
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZwitterlation mitigates protein bioactivity loss in vitro over PEGylation†
Yanjiao
Han
 a,
Zhefan
Yuan
b,
Peng
Zhang
b and
Shaoyi
Jiang
*ab
a,
Zhefan
Yuan
b,
Peng
Zhang
b and
Shaoyi
Jiang
*ab
aMolecular Engineering and Science Institute, University of Washington, Seattle, WA 98195, USA. E-mail: sjiang@uw.edu
bDepartment of Chemical Engineering, University of Washington, Seattle, WA 98195, USA
First published on 14th September 2018
Abstract
Conjugation with poly(ethylene glycol) (PEG) or PEGylation is a widely used tool to overcome the shortcomings of native proteins, such as poor stability, inadequate pharmacokinetic (PK) profiles, and immunogenicity. However, PEGylation is often accompanied by an unwanted detrimental effect on bioactivity, particularly, resulting from the amphiphilic nature of PEG. This is especially true for PEGylated proteins with large binding targets. Pegasys, a PEGylated interferon alpha-2a (IFN-α2a) bearing a 40 kDa branched PEG, is a typical example that displays only 7% in vitro activity of the unmodified IFN-α2a. In this work, by employing IFN-α2a as a model protein, we demonstrated that a protein conjugated with zwitterionic polymers (or zwitterlation) could significantly mitigate the antiproliferative bioactivity loss in vitro after polymer conjugation. The retained antiproliferative activity of zwitterlated IFN-α2a is 4.4-fold higher than that of the PEGylated IFN-α2a with the same polymer molecular weight, or 3-fold higher than that of the PEGylated IFN-α2a with a similar hydrodynamic size. It is hypothesized that nonspecific interactions between zwitterionic polymers and IFN-α2a/IFN-α2a receptors can be mitigated due to the super-hydrophilic nature of zwitterionic polymers. This, in turn, reduces the ‘nonspecific blocking’ between IFN-α2a and IFN-α2a receptors. In addition, we demonstrated that zwitterlated IFN-α2a showed a prolonged circulation time and a mitigated accelerated blood clearance after repeated injections in rats.
Introduction
Advances in effective protein therapy are hindered by the poor stability, short circulation half-time, and immunogenicity of proteins.1,2 The covalent conjugation of poly(ethylene glycol) (PEG) to a protein or PEGylation is clinically proven to favorably alter protein pharmacokinetics by prolonging its circulation time and decreasing its toxicity profile, especially by reducing immunogenicity.3–7 Additional advantages of PEGylation include the potential to increase protein solubility and its storage stability. At present, more than 10 PEGylated proteins have been brought to market, and more than 15 are under clinical trials.8 Although PEGylation has addressed several limitations and is clinically proven, there are a variety of obstacles and pitfalls to overcome in the continued development of PEGylated protein drugs. In almost all cases, the in vitro biological activity of PEGylated proteins is lower, sometimes significantly lower, than that of the unmodified proteins.4,5 This is particularly true for those proteins, which interact with large binding targets whose size is bigger than 900 Da.9 For example, Pegasys, a PEGylated interferon alpha-2a (IFN-α2a) bearing a 40 kDa branched PEG, is a typical example of a popular protein drug that displays only 7% in vitro activity of the unmodified IFN-α2a.10 Even for enzymes dealing with small substrates, a significant bioactivity loss can also be observed. For example, Adagen, for which PEGylation is used to decrease the immunogenicity of the non-human protein, can retain only 18% bioactivity when compared with the unmodified protein.11 PEG is an amphiphilic polymer with both hydrophilic and hydrophobic characters. It was shown previously from both experimental and simulation studies that protein bioactivity decreases after PEGylation due to the hydrophobic nature of PEG.12,13 Although various efforts have been made to minimize this effect, such as site-specific PEGylation,14,15 the material issue remains unresolved over the last 40 years.16In contrast, zwitterionic polymers, bearing simultaneously a pair of oppositely charged ions in the