
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise supramolecular control of surface coverage densities on polymer micro- and nanoparticles†
Shuai
Zhang
a,
Zoe
Domínguez
b,
Khaleel I.
Assaf
 a,
Mohamed
Nilam
a,
Thomas
Thiele
c,
Uwe
Pischel
b,
Uwe
Schedler
c,
Werner M.
Nau
a and
Andreas
Hennig
*a
a,
Mohamed
Nilam
a,
Thomas
Thiele
c,
Uwe
Pischel
b,
Uwe
Schedler
c,
Werner M.
Nau
a and
Andreas
Hennig
*a
aDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, D-28759 Bremen, Germany. E-mail: a.hennig@jacobs-university.de
bPolyAn GmbH, Rudolf-Baschant-Strasse 2, D-13086 Berlin, Germany
cCIQSO – Center for Research in Sustainable Chemistry, Department of Chemistry, University of Huelva, Campus de El Carmen, E-21071 Huelva, Spain
First published on 18th September 2018
Abstract
We report herein the controlled surface functionalization of micro- and nanoparticles by supramolecular host–guest interactions. Our idea is to exploit the competition of two high-affinity guests for binding to the surface-bound supramolecular host cucurbit[7]uril (CB7). To establish our strategy, surface azide groups were introduced to hard-sphere (poly)methylmethacrylate particles with a grafted layer of poly(acrylic acid), and a propargyl derivative of CB7 was coupled to the surface by click chemistry. The amount of surface-bound CB7 was quantified with the high-affinity guest aminomethyladamantane (AMADA), which revealed CB7 surface coverage densities around 0.3 nmol cm−2 indicative of a 3D layer of CB7 binding sites on the surface. The potential for surface functionalization was demonstrated with an aminoadamantane-labeled rhodamine (Ada-Rho) as a second high-affinity guest. Simultaneous incubation of CB7-functionalized particles with both high-affinity guests, AMADA and Ada-Rho, revealed a simple linear relationship between the resulting surface coverage densities of the model fluorescent dye and the mole fraction of Ada-Rho in the incubation mixture. This suggests a highly modular supramolecular strategy for the stable immobilization of application-relevant molecules on particle surfaces and a precise control of their surface coverage densities.
Introduction
The possibility to precisely control the attachment of application-relevant molecules to the surfaces of micro- and nanoparticles creates a powerful platform technology with a large number of conceivable applications. A vast number of different methods have therefore been explored, but the performance of these materials is still limited by the shortcomings of existing surface functionalization methods.1–3 For example, the highly specific and strong (Ka ∼ 1015 M−1) binding interaction between the small-molecule ligand biotin with the proteins avidin or streptavidin is popular for surface attachment in initial proof-of-principle