tolbert, ACS Comb. Sci., 2011, 13, 214–217 CrossRef CAS PubMed.
- A. Kumagai, R. Ando, H. Miyatake, P. Greimel, T. Kobayashi, Y. Hirabayashi, T. Shimogori and A. Miyawaki, Cell, 2013, 153, 1602–1611 CrossRef CAS PubMed.
- A. N. Glazer, Mol. Cell. Biochem., 1977, 18, 125–140 CrossRef CAS PubMed.
- V. T. Oi, A. N. Glazer and L. Stryer, J. Cell Biol., 1982, 93, 981–986 CrossRef CAS PubMed.
- W. A. Sidler, in The Molecular Biology of Cyanobacteria, ed. D. A.Bryant, Springer Netherlands, Dordrecht, 1994, pp. 139–216, DOI:10.1007/978-94-011-0227-8_7.
- J. A. He, Y. Z. Hu and L. J. Jiang, J. Am. Chem. Soc., 1996, 118, 8957–8958 CrossRef CAS.
- A. N. Glazer, J. Appl. Phycol., 1994, 6, 105–112 CrossRef CAS.
- S. Hayashi and Y. Toda, Fish. Sci., 2009, 75, 1461–1469 CrossRef CAS.
- J. T. H. Yeh, K. Nam, J. T. H. Yeh and N. Perrimon, Sci. Rep., 2017, 7, 41619 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- D. A. Case et al. , AMBER 16, University of California, San Francisco, 2016 Search PubMed.
- J. M. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- L. X. Dang and P. A. Kollman, J. Phys. Chem., 1995, 99, 55–58 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Bonnett, J. E. Davies, M. B. Hursthouse and G. M. Sheldrick, Proc. R. Soc. London, Ser. B, 1978, 202, 249–268 CrossRef CAS.
- S. E. Braslavsky, A. R. Holzwarth and K. Schaffner, Angew. Chem., Int. Ed., 1983, 22, 656–674 CrossRef.
- R. V. Person, B. R. Peterson and D. A. Lightner, J. Am. Chem. Soc., 1994, 116, 42–59 CrossRef CAS.
- D. A. Lightner, D. L. Holmes and A. F. McDonagh, J. Biol. Chem., 1996, 271, 2397–2405 CrossRef CAS PubMed.
- D. W. J. Cruickshank, Acta Crystallogr., 1956, 9, 747–753 CrossRef CAS.
- N. Reuter, H. Lin and W. Thiel, J. Phys. Chem. B, 2002, 106, 6310–6321 CrossRef CAS.
- P. A. Janowski, D. S. Cerutti, J. Holton and D. A. Case, J. Am. Chem. Soc., 2013, 135, 7938–7948 CrossRef CAS PubMed.
- E. Lee, J. H. Choi and M. Cho, Phys. Chem. Chem. Phys., 2017, 19, 20008–20015 RSC.
- M. Feliks, C. Lafaye, X. K. Shu, A. Royant and M. Field, Biochemistry, 2016, 55, 4263–4274 CrossRef CAS PubMed.
- Y. Shitashima, T. Shimozawa, A. Kumagai, A. Miyawaki and T. Asahi, Biophys. J., 2017, 113, 2805–2814 CrossRef CAS PubMed.
- M. Levitt and B. H. Park, Structure, 1993, 1, 223–226 CrossRef CAS PubMed.
- J. E. Ladbury, Chem. Biol., 1996, 3, 973–980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03558j |
| This journal is © The Royal Society of Chemistry 2018 |