same moiety while maintaining the overall neutral charge, have been identified recently as a class of extremely hydrophilic materials.17 Taking poly(carboxybetaine) (pCB) as an example, its strong electrostatically induced hydration confers an ability to effectively resist nonspecific protein adsorption in complex media.18 Due to their nonfouling characteristics, zwitterionic polymers have been used for inhibiting foreign body reactions,19 maintaining stem cell phenotypes,20 sustaining protein delivery21 and stabilizing proteins.22,23 In addition, zwitterionic polymer layers significantly prolong the in vivo circulation half-life of nanoparticles including liposome, protein and gold particles.24–28 Among zwitterionic polymers, pCB is particularly unique because it is composed of glycine betaine, which is a well-known protein stabilizer. In a previous study, pCB has been conjugated to chymotrypsin (CT) to test its protein stabilizing effect.12 It has been shown that pCB conjugation (or zwitterlation) not only preserves enzyme bioactivity, but also slightly increases its binding affinity to peptide substrates. At the same time, it is found that PEGylation decreases the binding affinity of the enzyme. A study on the fundamental understanding of the difference between PEGylation and zwitterlation was performed before.17 For proteins, hydrophobic–hydrophobic interactions are significantly important for enzyme–substrate specific binding (or bioactivity). With PEGylation, the amphiphilicity of PEG reduces the hydrophobic–hydrophobic driving force of enzyme–substrate interactions as confirmed from our molecular simulations.17 In contrast, with zwitterlation, its binding affinity is either unaltered or even improved when the binding target of the protein is a small molecule.12 Owing to the super-hydrophilicity of pCB polymers, water is drawn away from the hydrophobic regions of the protein, shifting the equilibrium to allow the substrate and binding site to interact. All these results make pCB a competitive candidate for protein conjugates. Previously, we have demonstrated that zwitterionic pCB conjugates are able to retain the bioactivity of enzymes with relatively small substrates. Here, we performed a systematic study of the effect of zwitterlation on the bioactivity of proteins whose binding target is substantially larger than Keefe studied previously.12
Experimental section
Synthesis of N-hydroxysuccinimide poly(carboxybetaine acrylamide) (NHS-pCBAA)
pCB-NHS was synthesized based on our previous procedure.29 Briefly, we firstly synthesized 3-acrylamido-N-(2-(tert-butoxy)-2-oxoethyl)-N,N-dimethylpropane-1-aminium (CBAAM-1-tBu). Then, a typical reversible addition–fragmentation chain-transfer (RAFT) polymerization reaction was performed to yield the 10 kDa and 20 kDa SH-pCB polymer. The final colorless NHS-activated polymer was formed by reaction with AMAS at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio in DI water (pH 6) for 30 min, followed by removal of unreacted AMAS via Amicon spin dialysis tubes and freeze-drying for 48 h.
:10 molar ratio in DI water (pH 6) for 30 min, followed by removal of unreacted AMAS via Amicon spin dialysis tubes and freeze-drying for 48 h.
Synthesis of protein conjugates
Conjugates of pCB-IFN and PEG-IFN were synthesized by reacting the NHS ester groups of the polymer with available amine groups on the protein. In a typical conjugation reaction, IFN-α2a and NHS-pCB at a 1:3 molar ratio were dissolved in 50 mM sodium borate buffer, pH 9.0. The final protein concentration was ∼5 mg mL−1. The reaction mixture was stirred for 2 hours at 4 °C and stopped by adjusting the pH of the mixture to 4.5 with glacial acetic acid. The polymer–protein conjugate was isolated via a molecular weight cut-off (MWCO) spin dialysis membrane followed by ion exchange chromatography. The same conjugation and purification methods were used to generate PEGylated IFN-α2a. The protein concentrations of the prepared conjugates were determined with the Pierce™ 660 nm protein assay reagent (ThermoFisher Scientific) and confirmed with a Human IFNα (multiple subtypes) bioluminescent ELISA kit (InvivoGen).