studies with nanoparticles,1 but it has also been noted that the tetrameric structure of the proteins and their large size (i) may induce crosslinking, (ii) prevent a precise control of conjugate stoichiometry, and (iii) limit the maximum achievable surface coverage densities.4–6 Alternative and refined strategies to reliably functionalize particle surfaces with molecular components are thus highly desired to achieve the transition from proof-of-principle to real applications of nanobioconjugate materials.1–3Numerous supramolecular host–guest systems have been previously investigated with the goal to equip nanoparticles with molecular recognition capabilities, and this has enabled a large variety of potential applications.7–22 The typically μM to mM affinities of most synthetic host–guest systems ensure reversible binding, which is highly desirable, e.g., in nanoparticle polymer composites, to control nanoparticle assembly, or for drug delivery. However, for surface functionalization aiming towards bioanalytical applications, host–guest complexes are required, which are sufficiently stable at much lower concentrations. To account for the low binding affinity, multivalent systems have been explored, but this strategy sacrifices – similar to the (strept)avidin/biotin system – control over the binding stoichiometry and induces particle crosslinking.11,14,23,24
Cucurbit[n]urils (CBn, n = 5–8, 10, and 14) composed of n glycoluril units comprise a class of biocompatible macrocycles, which stand out from all other supramolecular host molecules by remarkably high binding affinities (Ka > 1017 M−1)25–30 towards certain guest molecules. This exceeds the affinity of biotin with (strept)avidin and clearly suggests CBn hosts as a complementary tool in bioconjugation.4,31 Moreover, CBs are highly biocompatible as shown in various applications, e.g., in enzyme and membrane transport assays,32–36 for immobilization of proteins and cells on planar surfaces,37–39 for enrichment and isolation of proteins by affinity-beads,40–42 for supramolecular PEGylation of biopharmaceuticals,43 for multi-stimuli-responsive release,44 and for protein imaging.45 CBn hosts were also adsorptively attached to metal surfaces such as planar46,47 and spherical48–50 gold surfaces, and to iron oxide nanoparticles through multidentate binding of their carbonyl-fringed portals.51,52 However, this adsorptive surface functionalization affects the host–guest recognition properties of the cavity since one of the portals is involved in surface binding. Moreover, the presence of two carbonyl-fringed portals may lead to particle aggregation.46,51–53
