
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous nitrosylation and N-nitrosation of a Ni-thiolate model complex of Ni-containing SOD†
Phan T.
Truong
 a,
Ellen P.
Broering
a,
Stephen P.
Dzul
b,
Indranil
Chakraborty
c,
Timothy L.
Stemmler
b and
Todd C.
Harrop
*a
a,
Ellen P.
Broering
a,
Stephen P.
Dzul
b,
Indranil
Chakraborty
c,
Timothy L.
Stemmler
b and
Todd C.
Harrop
*a
aDepartment of Chemistry, Center for Metalloenzyme Studies, The University of Georgia, Athens, Georgia 30602, USA. E-mail: tharrop@uga.edu
bDepartments of Pharmaceutical Sciences, Biochemistry, and Molecular Biology, Wayne State University, Detroit, Michigan 48201, USA
cDepartment of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, USA
First published on 17th September 2018
Abstract
Nitric oxide (NO) is used as a substrate analogue/spectroscopic probe of metal sites that bind and activate oxygen and its derivatives. To assess the interaction of superoxide with the Ni center in Ni-containing superoxide dismutase (NiSOD), we studied the reaction of NO+ and NO with the model complex, Et4N[Ni(nmp)(SPh-o-NH2-p-CF3)] (1; nmp2− = dianion of N-(2-mercaptoethyl)picolinamide; −SPh-o-NH2-p-CF3 = 2-amino-4-(trifluoromethyl)benzenethiolate) and its oxidized analogue 1ox, respectively. The ultimate products of these reactions are the disulfide of −SPh-o-NH2-p-CF3 and the S,S-bridged tetrameric complex [Ni4(nmp)4], a result of S-based redox activity. However, introduction of NO to 1 affords the green dimeric {NiNO}10 complex (Et4N)2[{Ni(κ2-SPh-o-NNO-p-CF3)(NO)}2] (2) via NO-induced loss of nmp2− as the disulfide and N-nitrosation of the aromatic thiolate. Complex 2 was characterized by X-ray crystallography and several spectroscopies. These measurements are in-line with other tetrahedral complexes in the {NiNO}10 classification. In contrast to the established stability of this metal-nitrosyl class, the Ni–NO bond of 2 is labile and release of NO from this unit was quantified by trapping the NO with a CoII–porphyrin (70–80% yield). In the process, the Ni ends up coordinated by two o-nitrosaminobenzenethiolato ligands to result in the structurally characterized trans-(Et4N)2[Ni(SPh-o-NNO-p-CF3)2] (3), likely by a disproportionation mechanism. The isolation and characterization of 2 and 3 suggest that: (i) the strongly donating thiolates dominate the electronic structure of Ni-nitrosyls that result in less covalent Ni–NO bonds, and (ii) superoxide undergoes disproportionation via an outer-sphere mechanism in NiSOD as complexes in the {NiNO}9/8 state have yet to be isolated.
Introduction
Nitric oxide (NO) and its derivatives (termed reactive nitrogen species or RNS) play a vital role in a variety of mammalian (and in some cases bacterial) physiological and pathological processes.1–4 Additionally, this gaseous free radical has applications in fundamental research, especially in bioinorganic chemistry, where it is utilized as a structural/spectroscopic probe of O2 (and other reactive oxygen species, e.g., O2˙− and H2O2) binding/activating metalloenzymes.5–9 In general, this approach is employed because metal-nitrosyl (MNO) bonds are highly covalent, and hence more stable, than metal–dioxygen (M–O2) adducts.10 The use of NO as an O2 analogue is based on similar electronic structures between these diatoms and their reduced derivatives.3 For example, 3NO− (termed the nitroxyl anion), the one electron reduced analogue of NO, is isoelectronic with O2 with two unpaired π* electrons in the HOMO. Additionally, NO, while not isoelectronic with O2˙−, has the same ground state electronic structure with a singly occupied π* MO. Thus, NO interactions with the active sites of O2-activating/ROS-breakdown enzymes report coordination (inner-sphere substrate binding) and the extent of substrate bond activation from vibrational spectroscopic measurements of the N–O and M–NO stretching frequencies.Since 2009, our lab has designed and constructed numerous low molecular weight models of the active site of Ni-containing superoxide dismutase (NiSOD).11–18 NiSOD is an unprecedented SOD due to NiIII/II-coordination to cysteinato-S (CysS) and peptido-N donors (Chart 1), the former of which is susceptible to oxidative modification by the substrate (O2˙−) and products (O2 and H2O2) of the SOD catalyzed reaction.19,20 Few models employ ligands with the correct spatial disposition and electronic nature of the unique N3S2 donor set found in the active site.21–23 Moreover, fewer report reversible electrochemical and/or spectroscopic evidence for the NiIII oxidation state due to redox associated with the coordinated thiolates. One model from our lab, namely Et4N[Ni(nmp)(SPh-o-NH2-p-CF3)] (1; nmp2− = dianion of the N2S ligand N-(2-mercaptoethyl)picolinamide; see Chart 1) displays a reversible redox-event at −0.43 V (vs. Fc/Fc+ in DMF) that, based on EPR, UV-vis, MCD, and DFT computations, represents the electrochemical conversion from NiII in 1 to a NiII-thiyl ↔ NiIII-thiolate resonance species termed 1ox.16 Because substrate binding to Ni in NiSOD has not been defined, although most reports favor an outer-sphere mechanism,15,24 we were curious to use NO as an O2˙− probe to define potential intermediates that may be traversed in the NiSOD mechanism. We report here, for the first time, the reactions and product characterization of NO (and NO+) with 1 and the well-defined analogue of NiSODox (1ox). NO/NO+ oxidize the aromatic thiolate ligand in 1ox and 1, respectively. However, introduction of NO to 1 affords the green dimeric {NiNO}10 complex (Et4N)2[{Ni(κ2-SPh-o-NNO-p-CF3)(NO)}2] (2) via NO-induced loss of nmp2− as the disulfide and N-nitrosation of the aromatic thiolate (Chart 1). While 2 bears little resemblance to NiSOD, its formation indicates how reactive NiSOD models such as 1 are in the presence of redox-active diatoms and suggest similar paths for other biological Ni-thiolate sites. Additionally, 2 contains a labile Ni–NO bond, a new feature for the {NiNO}10 formulation that appears to be controlled by the presence of the thiolate ligands. We describe the synthesis, spectroscopy, electronic structure, reactivity and mechanistic insight into the formation of the Ni-nitrosyl in this account.