In vitro antiproliferative activity
The procedures of in vitro proliferative activity measurement were employed with lymphoblast cells, Daudi. Daudi cells at passage 4 or 5 were cultured in RPMI 1640 medium containing 10% (vol/vol) fetal bovine serum (FBS) and antibiotics at 37 °C in a humidified, 5% CO2 atmosphere. For bioassays, the cells were suspended in 2 × 105 cells per mL and seeded in a 96-well plate (Corning) (50 μL, 1 × 104 cells per well). Conjugated protein samples were diluted in culture medium at twice the desired concentrations (5, 10, 20, 50, 100, 500, 1000, and 10000 pg mL−1) and 50 μL was added to each test cell. Wells without IFN-α2a were used as the negative control, defined as 100% cell viability. After incubation for 72 h, the viability of the cells was determined by the MTT assay.Pharmacokinetics
The pharmacokinetics of native and conjugated IFN-α2a were studied using Sprague Dawley rats (female, body weight 101–125 g) as the animal model. All animal experiments adhered to federal guidelines and were approved by the University of Washington Institutional Animal Care and Use Committee (IACUC) under protocol #4203-01. The rats were randomly distributed to six groups (3 rats per group) and injected with IFN-α2a, pCB10K-IFN, pCB20K-IFN, PEG10K-IFN, PEG20K-IFN, and PEG40K-IFN at a dosage of 100 μg IFN-equivalent per kg body weight via the tail vein. Blood samples were collected from the tail vein at 5 min, 4 h, 8 h, 24 h, 28 h and 72 h post injection to generate a circulation profile of the different formulations. A pre-dose sample (0 h) was drawn before the injection of the test proteins as a control sample. The concentration of IFN-α2a was determined by the ELISA assay according to the instructions of the human IFNα (multiple subtypes) bioluminescent ELISA kit (InvivoGen). Pharmacokinetic parameters were analyzed by PKSolver following the instruction. The IV injections and bleeding procedure were repeated 3 times, with one week between each injection. Five weeks after the first injection, 5 mL blood was drawn by using cardiac punch, and serum was prepared for antibody detection.Results and discussion
IFN-α2a is a small protein (19.2 kDa) which targets large cellular receptors on the cell membrane. In clinical applications, unconjugated IFN-α2a can be rapidly cleared from the body through renal filtration due to its small size. PEGylation of IFN-α2a dramatically increases its circulation time. However, the bioactivity of PEGylated IFN-α2a is only 7% of its native form. In this work, as illustrated in Fig. 1a and b, zwitterionic polymers with different molecular weights were conjugated to IFN-α2a through a two-step method as we reported before,29 and the schematic is illustrated in Fig. 1a. To make a fair comparison between zwitterlated and PEGylated conjugates, PEGylated IFN-α2a with both similar molecular weights and hydrodynamic sizes to zwitterlated IFN-α2a were prepared and compared. The size differences of various polymers and IFN-α2a conjugates are presented in Fig. S1† and Fig. 1c. After purification, pCB-IFN and PEG-IFN with single-polymer conjugation were obtained. The successful synthesis of mono-PEGylated or mono-zwitterlated IFN was confirmed by SDS-PAGE analysis (Fig. S2†). The hydrodynamic size and polydispersity index of the polymer-IFN conjugates (Table S1†) further confirmed the successful preparation and the monodispersity of the conjugates. The conversions and yields of the conjugate reactions are shown in Table S2.† | ||
| Fig. 1 Synthesis and characterization of zwitterionic polymer–IFN conjugates. (a) Synthetic route to conjugates; (b) schematic illustration of conjugates of different molecular weights; (c) representative size-exclusion chromatograms for conjugates of different molecular weights. | ||
IFNs are highly pleiotropic cytokines and exhibit potent antiproliferative properties.30 To demonstrate whether zwitterlation could reduce the bioactivity loss of IFN-α2a, we employed a widely accepted antiproliferation assay to study the in vitro bioactivity change of zwitterlated and PEGylated IFN-α2a conjugates. In brief, the antiproliferation assay was performed by serial dilutions in 96-well microtiter plates seeded with Daudi cells, a human lymphoblastoid cell line. After 72 h incubation, cells were treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and the microplate reader was analyzed for viability. Antiproliferative activity, expressed as IC50, was calculated by a nonlinear regression method (Fig. 2a). As presented in Fig. 2b, the antiproliferative activity of pCB-IFNs is much higher than that of PEGylated IFNs. Notably, pCB20k-IFN and PEG20k-IFN have a similar molecular weight, but the antiproliferative activity of pCB20k-IFN (62.1%) is 4.4-fold higher than that of PEG20k-IFN (14.2%). Considering the hydrodynamic volume difference, we also prepared PEG10k-IFN, which has a similar hydrodynamic size to pCB20k-IFN. The antiproliferative activity of PEG10k-IFN is 20.9% and is about 1/3 of pCB20k-IFN. These results indicate that pCB conjugation can better mitigate bioactivity loss than PEGylation.