Herein, we present for the first time the covalent surface modification of small, hard-sphere core–shell particles with cucurbit[7]uril (CB7). To demonstrate this, we use polymer microparticles, also known as beads or microspheres, as well as nanoparticles. These particles play important roles, e.g., in optical tweezers,54 drug delivery,55 medical imaging,56,57 and in diagnostic, multiplexing bead-based assays,58–60 as well as lateral flow immunoassays.61,62 Our approach affords, similar to the previously established planar surfaces37–39 and porous resins of large (>40 μm) sepharose beads,40–42 a suitable supramolecular strategy to subsequently immobilize application-relevant molecules. Moreover, we demonstrate herein, that host–guest chemistry allows an unprecedented control of surface functionalization of particles in an easily quantifiable manner.
Results and discussion
Particle synthesis
For the synthesis of CB7-functionalized particles, we decided to use copper-catalyzed azide–alkyne click chemistry (Fig. 1). To obtain the required propargyl-CB7 (CB7-OPr), a different procedure than the recently reported method by Zhang and coworkers was established.63 First, monohydroxylated-CB7 was synthesized according to the method of Bardelang and coworkers.64 Quantitative conversion into CB7-OPr was then achieved by repeated cycles of crude product resubmission to propargyl bromide and sodium hydride in DMSO, and the identity and purity of CB7-OPr was confirmed by comparison with the reported data (see ESI†).63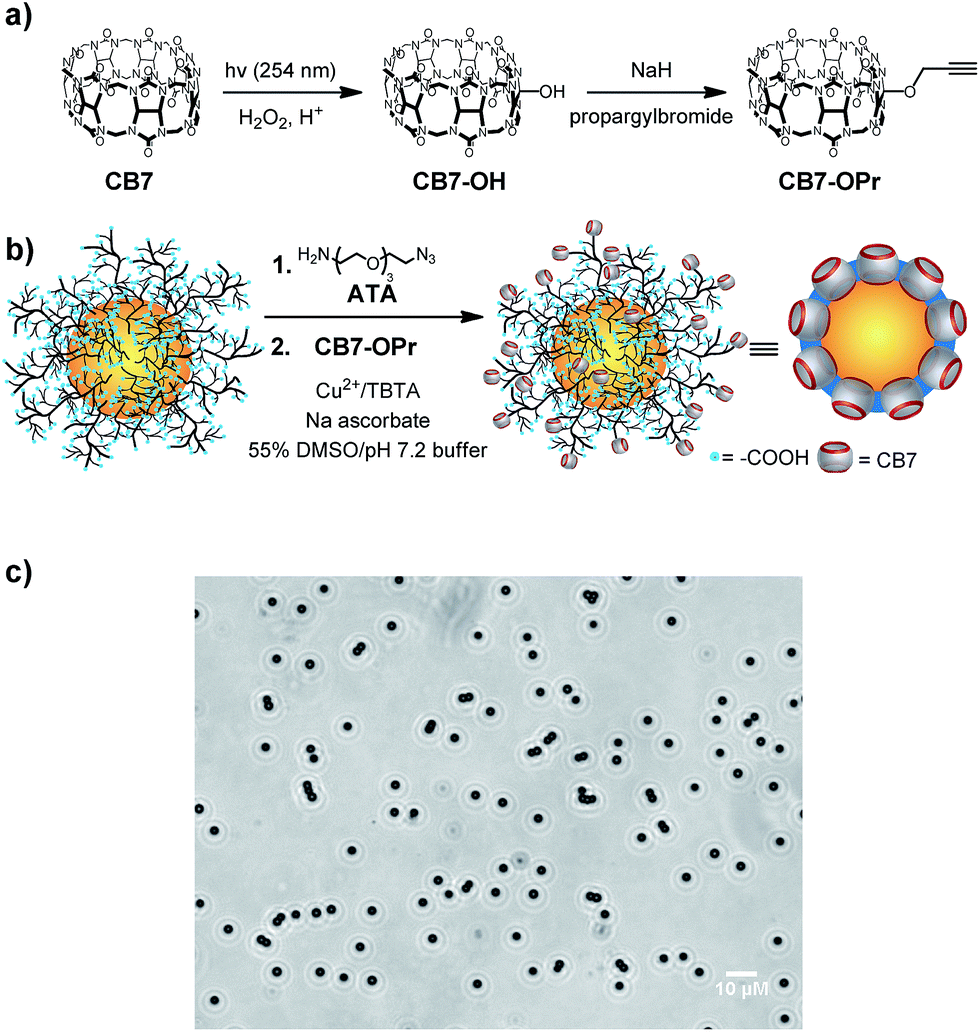 | ||
| Fig. 1 (a) Synthesis of propargyl-CB7 (CB7-OPr). (b) Synthesis of CB7-functionalized PMMA microparticles. (c) Optical microscopy image (at 40-fold magnification) of 5 mg mL−1 CB7-functionalized PMMA particles in 10 mM (NH4)2HPO4, pH 7.2. | ||
As particles, we used microparticles (mean diameter of 2.55 μm) composed of a compact, hard-sphere poly(methylmethacrylate) (PMMA) core and a grafted layer (111 μmol g−1) of poly(acrylic acid) (PAA). These particles were chosen for their ease of handling, optical transparency, and the possibility for microscopic observation, and they were previously characterized by us in detail.6,65–69 In addition, commercially available nanoparticles (mean diameter of 110 nm) with a hard-sphere polystyrene core and surface carboxylic acids were also tested.
Azide groups were introduced by reaction of surface COOH groups with 11-azido-3,6,9-trioxaundecan-1-amine (ATA) using standard amide coupling protocols,6,65 and CB7 was finally covalently bound to the surface by Cu-catalyzed click chemistry (Fig. 1). The resulting particles were washed into 10 mM (NH4)2HPO4, pH 7.2, which was also used for all subsequent experiments. Inspection by optical microscopy indicated no increased tendency for aggregation compared to the PAA- and ATA-functionalized particles (Fig. 1c and S7†), and IR spectroscopic results were in accordance with surface-immobilized CB7 (Fig. S8†).