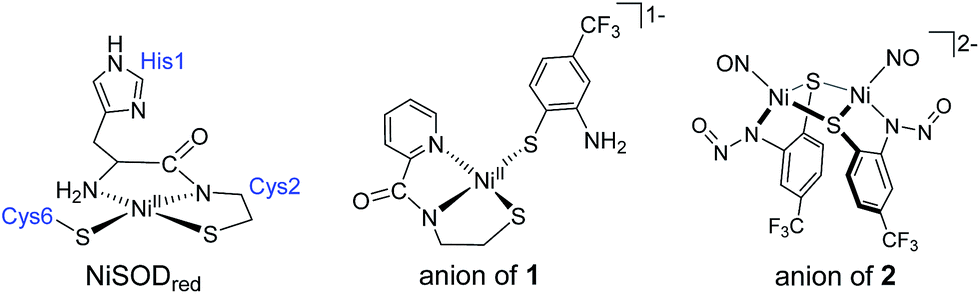 | ||
| Chart 1 Structures of the active site of NiSODred (left; His1 coordinates to NiIII in NiSODox), the anion of the NiSOD model complex Et4N[Ni(nmp)(SPh-o-NH2-p-CF3)] (1) (center; nmp2− = dianion of the N2S ligand N-(2-mercaptoethyl)picolinamide), and the anion of the {NiNO}10 complex (Et4N)2[{Ni(κ2-SPh-o-NNO-p-CF3)(NO)}2] (2). | ||
Results and discussion
In the anticipation of isolating a Ni-nitrosyl as an analogue of a potential Ni-superoxo/peroxo catalytic intermediate of NiSOD, we examined the reaction of 1 with NOBF4 and in situ prepared 1ox with NO (Scheme 1). In theory, both reactions yield the same product. For example, nitrosonium (NO+; a strong oxidant, E = +0.56 V vs. Fc/Fc+ in DMF25) will oxidize 1 to 1ox and form NO in the process. The newly generated 1ox (S = 1/2) then reacts with NO to form the Ni-nitrosyl, formally a {NiNO}8 complex, assuming binding of NO and no other coordination sphere changes, using the notation defined by Enemark and Feltham.26 Likewise, NO will readily intercept paramagnetic 1ox to generate the same species. Mixing a DMF solution of 1 with NOBF4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulted in instantaneous bleaching of the solution, consistent with oxidation of the RS− ligand to disulfide (RSSR), and the appearance of a dark-red precipitate that was spectroscopically identified to be the neutral S,S-bridged tetramer [Ni4(nmp)4] (Scheme 1).16 This outcome is typical for all [Ni(nmp)(SR)]− complexes when treated with chemical oxidants, i.e., S-oxidation of the coordinated monodentate thiolate to RSSR.15 Incidentally, the same result was obtained when introducing NO(g) into a DMF solution of in situ generated 1ox. In this case, formation of the disulfide may traverse a fleeting, and yet to be characterized, RSNO intermediate that releases NO via homolytic cleavage of the RS–NO bond (Scheme 1).27 Overall, a Ni-nitrosyl was not isolated. This result may not be too surprising considering that all known Ni-nitrosyls are in the {NiNO}10 Enemark–Feltham (EF) classification,28 although a {NiNO}9/8 species is not entirely unrealistic in light of the strong donors present in 1 and in NiSOD, i.e., peptido-N and alkyl-thiolato-S.
:1) resulted in instantaneous bleaching of the solution, consistent with oxidation of the RS− ligand to disulfide (RSSR), and the appearance of a dark-red precipitate that was spectroscopically identified to be the neutral S,S-bridged tetramer [Ni4(nmp)4] (Scheme 1).16 This outcome is typical for all [Ni(nmp)(SR)]− complexes when treated with chemical oxidants, i.e., S-oxidation of the coordinated monodentate thiolate to RSSR.15 Incidentally, the same result was obtained when introducing NO(g) into a DMF solution of in situ generated 1ox. In this case, formation of the disulfide may traverse a fleeting, and yet to be characterized, RSNO intermediate that releases NO via homolytic cleavage of the RS–NO bond (Scheme 1).27 Overall, a Ni-nitrosyl was not isolated. This result may not be too surprising considering that all known Ni-nitrosyls are in the {NiNO}10 Enemark–Feltham (EF) classification,28 although a {NiNO}9/8 species is not entirely unrealistic in light of the strong donors present in 1 and in NiSOD, i.e., peptido-N and alkyl-thiolato-S.
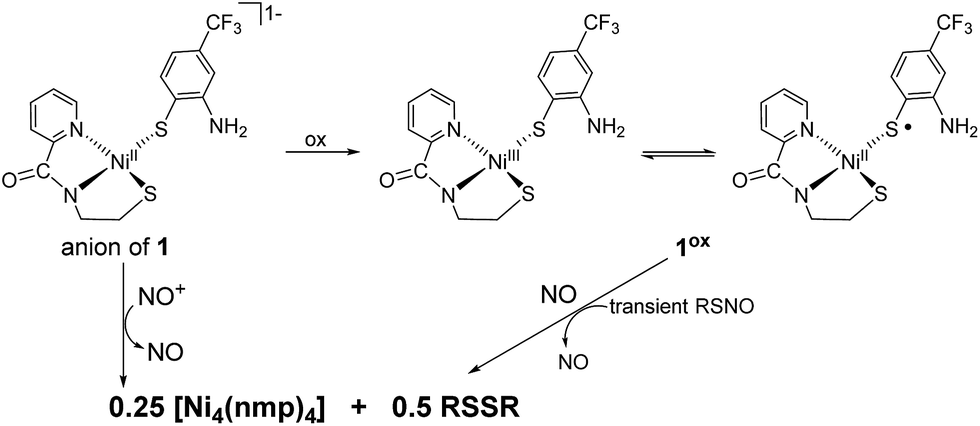 | ||
| Scheme 1 NO and NO+ reactions of 1 and 1ox, respectively. R = 2-amino-4-(trifluoromethyl)benzenethiolate. | ||
As a control, we also explored the reaction of NiII complex 1 with NO. In general, NO does not react with square-planar [Ni(nmp)(SR)]− (R = simple aryl or alkyl groups) complexes due to their diamagnetic nature. However, when R contains a potentially bidentate chelate, as in 1, a different course takes place. For instance, exposing a DMF solution of 1 with NO(g) for 30 s resulted in a gradual change of the solution from dark-red to green over several minutes. Workup of this reaction indicated a Ni-nitrosyl based on the strong double-humped peak in the N–O stretching (νNO) region of the IR spectrum (vide infra). Subsequent crystallization of the bulk material from MeCN/Et2O at −20 °C resulted in green crystals of a dinuclear thiolate-bridged {NiNO}10 complex (Et4N)2[{Ni(κ2-SPh-o-NNO-p-CF3)(NO)}2] (2) as depicted in Fig. 1 with selected metric parameters listed in Table 1. The Ni centers in 2 are distorted tetrahedral (τ4 = 0.73 (ref. 29)) resulting from N2S2 coordination of the thiolato-S/deprotonated amine-N of the S-bridged o-nitrosaminobenzenethiolate and a terminal nitrosyl. To our knowledge, complex 2 represents the first example of a structurally characterized first-row metal complex with both a coordinated nitrosyl and amine-N-bound nitrosamine. In accord with other tetrahedral {NiNO}10 complexes,28 the Ni–N(O) distance is short (1.659 Å), the N–O bond (1.182 Å) is intermediate between free NO˙ (1.15 Å) and 1HNO (1.21 Å),10 and the Ni–N–O bond angle is close to linear albeit slightly bent (167.8°) (see Table 1). Complex 2 is analogous to the limited number of four-coordinate/S-bound Ni-nitrosyls,30–34 fewer of which contain Ni–Sthiolate bonds31,33 which display Ni–N(O) (1.663–1.683 Å), N–O (1.131–1.173 Å), and Ni–N–O (156.6–173.9°) distances/angles in similar ranges. Even neutral/cationic P-35–40 and N-bound34,41–43 L3Ni–NO/L2XNi–NO complexes exhibit similar metric parameters. The coordinated nitrosamine is bent (N–N–O: 115.4°), i.e., sp2-hybridized nitroso-N, with N–N and N–O distances of 1.299 and 1.269 Å, respectively. These values suggest a small degree of delocalization in the R–N–N–O unit. However, the structure is more biased towards the nitrosamino