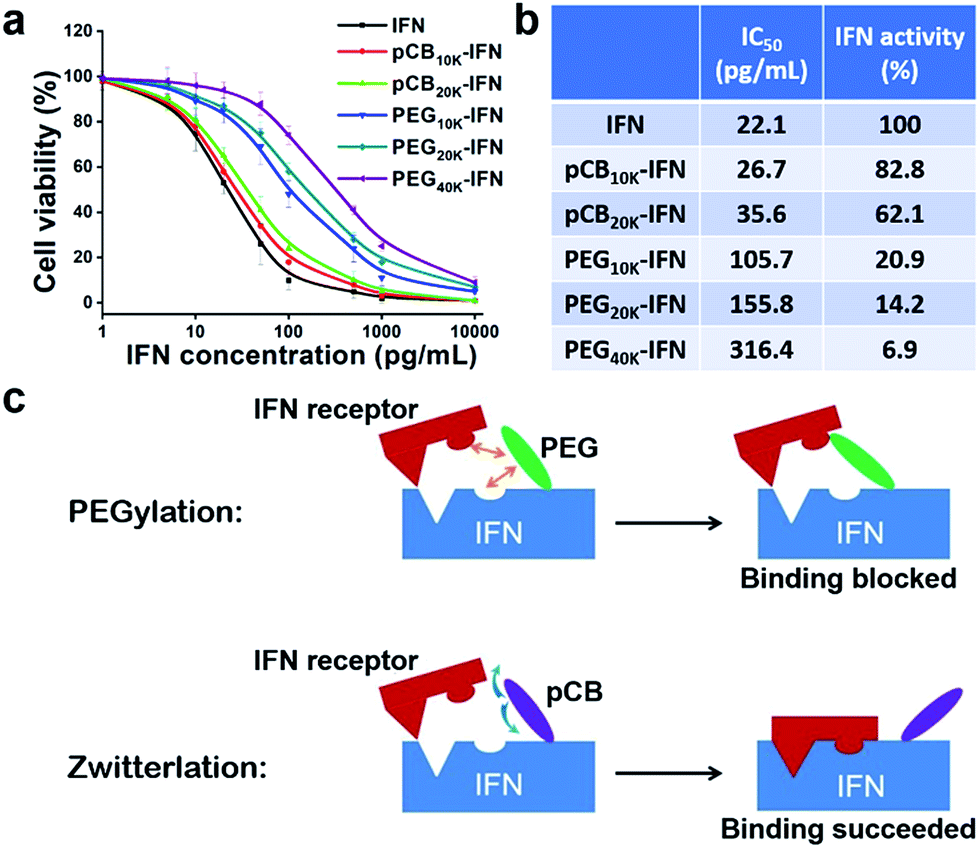 | ||
| Fig. 2 Zwitterlation retains the most bioactivity of conjugated IFN. (a) Conjugates with serial dilutions were incubated with human Daudi cells for 4 days at 37 °C in 96-well tissue culture plates. MTT assay was used for the cell viability assay; (b) antiproliferative activity; (c) schematic illustration of how a zwitterionic conjugate avoids bioactivity loss by effectively eliminating nonspecific interactions. | ||
As presented in Fig. 2c, we hypothesize that the bioactivity loss after PEGylation is mainly attributed to the steric effect and nonspecific interactions of amphiphilic PEG with both the interferon binding domain and interferon receptor. This nonspecific blocking effect sometimes does not significantly affect the bioactivity when the protein size is large, and the substrate size is small (e.g., uricase). However, for conjugated proteins whose receptors have a similar or larger size, this nonspecific blocking usually results in a significant bioactivity loss (e.g., interferon). In contrast to PEGylation, we hypothesize that zwitterionic conjugation could mitigate this bioactivity loss due to the reduced nonspecific interactions of pCB polymers with both the IFN-α2a binding domain and IFN-α2a receptor (Fig. 2c). Due to the tightly bound water layer around zwitterionic polymers, nonspecific interactions between either the IFN-α2a or IFN-α2a receptor and zwitterionic polymers can be minimized. As a result, the bioactivity loss of pCB conjugated IFN-α2a can be reduced.