Quantification of surface-bound CB7
The most compelling evidence for surface functionalization with CB7 was obtained by successful extraction of aminomethyladamantane (AMADA) from 10 mM (NH4)2HPO4, pH 7.2 using CB7-functionalized particles (Fig. 2). The ultra-strong affinity of AMADA to CB7 (Kaca. 1015 M−1)27 was previously exploited by us to evaluate various surface quantification methods.6,65–69 Therein, AMADA-putrescine was used as a chemical labelling agent for surface COOH groups by amide formation and the number of surface-bound AMADA was determined by extraction of CB7 and subsequent quantification of remaining CB7 by the fluorescent dye acridine orange (AO).6,64 Similarly, remaining AMADA is now quantified by addition of a known concentration of CB7 and AO to the supernatant.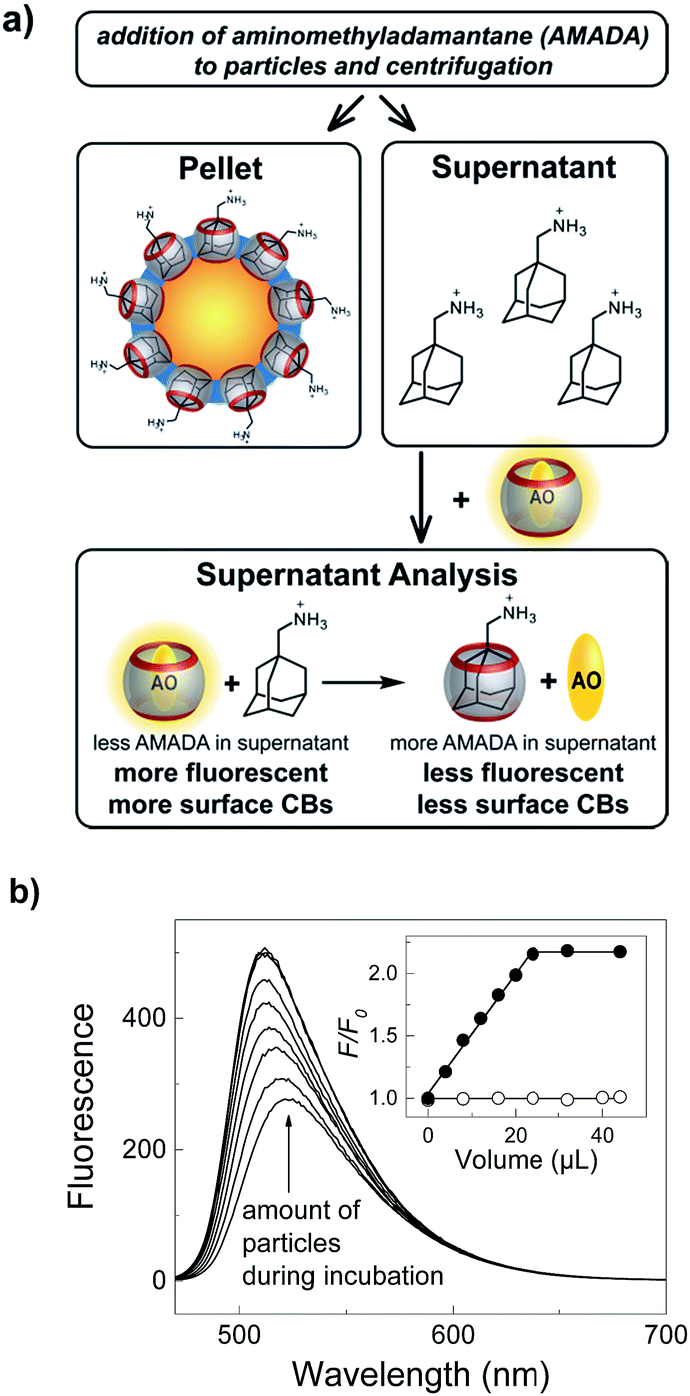 | ||
| Fig. 2 Quantification of CB7 on particle surfaces. (a) Incubation of CB7-functionalized particles with AMADA and subsequent centrifugation gives a supernatant, which can be analyzed to afford the concentration of remaining AMADA by addition of the fluorescent dye acridine orange (AO) and CB7. (b) Dependence of fluorescence spectral changes (λexc = 450 nm, λobs = 510 nm) of the supernatant on the volume of added CB7-functionalized particles stock solution (10 mg mL−1) during incubation with 25 μM AMADA. The inset compares CB7-functionalized (filled circles) and ATA-functionalized particles as control (open circles) in 10 mM (NH4)2HPO4, pH 7.2. | ||
In accordance with our expectations, an increase of the fluorescence with increasing volume of the particle stock solution immediately indicated the particle-dependent extraction of AMADA and thus successful immobilization of CB7 on the particle surface (Fig. 2b). The clearly linear dependence with no indications of the typical curvature of a reversible binding isotherm is consistent with quantitative binding between surface-bound CB7 and AMADA, as well as with quantitative binding in the supernatant analysis (Fig. S9†). As a consequence, we can determine the average (bulk) CB7 surface coverage densities from the intersection between the linear increase and the plateau region in the inset of Fig. 2, which indicates the amount of particles required to extract all AMADA from solution (see ESI†).65,66
This simple method to determine the resulting CB7 loading capacities and surface coverage densities was highly reproducible (Table S1,† coefficient of variation of ca. 2% for n = 7) and enabled us to evaluate various reaction conditions and their reproducibility during surface functionalization in a straightforward manner (Table S2†). For example, we could show that washing the particles after click reaction with buffer containing EDTA or not had no influence on the resulting surface coverage, which suggests that no copper ions remained on the particle surface (cf. entry #1 and #2 in Table S2†). Furthermore, we could establish that the click reaction is considerably reproducible. The resulting loading capacities of three different reaction batches varied by less than 1% (Table S2,† entry #1).