R–N−–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O versus diazoate R–NN–O− resonance form. To compare, the structure of syn-methanediazoate (N–N: 1.246 Å, N–O: 1.306 Å) reflects the true double bond character in an authentic R–NN–O unit.44 These values are somewhat comparable to other N-bound nitrosamine complexes,45,46 especially [CpNi(PPh3)(ON2Ph-p-NO2)] (I)47 (N–N: 1.327 Å, N–O: 1.249 Å, N–N–O: 113.1°). Structures of coordinated nitroso-N-metal complexes (vs. amine-N as in 2) also afford similar structural parameters in the RNNO.48 In contrast, O-bound nitrosamine complexes appear to favor more of a resonance delocalized structure as the N–N (1.275–1.288 Å) and N–O (1.251–1.275 Å) distances in a series of [FeIII(P)(ONNR2)2]+ (P = porphyrin) complexes are nearly identical and result in a single 15N-sensitive peak in the IR due to overlapping νNN/νNO modes.49–51
O versus diazoate R–NN–O− resonance form. To compare, the structure of syn-methanediazoate (N–N: 1.246 Å, N–O: 1.306 Å) reflects the true double bond character in an authentic R–NN–O unit.44 These values are somewhat comparable to other N-bound nitrosamine complexes,45,46 especially [CpNi(PPh3)(ON2Ph-p-NO2)] (I)47 (N–N: 1.327 Å, N–O: 1.249 Å, N–N–O: 113.1°). Structures of coordinated nitroso-N-metal complexes (vs. amine-N as in 2) also afford similar structural parameters in the RNNO.48 In contrast, O-bound nitrosamine complexes appear to favor more of a resonance delocalized structure as the N–N (1.275–1.288 Å) and N–O (1.251–1.275 Å) distances in a series of [FeIII(P)(ONNR2)2]+ (P = porphyrin) complexes are nearly identical and result in a single 15N-sensitive peak in the IR due to overlapping νNN/νNO modes.49–51
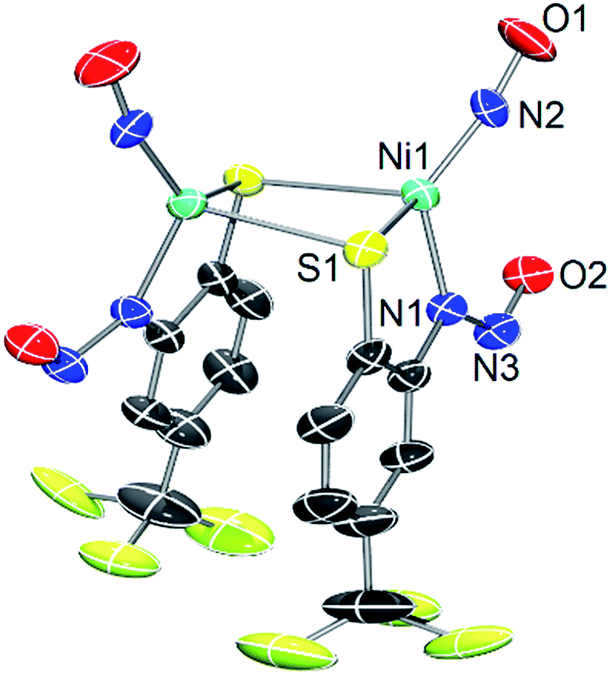 | ||
| Fig. 1 X-ray structure of the anionic portion of 2 with the atom labeling scheme (50% thermal probability). H atoms and Et4N+ counterions are omitted for clarity. | ||
| X-ray structure 2 | DFT (BP86/def2-TZVPP) optimized structure 2* | |
|---|---|---|
| Ni1–S1 | 2.3169(7) | 2.294 |
| Ni1–S1′ | 2.3555(6) | 2.344 |
| Ni1–N2 | 1.659(7) | 1.648 |
| Ni1–N1 | 1.971(2) | 1.974 |
| N2–O1 | 1.182(8) | 1.191 |
| N1–N3 | 1.299(3) | 1.324 |
| N3–O2 | 1.269(3) | 1.254 |
| S1–Ni–S1′ | 96.66(2) | 90.05 |
| S1–Ni–N1 | 86.21(6) | 86.67 |
| S1′–Ni–N1 | 99.10(6) | 98.40 |
| S1–Ni–N1 | 123.8(5) | 125.92 |
| N2–Ni–S1′ | 109.8(6) | 113.43 |
| N1–Ni–N2 | 133.6(5) | 132.15 |
| Ni1–N2–O1 | 167.8(12) | 171.42 |
| N1–N3–O3 | 114.2(2) | 115.80 |
| τ 4 | 0.73 | 0.72 |
Complex 2 was characterized by a variety of spectroscopic methods. The solid-state IR spectrum (KBr matrix) of 2 exhibits two closely spaced, but well-resolved, νNO at 1759 and 1743 cm−1 (1724, 1708 cm−1 for 2-15NO; ΔνNO: 35 cm−1; see Fig. 2). These values fall in the range of known tetrahedral, neutral, and anionic {NiNO}10 complexes.28 Because 2 is of C2 symmetry (cis NO, syn bridging thiolates), two IR-active N–O vibrational modes are expected. The other feasible isomer of 2 would be of Ci symmetry (trans NO, anti bridging thiolates) and would display one IR-active N–O stretch. Indeed, the IR spectrum of 2 in DMSO exhibits one νNO at 1784 cm−1 suggesting possible cis/trans-NO conversion in solution (or an averaged νNO value due to rapid tumbling) or thiolate-bridge splitting to yield a four-coordinate mononuclear {NiNO}10 with DMSO as the fourth ligand, i.e., [Ni(κ2-SPh-o-NNO-p-CF3)(DMSO)(NO)]−. The 1H NMR spectrum of 2 in CD3CN (Fig. S6†) or DMSO-d6 (not shown) are similar and thus do not distinguish any of the proposed structures. Comparable IR spectral changes in the opposite direction are observed for the one other thiolate-supported anionic dinuclear {NiNO}10, (Et4N)2[Ni2(NO)2(μ-SPh)2(SPh)2] (II), with trans NO ligands (νNO: 1709 cm−1 in KBr; 1751, 1721 cm−1 in THF).33 A similar situation is described for a pyrazolate-bridged anionic dinuclear {NiNO}10 complex.52 IR peaks arising from the nitrosamine were not as obvious due to multiple overlapping peaks in the region (Fig. S5†). However, 15N-sensitive peaks in the IR of 2 at 1342 and 1258 cm−1 (1326, 1249 cm−1 in 2-15NO) are assigned as νNO and νNN, respectively. In comparison, a series of secondary nitrosamines display νNO: 1428–1463 cm−1 and νNN: 1035–1154 cm−1 in CCl4.53 Therefore, a significant degree of delocalization occurs in the RNNO unit of 2 to cause the corresponding downshift in νNO/upshift in νNN. While no paramagnetically shifted resonances are observed in the 1H NMR (CD3CN) of 2, several species are indicated in freshly prepared solutions (Fig. S6†) that are likely caused by the lability of the Ni–NO bond and presence of nmpS2 (vide infra). The 15N NMR spectrum of 2-15NO confirms multiple solution speciation with four major peaks in the range for nitrosamines and linearly coordinated NO (δ: 40–190 ppm in CD3CN, vs. CH3NO2, Fig. S7†).54–57 Moreover, the 1H NMR of thiolate-bridged dinuclear complex II displays broadened aryl-H resonances caused by rapid exchange of PhS− ligands because of disproportionation to the mononuclear (Et4N)2[Ni(NO)(SPh)3] (III) and an uncharacterized [Ni(NO)(SPh)] species.33 However, high-resolution electrospray ionization mass spectrometry (HR-ESI-MS; negative mode) displays one dominant compound with the formula and isotopic distribution consistent with the dianionic portion of 2 (m/z: 307.926, z = 2, Fig. 2, S8 and S9†) and 2-15NO (m/z: 309.920, z = 2; Fig. S10 and S11†), although this measurement does not discriminate against cis and trans NO conformers. Another minor peak in the HR-ESI-MS(−) is centered at m/z: 248.960 (z = 2; Fig. S8†) that suggests a new [Ni(N2S2)]2− species through loss of the Ni-coordinated NO and one Ni (vide infra).