Monomeric PEGylated IFNs suffer from accelerated blood clearance upon repeated injections in monkeys.31 In this work, encouraged by the in vitro results, we further examined the in vivo performance of zwitterlated, PEGylated and unconjugated IFN-α2a conjugates with the same doses injected every week for three weeks. The blood was harvested at different time points to examine the circulation time. As shown in Fig. 3a, for the circulation profile of the first injection, both PEGylation and zwitterlation significantly improve the circulation time when compared with unconjugated IFN-α2a. In brief, we observed a 47.8-fold increase in t1/2 when pCB20k was conjugated. At the same time, a 26.6-fold increase was observed when PEG with a similar molecular weight (PEG20k) was conjugated and a 11.9-fold increase was observed when PEG with a similar hydrodynamic size (PEG10k) was conjugated. Notably, the circulation time of pCB20k-IFN is comparable to that of PEG40k-IFN (Pegasys) while its bioactivity is 9-fold higher although pCB20k is half of PEG40K in its molecular weight (Fig. 2b). In this work, we found that the increased molecular weight of conjugated polymers could further improve the circulation time. As presented, the t1/2 of pCB20k-IFN shows a 2.3-fold increase over the t1/2 of pCB10k-IFN. A similar scenario was observed in PEG conjugation where a 2.2-fold increase was achieved when 10 kDa PEG was replaced with 20 kDa PEG.
 | ||
| Fig. 3 Zwitterlation mitigates the accelerated blood clearance. (a) In vivo circulation profiles of native IFN-α2a and pCB or PEG conjugated IFN-α2a; (b) pharmacokinetic parameters after repeated injections. | ||
Upon repeated injection, a significant difference was observed between pCB-IFNs and PEG-IFNs. As presented in Fig. 3a and b, for unconjugated IFN-α2a, we noticed a 15.6% and 36.2% decrease in t1/2 after the 2nd and 3rd injections, respectively when compared with the first injection. Despite PEG conjugation can significantly increase the circulation time of IFN-α2a after the 1st injection, the circulation profiles show a 20.0% and 46.3% decrease in t1/2 after the 2nd and 3rd injections respectively for PEG10k-IFN, and a 16.2% and 34.9% decrease in t1/2 after the 2nd and 3rd injections respectively for PEG20k-IFN. These results show the “accelerated blood clearance” effect32 of PEG conjugation after repeated injection. For this reduced circulation time, PEG10k-IFN is even more evident than unconjugated IFN-α2a. In sharp contrast to unconjugated IFN-α2a and PEGylated IFN-α2a, zwitterlated IFN-α2a does not show a significant decline in t1/2 after the 2nd and 3rd injections.
It is known that the surface attachment of hydrophilic polymers like PEG to a protein can significantly prolong the in vivo circulation time by increasing its hydrodynamic size to avoid rapid renal clearance (for small particles) and reducing interactions with both blood components (opsonization) and immune cells in early studies.4,8 However, several studies since then have found anti-PEG antibodies after treatment with PEGylated therapies, with titers strongly related to the modification density of PEG chains and the immunogenicity of the anchoring protein.33,34 The haptenic character of PEG is considered as the main culprit in the therapeutic efficacy loss of PEGylated products. Recent clinical studies of pegloticase (PEGylated uricase) in refractory chronic gout patients unequivocally demonstrated that the production of anti-PEG antibodies is responsible for the reduction in drug effectiveness—pegloticase loses efficacy in more than 40% of patients, and the presence of anti-PEG antibodies doubles the risk of infusion reactions.35,36 Similar results have also been observed for other PEGylated proteins in clinical use such as PEG-asparaginase.37 PEGylated nanoparticles such as liposomes also stimulate a strong anti-PEG response, and these antibodies are found to cross-react with different PEGylated products.38,39 Furthermore, there is a concern that the prevalence of anti-PEG antibodies was found to be 72% (273/377) in healthy individuals, attributed to daily exposure to consumer products.40 However, a cautious examination of whether these pre-existing antibodies have an equivalent effect on different PEGylated proteins is needed. Furthermore, antibodies toward proteins (e.g., peginterferon-alfa41) can also further reduce the therapeutic effect of PEGylated proteins. Altogether, these findings raise concerns regarding the toxicity and efficacy of PEGylated drugs. As a result, alternatives to this PEGylation strategy are urgently needed.42,43 Systematic studies have shown that PEG immunogenicity strongly results from the hydrophobic characteristics of this amphiphilic polymer.44–46 Due to the super-hydrophilic nature of zwitterionic pCB polymers, we hypothesized that zwitterlation could mitigate the immunogenicity of IFN-α2a. At the same time, zwitterionic polymers do not elicit a strong immune response in the body.