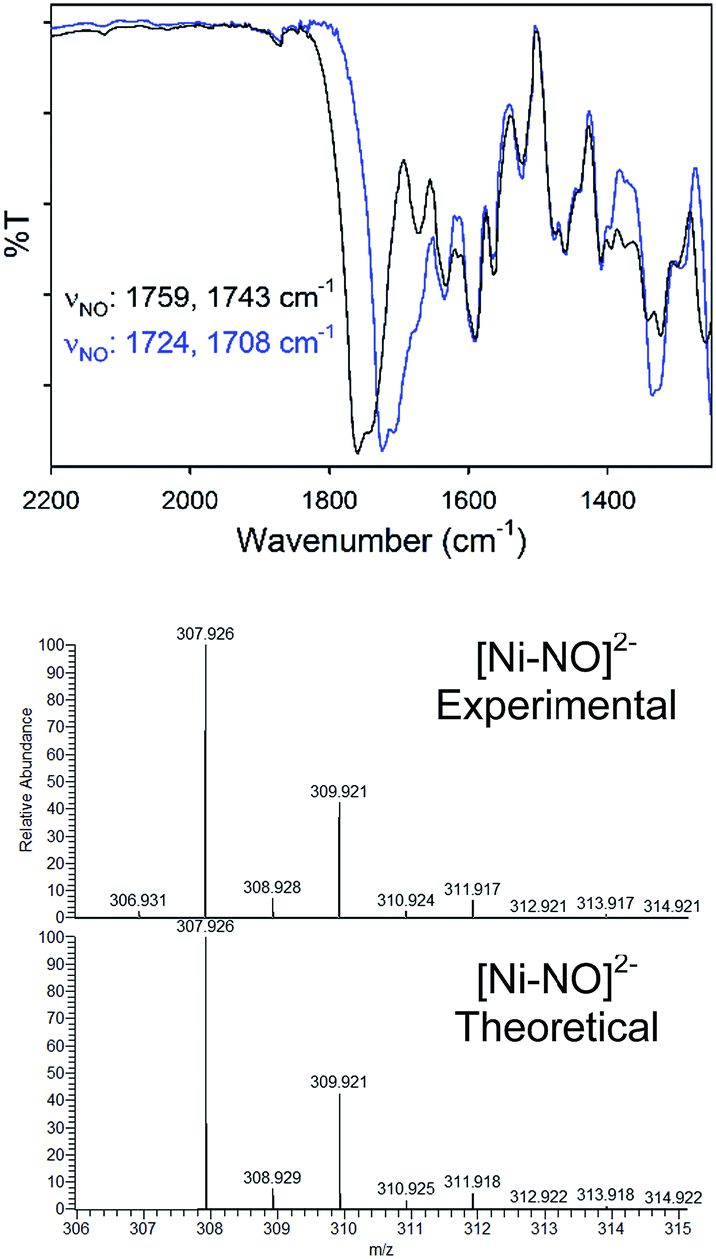 | ||
| Fig. 2 (Top) Solid-state IR of the νNO region for 2 (black) and 2-15NO (blue) in a KBr matrix. (Bottom) High resolution ESI-MS(−) of 2 with the theoretical isotopic distribution. | ||
Solutions of 2, especially in donor solvents such as MeCN or DMF, gradually lose their green color to give red-brown solutions more reminiscent of square-planar NiII–N2S2 complexes.15,58 Even freshly prepared CD3CN solutions of 2 exhibit multiple peaks in the 1H/15N NMR, and ESI-MS shows a new species with a Ni isotope pattern at m/z ∼ 249 (vide supra). This change is enhanced when vacuum is applied and FTIR spectra of these reaction mixtures lack any νNO suggesting the loss of coordinated NO from 2 to generate a new Ni species. Slow diffusion of Et2O into MeCN solutions of 2 that have been left standing for several weeks result in crystals (10–20% isolated yield from crystallization) of a square-planar (τ4 = 0.12) NiII compound where two N,S-chelating o-nitrosaminobenzenethiolato ligands bind to Ni in a trans configuration, viz. trans-(Et4N)2[Ni(SPh-o-NNO-p-CF3)2] (3) (Fig. 3). The bond lengths (Ni–S: 2.2072 Å, Ni–N: 1.896 Å) and angles (Table S3†) are similar to other planar NiII–N2S2 complexes that contain κ2-N,S-o-aminobenzenethiolate ligands.59–61 The Ni–N distance in 3 is shorter than the typical Ni–Namine bond and reflects the enhanced donor strength of the deprotonated nitrosamino-N, which is comparable to, although weaker than, a Ni–Ncarboxamido (∼1.86 Å).15,58 No evidence for a coordinated ligand radical is evident from the X-ray structure (i.e., short C–S, C–N distances of the coordinated o-aminobenzenethiolate62) and confirm the N,S-ligand is a closed-shell dianion. The R–N–N–O linkage in 3 (avg. of two crystallographically distinct molecules, N–N: 1.309 Å, N–O: 1.264 Å; avg. N–N–O: 115.2°) is unremarkable from 2. 1H and 15N NMR (RN15NO, δ: 194 ppm vs. CH3NO2) of crystals of 3 are consistent with the X-ray structure and analogous to other nitrosamines (Fig. S13 and S14†).55,57 As expected, the IR of 3 lacks the intense νNO from the NiNO of 2 (although IR and ESI-MS show that some 2 remains even in crystals of 3, Fig. S12†) and the νNO and νNN of the R–N–N–O unit is similar. HR-ESI-MS(−) confirm this formulation with peaks corresponding to [M–2Et4N]2− (m/z: 248.960, z = 2, for 3; m/z: 249.957, z = 2, for 3-15NO) as the prominent peak (Fig. S15–S18†).