To confirm this hypothesis, we further proceeded to analyze the production of IgM and IgG antibodies after repeated injections. The results are summarized in Fig. 4. As presented, both PEGylation and zwitterlation can mitigate the generation of IFN-α2a-specific antibodies after repeated injections. For PEGylation, at the lowest dilution, the protein-specific IgM titers for PEG10k-IFN and PEG20k-IFN groups are 36.7% and 31.1% of that of the unconjugated IFN-α2a group. The protein-specific IgG titers for PEG10k-IFN and PEG20k-IFN groups are 57.4% and 51.1% of that of the unconjugated IFN-α2a group. At the same time, it is observed that zwitterlation can further decrease the production of both protein-specific IgM and IgG. At the lowest dilution, the protein-specific IgM titers for pCB10k-IFN and pCB20k-IFN groups are 22.4% and 16.7% of that of the unconjugated IFN-α2a group. The protein-specific IgG titers for pCB10k-IFN and pCB20k-IFN groups are 26.9% and 23.5% of that of the unconjugated IFN-α2a group. These results also indicate that pCB polymers can better protect the conjugated IFN-α2a from immunosurveillance than PEG polymers. We further examined the production of specific IgM and IgG production against PEG and pCB polymers. As presented in Fig. 4c and d, we observed a dramatic difference between PEG and pCB conjugates for their antibody production specifically against polymers. At the lowest dilution, we found that pCB-IFNs induce negligible anti-polymer production of either IgM or IgG compared with PEG-IFNs. These results indicate that the accelerated blood clearance of PEG-IFN is mainly attributed to the production of antibodies against both IFN-α2a and the PEG polymer. For pCB-IFN, since pCB polymers can mitigate the antibody production against IFN-α2a and do not elicit specific antibody production against themselves in the body, the accelerated blood clearance can be significantly attenuated.
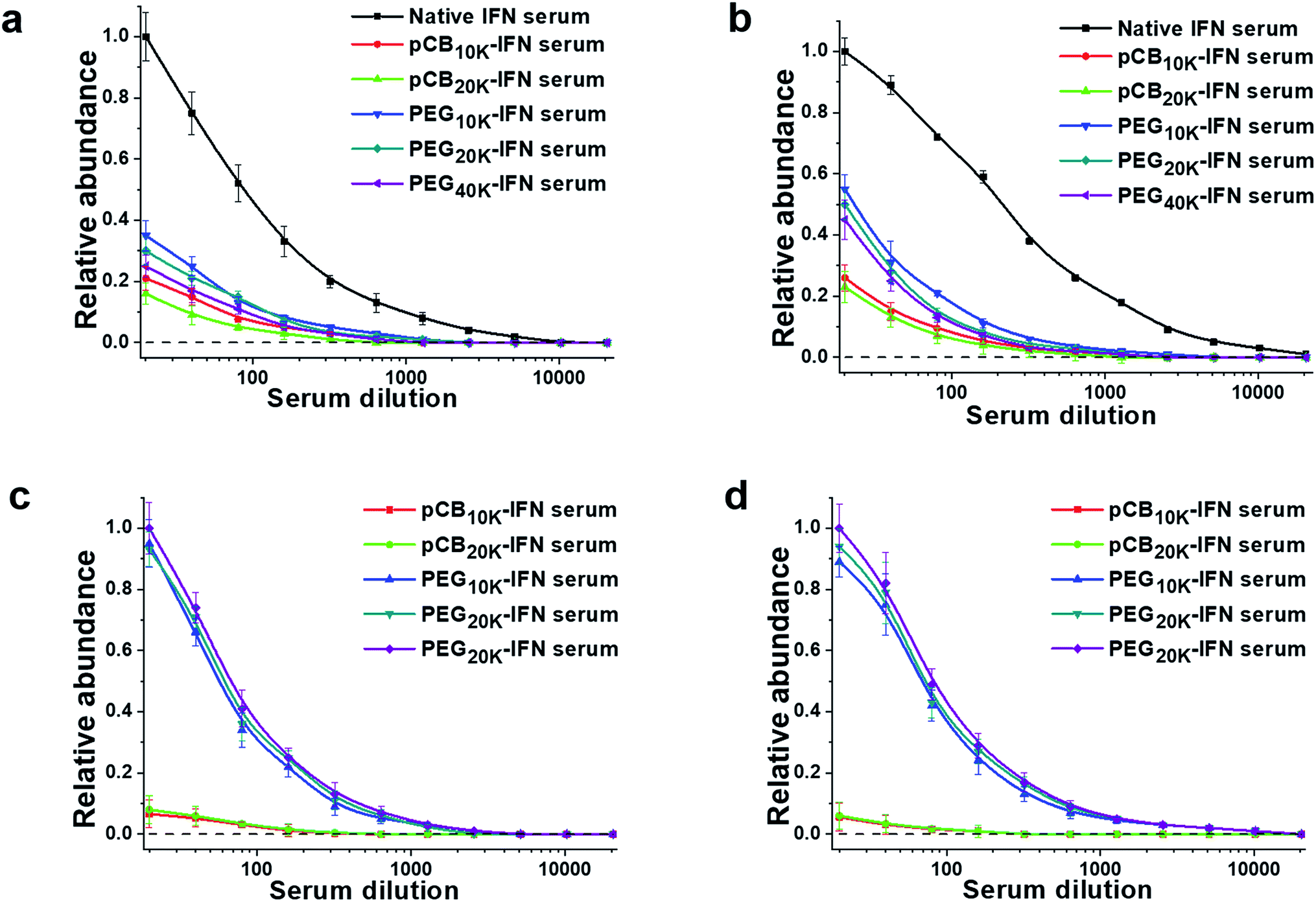 | ||
| Fig. 4 Zwitterlation mitigates the production of specific antibodies against the polymer and IFN. After 3 injections, IFN-specific IgM (a) and IgG (b) antibodies and polymer-specific IgM (c) and IgG (d) antibodies were analysed. | ||
Conclusions
In summary, protein–polymer conjugation has been demonstrated to be an effective method to improve the therapeutic result of protein drugs in vivo. However, for proteins whose receptors/substrates have a large size, PEG polymer conjugation often results in a significant loss in protein bioactivity. In this work, we selected IFN-α2a as a model protein whose in vitro bioactivity can be reduced by more than 90% after PEGylation. We demonstrate that zwitterionic conjugation can significantly