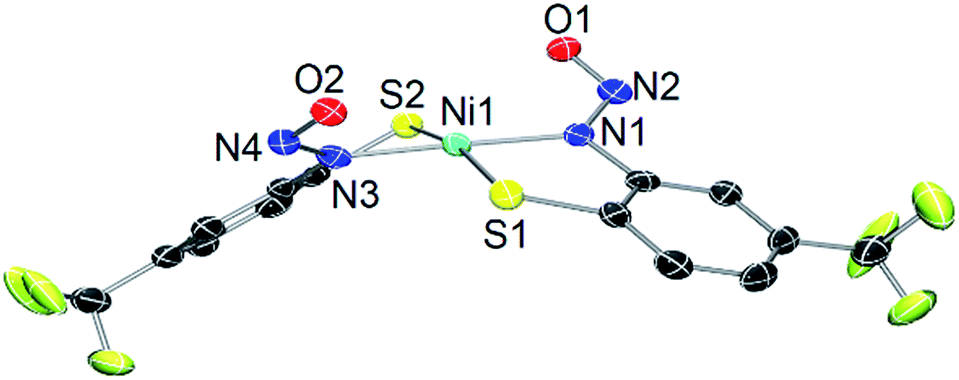 | ||
| Fig. 3 X-ray structure of the dianion of 3 with the atom-labeling scheme (50% thermal probability). One of two crystallographically distinct molecules shown. H atoms, Et4N+ counterions, and solvent of crystallization (Et2O) are omitted for clarity. | ||
To confirm that NO(g) is released from 2 (forming 3 among other products), solutions of 2 were mixed with the NO(g) trap [Co(T(-OMe)PP)] (T(-OMe)PP = 5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphine).63 For example, mixing 2 and the CoII–P (1:2) in CH2Cl2 at RT for 24 h resulted in the {CoNO}8 complex [Co(T(-OMe)PP)(NO)] in ∼70% avg. yield as quantified by 1H NMR (CD2Cl2) and further verified by IR spectroscopy using 2-15NO (Fig. S21–S23†). Notably, the reaction mixture becomes red over the course of the reaction. Workup of this solution after separating the Co–P compounds (MeOH-insoluble) reveals the presence of 3 (MeOH-soluble) via1H NMR to confirm the fate of the {NiNO}10 complex 2. To eliminate bimolecular NO-transfer via a putative Co⋯NO⋯Ni intermediate, NO(g) release was further verified by vial-to-vial trapping reactions wherein a CH2Cl2 solution of the CoII–P was separated from an MeCN solution of 2 (CoII–P in excess, see the ESI†). Carrying out this reaction confirmed that NO(g) is indeed released from 2 (or 2-15NO) to generate the {CoNO}8 porphyrin complex (80% avg. yield) as shown by 1H NMR and IR measurements (Fig. S24†). In contrast, no reaction takes place between THF solutions of 2 with [Fe(TPP)Cl] (1:2; TPP = 5,10,15,20-tetraphenylporphyrin), a common HNO (or NO−) trap.64 Although {NiNO}10 has not been characterized as a particularly labile EF notation, we note that the majority of these complexes are cationic or neutral without coordinated thiolate ligands.28 Indeed, the thiolate-ligated {NiNO}10 complex III photochemically releases NO to [Co(TPP)] in MeCN suggesting some lability in the Ni–NO bond. Furthermore, the RN–NO bond is quite stable (as noted by formation of 3) and the energetically stabilized MOs that contribute to the electronic structure of 2 and 3 where HOMO−3 represents a bonding MO with primary contributions from σ-NR and σ-NO orbitals (Fig. S25†).
Density functional theory (DFT) computations have provided a deeper understanding of the electronic structure of a variety of metal nitrosyls,65,66 and we have employed them here for 2 and 3 at the OLYP/def2-TZVPP level of theory. Pure functionals such as BP86 and OLYP were used for geometry optimization and single point energy calculations, respectively, as these functionals have been established to deliver better matches with experimental geometries in MNO systems.67–69 Geometry optimization of 2 was performed with coordinates from the crystal structure to yield DFT-optimized complex 2* (Fig. 4, Tables 1, S5 and S7 in the ESI†). Structurally, 2* replicates the metrics of 2 well, suggesting the computational model is reasonable. While the distances in 2* are within ±0.025 Å of experimental values, the bond angles (especially S–Ni–S: −6.6°, and Ni–N–O: +3.6° from 2) are slightly beyond the allowable tolerances for satisfactory DFT performance in small molecules (i.e., distances ±0.03 Å; angles ±1°).70 However, these rules may be broken to some degree because of the enhanced complexity arising from the covalent MNO unit in 2. The computations also reasonably match the two closely spaced N–O stretching frequencies for the symmetric and asymmetric νNO in the IR at 1730 and 1708 cm−1, respectively. The ∼30 cm−1 downshift from 2 is likely due to a slight overestimation of Ni–NO bond covalency arising from Ni-dπ backbonding. Previous calculations on three-71 and four-coordinate43,72 {NiNO}10 complexes support a NiII–3NO− (Stot = 0, antiferromagnetically coupled) oxidation state assignment. This is comparable to high-spin nonheme {FeNO}7 systems that are classified as FeIII–3NO− (Stot = 3/2).6,66,73 In the Fe case, 3NO− serves as a strong π-donor to afford a highly covalent Fe–NO bond.74 The strength of this interaction originates from the effective nuclear charge on the metal, which is controlled by the basicity of the supporting ligands.75 Thus, electron rich supporting ligands attenuate the π-basicity of 3NO− to result in diminished M–NO bond covalency. This property has been established in the {FeNO}7 case, but not yet for {NiNO}10. Indeed, examination of the frontier MOs of 2* show that, much like other {NiNO}10 systems with Tp ligands43,72 (Tp = tris(pyrazolyl)borate), the LUMO is a π* MO primarily comprised of antibonding interactions between Ni-dπ and NO-π* orbitals (Fig. S25†). On the other hand, the HOMO (Fig. 4) and HOMO−1 have little contribution from NO, but large contributions from Ni-dσ (38.0%) and S-pσ (19.3%) orbitals of the Ni(μ-SR)2Ni core. The HOMO is antibonding in nature and suggests a thermally unstable structure. As expected from analogous {FeNO}7 systems, based on the increased donor strength of the anionic nitrosamine-N/thiolate-S supporting ligands in 2*, the covalency in the Ni–N–O unit is less than in TpNi–NO complexes and rationalizes the observed lability of the Ni–NO bond and the Ni(μ-SR)2Ni core in 2.
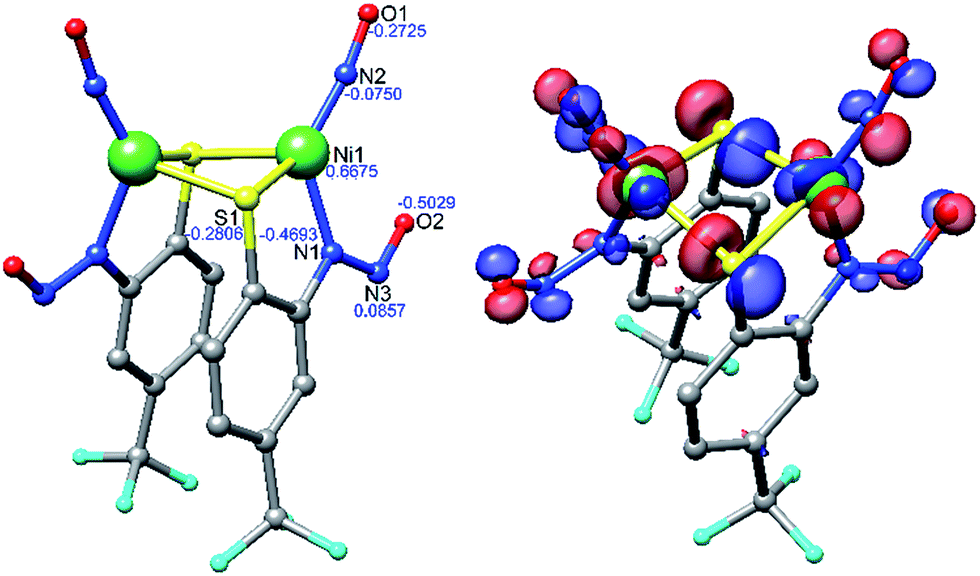 | ||
| Fig. 4 DFT (OLYP/def2-TZVPP) optimized structure of 2* (left) with natural population analysis charges in blue and HOMO (right). | ||
DFT computations on 3* were performed in the same fashion as for 2*. Geometry optimized 3* is square-planar (τ4 = 0.09) with metric parameters on-par with the X-ray structure of 3 and within the error of the DFT method (Table S9†). Unlike 2*, the π* HOMO of 3* is comprised primarily of Ni(dπ)/S(pπ) contributions (Fig. S26†), typical of planar NiII–N2S2 complexes with strong-field ligands and suggests a highly covalent Ni–SR bond.15
The formation of 2 likely follows a mechanistic path analogous to those observed in the reductive nitrosylation of Cu–amine systems, where one-equiv. of NO reacts with CuII–NR2 complexes to yield R2N–NO and deligated CuI.76,77 The difference here is that the nitrosated ligand remains coordinated and the resulting paramagnetic Ni binds NO radical. Our working model is depicted in Scheme 2. Complex 1 is likely in resonance with a distorted tetrahedral species which places the anilido-N in the coordination sphere. This proposal is supported by the presence of low intensity peaks in the 1H NMR spectrum of 1 and may explain the difficulty in crystallizing this complex.16 On the other hand, X-ray absorption spectroscopic (XAS) characterization of 1, not reported previously, suggests a four-coordinate planar NiII center (XANES analysis, see Fig. S3†) with two O/N- and S-ligands at 1.90 Å and 2.17 Å (EXAFS, Fig. S3, Table S4†), respectively. Thus, 1 is structurally analogous to other [Ni(nmp)(SR)]− complexes at least in the solid-state. Introduction of NO(g) can then result in either: (i) reduction of NiII to NiI and formation of NO+ that nitrosates the coordinated amine, or (ii) nitrosylation of Ni to yield {NiNO}10 with the electron originating from the coordinated thiolate of nmp2− to result in the disulfide. Our results do not differentiate either of these transformations, but the disulfide of nmp2− (i.e., nmpS21H NMR and IR of the reaction mixture, see Fig. S19 and S20†) is spectroscopically observed in the reaction mixture and checked against independently synthesized nmpS2. Thus, the fate of one proton and one electron is reasonably confirmed. At this point these intermediates can react with another equiv. of NO to yield the three-coordinate precursor to 2. Compound 3 forms through either disproportionation (shown in Scheme 2) to yield a Ni0 species or ligand rearrangement via the loss of a NiI–NO fragment (not shown). In ligand rearrangement, the products would be a NiI–N2S2 precursor to 3 (3-PC), an L–NiI–NO species (L = solvent), and free NO. Ultimately this NiI intermediate oxidizes 3-PC to generate NiII complex 3 and an L–Ni0–NO complex that would presumably release NO(g) as evidenced by the NO(g) trap experiments (vide supra). While the reaction mechanism for the conversion of 2-to-3 is likely more complex, similar chemistry has been proposed for N-heterocyclic carbene (NHC) Ni-nitrosyls.28,78 The details of this mechanism are still under investigation.
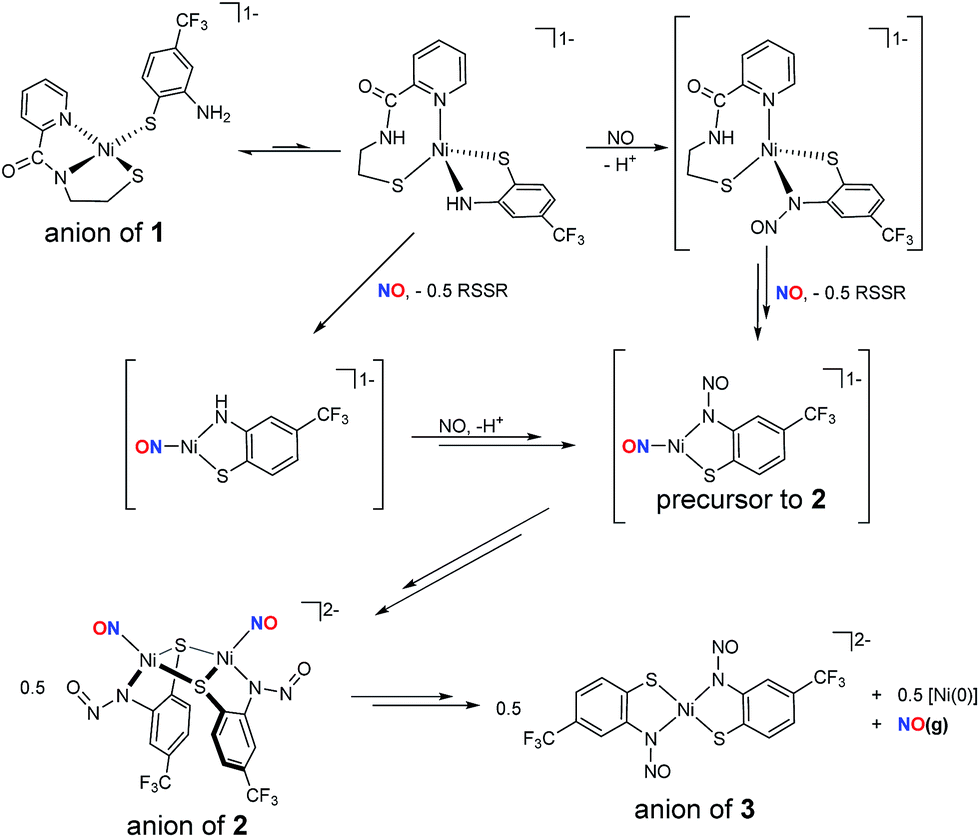 | ||
| Scheme 2 Working model for the formation of {NiNO}10 complex 2 and NiII–N2S2 complex 3 starting from NiII–N2S2 monomer 1. RSSR: disulfide of nmp2−, i.e., N-(2-mercaptoethyl)picolinamide. Intermediates represented in brackets have not been spectroscopically identified. The py-N of RSSR is a possible H+ receptor. | ||
Conclusions
In conclusion, NiSOD model complex 1 reacts with NO(g) in the NiII state to form the metastable {NiNO}10 dimeric complex 2via loss of the nmp2− ligand as the disulfide and N-nitrosation of the o-aminobenzenethiolate ligand. Reaction of NO with 1ox, or NO+ with 1, only yields the S,S-bridged tetrameric compound [Ni4(nmp)4] through oxidation of the aromatic thiolate ligand. While any reaction with NO (S = 1/2) is generally unexpected for square-planar (S = 0) NiII complexes, this Ni-nitrosyl likely forms due to an equilibrium mixture of 1 and a tetrahedral (S = 1) or five-coordinate derivative (Scheme 2). Even if NO were to result in an nmp-bound Ni–NO complex, the resulting {NiNO}9 (reaction of 1 with NO) or {NiNO}8 (reaction of 1ox with NO) oxidation levels have yet to be defined and support an outer-sphere superoxide interaction in NiSOD. Although these EF notations have yet to be accessed, one would propose that NiSOD mimetics, especially with strong-field carboxamido-N and alkyl-thiolato-S donors, would surely stabilize such an electron poor species. Furthermore, the properties of complexes such as 2 extend to biology, where analogous S-bridged mononitrosyl species, i.e., Fe–S clusters and tetrahedral (RS)3Fe–NO complexes are proposed as intermediates in the repair of NO-damaged clusters.79–81 Complex 2 is stable in the solid-state but breaks down slowly in solution causing rupture of the Ni(μ-SR)2Ni core and release of NO that was trapped in near quantitative yield with a CoII–porphyrin receptor. The resulting NiII–N2S2 complex 3 (coordination of two o-aminobenzenethiolate in trans configuration) was isolated and structurally/spectroscopically characterized as the ultimate Ni breakdown product with the nitrosamine unit still intact. This release may take place through a disproportionation mechanism (or through ligand rearrangement), as has been proposed in other Ni-nitrosyls, to a yet ill-defined Ni0 complex (see Scheme 2).28,78 Hence, thiolate-supported {NiNO}10 cores are reactive. While nitrosamines have been utilized as sources of NO, the RN–NO homolytic bond dissociation energy (BDE) is high (87.7 kcal mol−1 (ref. 82)) compared to more traditional small molecule sources of NO such as nitrosothiols (RSNO) that have RS–NO BDEs between 20–32 kcal mol−1.83,84 Overall, the electronic structures of {NiNO}10 complexes are modulated by the supporting ligands. Indeed, the majority are stable entities; however, a small number are reactive and result in release of NO (thiolate-supported/anionic complexes 2 and III) or generate other reactive intermediates of environmental significance such as hyponitrite (N2O22−) in five-coordinate {NiNO}10 species85 (highly reduced NO, with a severely bent Ni–N–O angle = 130°).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Science Foundation, Division of Chemistry, Chemistry of Life Sciences Program (CHE-1506375) to TCH. TLS and SPD were supported by the National Institutes of Health (NIH) DK068139 (T. L. S.), DK101230 (SPD). XAS studies were performed at the Stanford Synchrotron Radiation Lightsource (SSRL). SSRL is a national user facility operated by Stanford University on behalf of the Department of Energy (DOE), Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the DOE, Office of Biological and Environmental Research, and by the NIH, National Center for Research Resources, Biomedical Technology Program. We thank Dr Dennis R. Phillips and Dr Chau-Wen Chou from the Proteomics and Mass Spectrometry (PAMS) Core Facility at UGA for their assistance with MS measurements. Instrumentation in the PAMS facility was purchased in part with funds from NIH Grant 1S10RR028859-01. Funds for PAMS facility operations were provided by the UGA Office of the Vice President for Research (OVPR) and UGA Department of Chemistry. This study was supported in part by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between UGA's OVPR and Office of the Vice President for Information Technology. We also acknowledge Ms Yi Liu (UGA Department of Chemistry) for assistance with the NO(g) vial-to-vial trap experiments.Notes and references
- S. Moncada and E. A. Higgs, Br. J. Pharmacol., 2006, 147, S193–S201 CrossRef PubMed.