retain the in vitro antiproliferative bioactivity of conjugated IFN-α2a due to the super-hydrophilic nature of zwitterionic polymers, and thus the ‘nonspecific blocking’ issue in PEGylation can be mitigated. Furthermore, in vivo results show that zwitterionic conjugation prolongs the IFN-α2a circulation time and attenuates its accelerated blood clearance significantly. Further analysis reveals that zwitterlation mitigates the production of antibodies toward both IFN-α2a and the polymer. This work indicates that zwitterionic polymer conjugation may be particularly suitable for the conjugation of therapeutic proteins with large binding targets. Our next step will focus on the in vivo bioactivity study of zwitterlated proteins.Conflicts of interest
S. Jiang is a cofounder of Furtim Therapeutics LLC.Acknowledgements
This work was funded by the National Science Foundation (DMR-1708436).Notes and references
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef PubMed.
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef PubMed.
- F. M. Veronese and G. Pasut, Drug discovery today, 2005, 10, 1451–1458 CrossRef PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 CrossRef PubMed.
- C. S. Fishburn, J. Pharm. Sci., 2008, 97, 4167–4183 CrossRef PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef PubMed.
- G. Molineux, Cancer Treat. Rev., 2002, 28, 13–16 CrossRef PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef PubMed.
- T. J. Dougherty and M. J. Pucci, Antibiotic discovery and development, Springer Science & Business Media, 2011 Search PubMed.
- B. Podobnik, B. Helk, V. Smilovic, S. Skrajnar, K. Fidler, S. Jevsevar, A. Godwin and P. Williams, Bioconjugate Chem., 2015, 26, 452–459 CrossRef PubMed.
- M. S. Hershfield, R. H. Buckley, M. L. Greenberg, A. L. Melton, R. Schiff, C. Hatem, J. Kurtzberg, M. L. Markert, R. H. Kobayashi and A. L. Kobayashi, N. Engl. J. Med., 1987, 316, 589–596 CrossRef PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59 CrossRef PubMed.
- C. Leng, H.-C. Hung, S. Sun, D. Wang, Y. Li, S. Jiang and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 16881–16888 CrossRef PubMed.
- S. Shaunak, A. Godwin, J.-W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312 CrossRef PubMed.
- B. K. Lee, J. S. Kwon, H. J. Kim, S. Yamamoto and E. Lee, Bioconjugate Chem., 2007, 18, 1728–1734 CrossRef PubMed.
- Y. Qi and A. Chilkoti, Curr. Opin. Chem. Biol., 2015, 28, 181–193 CrossRef PubMed.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J.-R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553 CrossRef PubMed.
- T. Bai, F. Sun, L. Zhang, A. Sinclair, S. Liu, J. R. Ella-Menye, Y. Zheng and S. Jiang, Angew. Chem., 2014, 126, 12943–12948 CrossRef.