- L. J. Ignarro, Nitric Oxide Biology and Pathobiology, Academic Press, San Diego, CA, 2000 Search PubMed.
- J. M. Fukuto, S. J. Carrington, D. J. Tantillo, J. G. Harrison, L. J. Ignarro, B. A. Freeman, A. Chen and D. A. Wink, Chem. Res. Toxicol., 2012, 25, 769–793 Search PubMed.
- C. L. Bianco, J. P. Toscano, M. D. Bartberger and J. M. Fukuto, Arch. Biochem. Biophys., 2017, 617, 129–136 CrossRef PubMed.
- M. D. Clay, C. A. Cosper, F. E. Jenney, M. W. W. Adams and M. K. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3796–3801 CrossRef PubMed.
- C. A. Brown, M. A. Pavlosky, T. E. Westre, Y. Zhang, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1995, 117, 715–732 CrossRef.
- M. J. Nelson, J. Biol. Chem., 1987, 262, 12137–12142 Search PubMed.
- V. J. Chen, A. M. Orville, M. R. Harpel, C. A. Frolik, K. K. Surerus, E. Münck and J. D. Lipscomb, J. Biol. Chem., 1989, 264, 21677–21681 Search PubMed.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef PubMed.
- J. A. McCleverty, Chem. Rev., 2004, 104, 403–418 CrossRef PubMed.
- E. M. Gale, A. K. Patra and T. C. Harrop, Inorg. Chem., 2009, 48, 5620–5622 CrossRef PubMed.
- E. M. Gale, B. S. Narendrapurapu, A. C. Simmonett, H. F. Schaefer III and T. C. Harrop, Inorg. Chem., 2010, 49, 7080–7096 CrossRef PubMed.
- E. M. Gale, D. M. Cowart, R. A. Scott and T. C. Harrop, Inorg. Chem., 2011, 50, 10460–10471 CrossRef PubMed.
- E. M. Gale, A. C. Simmonett, J. Telser, H. F. Schaefer III and T. C. Harrop, Inorg. Chem., 2011, 50, 9216–9218 CrossRef PubMed.
- E. P. Broering, P. T. Truong, E. M. Gale and T. C. Harrop, Biochemistry, 2013, 52, 4–18 CrossRef PubMed.
- E. P. Broering, S. Dillon, E. M. Gale, R. A. Steiner, J. Telser, T. C. Brunold and T. C. Harrop, Inorg. Chem., 2015, 54, 3815–3828 CrossRef PubMed.
- R. A. Steiner, S. P. Dzul, T. L. Stemmler and T. C. Harrop, Inorg. Chem., 2017, 56, 2849–2862 CrossRef PubMed.
- P. T. Truong, E. M. Gale, S. P. Dzul, T. L. Stemmler and T. C. Harrop, Inorg. Chem., 2017, 56, 7761–7780 CrossRef PubMed.
- D. P. Barondeau, C. J. Kassmann, C. K. Bruns, J. A. Tainer and E. D. Getzoff, Biochemistry, 2004, 43, 8038–8047 CrossRef PubMed.
- A. T. Fiedler, P. A. Bryngelson, M. J. Maroney and T. C. Brunold, J. Am. Chem. Soc., 2005, 127, 5449–5462 CrossRef PubMed.
- J. Shearer and N. Zhao, Inorg. Chem., 2006, 45, 9637–9639 CrossRef PubMed.
- V. Mathrubootham, J. Thomas, R. Staples, J. McCraken, J. Shearer and E. L. Hegg, Inorg. Chem., 2010, 49, 5393–5406 CrossRef PubMed.
- D. Nakane, Y. Wasada-Tsutsui, Y. Funahashi, T. Hatanaka, T. Ozawa and H. Masuda, Inorg. Chem., 2014, 53, 6512–6523 CrossRef PubMed.
- J. Shearer, Acc. Chem. Res., 2014, 47, 2332–2341 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef . {MNO}n, n = total sum of electrons in the metal-d and NO-π* orbitals.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef.
- A. M. Wright and T. W. Hayton, Comments Inorg. Chem., 2012, 33, 207–248 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- P. J. Schebler, C. G. Riordan, I. A. Guzei and A. L. Rheingold, Inorg. Chem., 1998, 37, 4754–4755 CrossRef PubMed.
- W.-F. Liaw, C.-Y. Chiang, G.-H. Lee, S.-M. Peng, C.-H. Lai and M. Y. Darensbourg, Inorg. Chem., 2000, 39, 480–484 CrossRef PubMed.
- L. S. Maffett, K. L. Gunter, K. A. Kreisel, G. P. A. Yap and D. Rabinovich, Polyhedron, 2007, 26, 4758–4764 CrossRef.
- A. G. Tennyson, S. Dhar and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 15087–15098 CrossRef PubMed.
- A. M. Wright, H. T. Zaman, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3207–3216 CrossRef PubMed.
- J. H. Enemark, Inorg. Chem., 1971, 10, 1952–1957 CrossRef.
- J. Kriege-Simondsen, G. Elbaze, M. Dartiguenave, R. D. Feltham and Y. Dartiguenave, Inorg. Chem., 1982, 21, 230–236 CrossRef.
- A. F. M. M. Rahman, G. Salem, F. S. Stephens and S. B. Wild, Inorg. Chem., 1990, 29, 5225–5230 CrossRef.