- G. Sener and M. D. Krebs, RSC Adv., 2016, 6, 29608–29611 RSC.
- R. Rajan and K. Matsumura, Sci. Rep., 2017, 7, 45777 CrossRef PubMed.
- J.-H. Seo, R. Matsuno, Y. Lee, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 4859–4867 CrossRef PubMed.
- S. Bhattacharjee, W. Liu, W. H. Wang, I. Weitzhandler, X. Li, Y. Qi, J. Liu, Y. Pang, D. F. Hunt and A. Chilkoti, ChemBioChem, 2015, 16, 2451–2455 CrossRef PubMed.
- A. Lewis, Y. Tang, S. Brocchini, J.-w. Choi and A. Godwin, Bioconjugate Chem., 2008, 19, 2144–2155 CrossRef PubMed.
- K. P. García, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2505 CrossRef.
- P. Zhang, F. Sun, C. Tsao, S. Liu, P. Jain, A. Sinclair, H.-C. Hung, T. Bai, K. Wu and S. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046–12051 CrossRef PubMed.
- J. Xie, Y. Lu, W. Wang, H. Zhu, Z. Wang and Z. Cao, Adv. Healthcare Mater., 2017, 6, 1601428 CrossRef PubMed.
- S. Liu and S. Jiang, Nano Today, 2016, 11, 285–291 CrossRef.
- M. R. Walter, R. Bordens, T. L. Nagabhushan, B. R. Williams, R. B. Herberman, C. A. Dinarello, E. C. Borden, P. P. Trotta, S. Pestka and L. M. Pfeffer, Cancer Biother.Radiopharm., 1998, 13, 143–154 CrossRef PubMed.
- X. Hu, K. Olivier, E. Polack, M. Crossman, K. Zokowski, R. S. Gronke, S. Parker, Z. Li, I. Nestorov and D. P. Baker, J. Pharmacol. Exp. Ther., 2011, 338, 984–996 CrossRef PubMed.
- Q. Yang and S. K. Lai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 655–677 Search PubMed.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef PubMed.
- A. Richter and E. Åkerblom, Int. Arch. Allergy Appl. Immunol., 1983, 70, 124–131 CrossRef PubMed.
- M. S. Hershfield, N. J. Ganson, S. J. Kelly, E. L. Scarlett, D. A. Jaggers and J. S. Sundy, Arthritis Res. Ther., 2014, 16, R63 CrossRef PubMed.
- P. E. Lipsky, L. H. Calabrese, A. Kavanaugh, J. S. Sundy, D. Wright, M. Wolfson and M. A. Becker, Arthritis Res. Ther., 2014, 16, R60 CrossRef PubMed.
- J. K. Armstrong, G. Hempel, S. Koling, L. S. Chan, T. Fisher, H. J. Meiselman and G. Garratty, Cancer, 2007, 110, 103–111 CrossRef PubMed.
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354, 56–62 CrossRef PubMed.
- K. Shiraishi, M. Hamano, H. Ma, K. Kawano, Y. Maitani, T. Aoshi, K. J. Ishii and M. Yokoyama, J. Controlled Release, 2013, 165, 183–190 CrossRef PubMed.
- Q. Yang, T. M. Jacobs, J. D. McCallen, D. T. Moore, J. T. Huckaby, J. N. Edelstein and S. K. Lai, Anal. Chem., 2016, 88, 11804–11812 CrossRef PubMed.
- A. A. van der Eijk, J. M. Vrolijk and B. L. Haagmans, N. Engl. J. Med., 2006, 354, 1323–1324 CrossRef PubMed.
- E. M. Pelegri-O’Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- T. J. Styslinger, N. Zhang, V. S. Bhatt, N. Pettit, A. F. Palmer and P. G. Wang, J. Am. Chem. Soc., 2012, 134, 7507–7515 CrossRef PubMed.
- M. G. Saifer, L. D. Williams, M. A. Sobczyk, S. J. Michaels and M. R. Sherman, Mol. Immunol., 2014, 57, 236–246 CrossRef PubMed.
- E. Schneck, I. Berts, A. Halperin, J. Daillant and G. Fragneto, Biomaterials, 2015, 46, 95–104 CrossRef PubMed.
- M. R. Sherman, L. D. Williams, M. A. Sobczyk, S. J. Michaels and M. G. Saifer, Bioconjugate Chem., 2012, 23, 485–499 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01777h |
| This journal is © The Royal Society of Chemistry 2018 |