- D. J. Darensbourg, T. J. Decuir, N. W. Stafford, J. B. Robertson, J. D. Draper, J. H. Reibenspies, A. Kathó and F. Joó, Inorg. Chem., 1997, 36, 4218–4226 CrossRef.
- C. E. MacBeth, J. C. Thomas, T. A. Betley and J. C. Peters, Inorg. Chem., 2004, 43, 4645–4662 CrossRef PubMed.
- V. M. Iluc, A. J. M. Miller and G. L. Hillhouse, Chem. Commun., 2005, 5091–5093 RSC.
- V. K. Landry, K. Pang, S. M. Quan and G. Parkin, Dalton Trans., 2007, 820–824 RSC.
- A. M. Wright, H. T. Zaman, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 3108–3116 CrossRef PubMed.
- S. Soma, C. Van Stappen, M. Kiss, R. K. Szilagyi, N. Lehnert and K. Fujisawa, J. Biol. Inorg. Chem., 2016, 21, 757–775 CrossRef PubMed.
- R. Huber, R. Langer and W. Hoppe, Acta Crystallogr., 1965, 18, 467–473 CrossRef.
- L. K. Keefer, S. M. Wang, T. Anjo, J. C. Fanning and C. S. Day, J. Am. Chem. Soc., 1988, 110, 2800–2806 CrossRef.
- J. Lee, L. Chen, A. H. West and G. B. Richter-Addo, Chem. Rev., 2002, 102, 1019–1066 CrossRef PubMed.
- F. J. Lalor, T. J. Desmond, G. Ferguson and P. Y. Siew, J. Chem. Soc., Dalton Trans., 1982, 1981–1985 RSC.
- F. Doctorovich and F. Di Salvo, Acc. Chem. Res., 2007, 40, 985–993 CrossRef PubMed.
- G.-B. Yi, M. A. Khan and G. B. Richter-Addo, J. Am. Chem. Soc., 1995, 117, 7850–7851 CrossRef.
- G. B. Richter-Addo, Acc. Chem. Res., 1999, 32, 529–536 CrossRef.
- F. Di Salvo, D. A. Estrin, G. Leitus and F. Doctorovich, Organometallics, 2008, 27, 1985–1995 CrossRef.
- K. S. Chong, S. J. Rettig, A. Storr and J. Trotter, Can. J. Chem., 1979, 57, 3099–3106 CrossRef.
- R. L. Williams, R. J. Pace and G. J. Jeacocke, Spectrochim. Acta, 1964, 20, 225–236 CrossRef.
- J. Mason, L. F. Larkworthy and E. A. Moore, Chem. Rev., 2002, 102, 913–934 CrossRef PubMed.
- M. H. Lim, B. A. Wong, W. H. Pitcock Jr, D. Mokshagundam, M.-H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef PubMed.
- M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef PubMed.
- R. Bonnett, R. Holleyhead, B. L. Johnson and E. W. Randall, J. Chem. Soc., Perkin Trans. 1, 1975, 2261–2264 RSC.
- T. C. Harrop and P. K. Mascharak, Coord. Chem. Rev., 2005, 249, 3007–3024 CrossRef.
- T. Kawamoto, H. Kuma and Y. Kushi, Bull. Chem. Soc. Jpn., 1997, 70, 1599–1606 CrossRef.
- T. Kawamoto and Y. Kushi, Inorg. Chim. Acta, 1998, 282, 71–75 CrossRef.
- S. Inoue, M. Mitsuhashi, T. Ono, Y.-N. Yan, Y. Kataoka, M. Handa and T. Kawamoto, Inorg. Chem., 2017, 56, 12129–12138 CrossRef PubMed.
- D. Herebian, E. Bothe, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 10012–10023 CrossRef PubMed.
- M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Inorg. Chem., 2012, 51, 7004–7006 CrossRef PubMed.
- S. E. Bari, M. A. Martí, V. T. Amorebieta, D. A. Estrin and F. Doctorovich, J. Am. Chem. Soc., 2003, 125, 15272–15273 CrossRef PubMed.
- A. Ghosh, Acc. Chem. Res., 2005, 38, 943–954 CrossRef PubMed.
- T. C. Berto, A. L. Speelman, S. Zheng and N. Lehnert, Coord. Chem. Rev., 2013, 257, 244–259 CrossRef.
- J. Conradie and A. Ghosh, J. Phys. Chem. B, 2007, 111, 12621–12624 CrossRef PubMed.
- K. H. Hopmann, L. Noodleman and A. Ghosh, Chem.–Eur. J., 2010, 16, 10397–10408 CrossRef PubMed.
- L. E. Goodrich, F. Paulat, V. K. K. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293–6316 CrossRef PubMed.
- J.-F. Guillemoles, V. Barone, L. Joubert and C. Adamo, J. Phys. Chem. A, 2002, 106, 11354–11360 CrossRef.
- J. Conradie and A. Ghosh, Inorg. Chem., 2014, 53, 4847–4855 CrossRef PubMed.
- N. C. Tomson, M. R. Crimmin, T. Petrenko, L. E. Rosebrugh, S. Sproules, W. C. Boyd, R. G. Bergman, S. DeBeer, F. D. Toste and K. Wieghardt, J. Am. Chem. Soc., 2011, 133, 18785–18801 CrossRef PubMed.
- J. Conradie, D. A. Quarless, H.-F. Hsu, T. C. Harrop, S. J. Lippard, S. A. Koch and A. Ghosh, J. Am. Chem. Soc., 2007, 129, 10446–10456 CrossRef PubMed.
- A. L. Speelman and N. Lehnert, Angew. Chem., Int. Ed., 2013, 52, 12283–12287 CrossRef PubMed.
- T. C. Berto, M. B. Hoffman, Y. Murata, K. B. Landenberger, E. E. Alp, J. Zhao and N. Lehnert, J. Am. Chem. Soc., 2011, 133, 16714–16717 CrossRef PubMed.
- P. C. Ford, B. O. Fernandez and M. D. Lim, Chem. Rev., 2005, 105, 2439–2456 CrossRef PubMed.
- K. Tsuge, F. DeRosa, M. D. Lim and P. C. Ford, J. Am. Chem. Soc., 2004, 126, 6564–6565 CrossRef PubMed.
- M. S. Varonka and T. H. Warren, Organometallics, 2010, 29, 717–720 CrossRef.
- C. T. Tran, P. G. Williard and E. Kim, J. Am. Chem. Soc., 2014, 136, 11874–11877 CrossRef PubMed.
- J. Fitzpatrick and E. Kim, Acc. Chem. Res., 2015, 48, 2453–2461 CrossRef PubMed.
- J. Fitzpatrick and E. Kim, Inorg. Chem., 2015, 54, 10559–10567 CrossRef PubMed.
- X.-Q. Zhu, J.-Q. He, Q. Li, M. Xian, J. Lu and J.-P. Cheng, J. Org. Chem., 2000, 65, 6729–6735 CrossRef PubMed.
- M. D. Bartberger, J. D. Mannion, S. C. Powell, J. S. Stamler, K. N. Houk and E. J. Toone, J. Am. Chem. Soc., 2001, 123, 8868–8869 CrossRef PubMed.
- J.-M. Lü, J. M. Wittbrodt, K. Wang, Z. Wen, H. B. Schlegel, P. G. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2001, 123, 2903–2904 CrossRef.
- A. M. Wright, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 9930–9933 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and spectroscopic/reactivity details, and full crystallographic information for 2 and 3. CCDC 1850485 and 1850488. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03321h |
| This journal is © The Royal Society of Chemistry 2018 |