The resulting CB7 surface coverage densities (ca. 0.3 nmol cm−2, i.e. 1.8 CB7 molecules per nm2 or a loading capacity of ca. 5.7 μmol g−1) were significantly higher than the value for a CB7 monolayer on planar gold surfaces (ca. 0.08 nmol cm−2),38 which is in accordance with a grafted 3D layer of PAA on the particle surface and thus a 3D layer of surface-bound CB7. Interestingly, the overall coupling yields in our two-step functionalization protocol were in very good agreement with typical coupling yields for amide formation only (ca. 5% with respect to COOH groups of surface PAA),6,66 which suggests that the second step, the click reaction to attach CB7 onto the surface, is nearly quantitative. As controls, azide-functionalized particles lacking CB7 gave no change in fluorescence intensity (inset of Fig. 2b), which excluded unspecific binding of AMADA to the particle surface. As additional controls, identical values for the CB7 surface coverage density were determined by extraction of (dimethylaminomethyl)ferrocene (Fig. S10†), which further excludes any unspecific binding. Moreover, poly(styrene) nanoparticles with surface carboxylic acid groups could be similarly surface-functionalized and analyzed, which afforded CB7 surface coverage densities of ca. 0.1 nmol cm−2 (Fig. S11†).
Supramolecular surface functionalization
The possibility to reliably immobilize application-relevant molecules on the surface of CB7-functionalized particles was demonstrated with an aminoadamantyl-labeled rhodamine (Ada-Rho, see ESI† for synthesis) as a model fluorescent dye (Fig. 3). Immobilization was simply achieved by addition of CB7-functionalized particles to a buffered aqueous solution containing Ada-Rho and washing, which gave a bright red fluorescence from surface-bound Ada-Rho in fluorescence microscopy, whereas the size of the particles remained same as judged by comparison of the bright-field images of particles with and without Ada-Rho. The absorbance as well as the fluorescence of the supernatant decreased linearly with increasing amounts of CB7-functionalized particles indicating quantitative binding (Fig. S12†). As controls, when the CB7 cavity was blocked by pre-incubation with the stronger binder AMADA,70 the particles were unable to extract Ada-Rho. Similarly, no Ada-Rho extraction was observed with ATA-functionalized particles (Fig. S13 and S14†).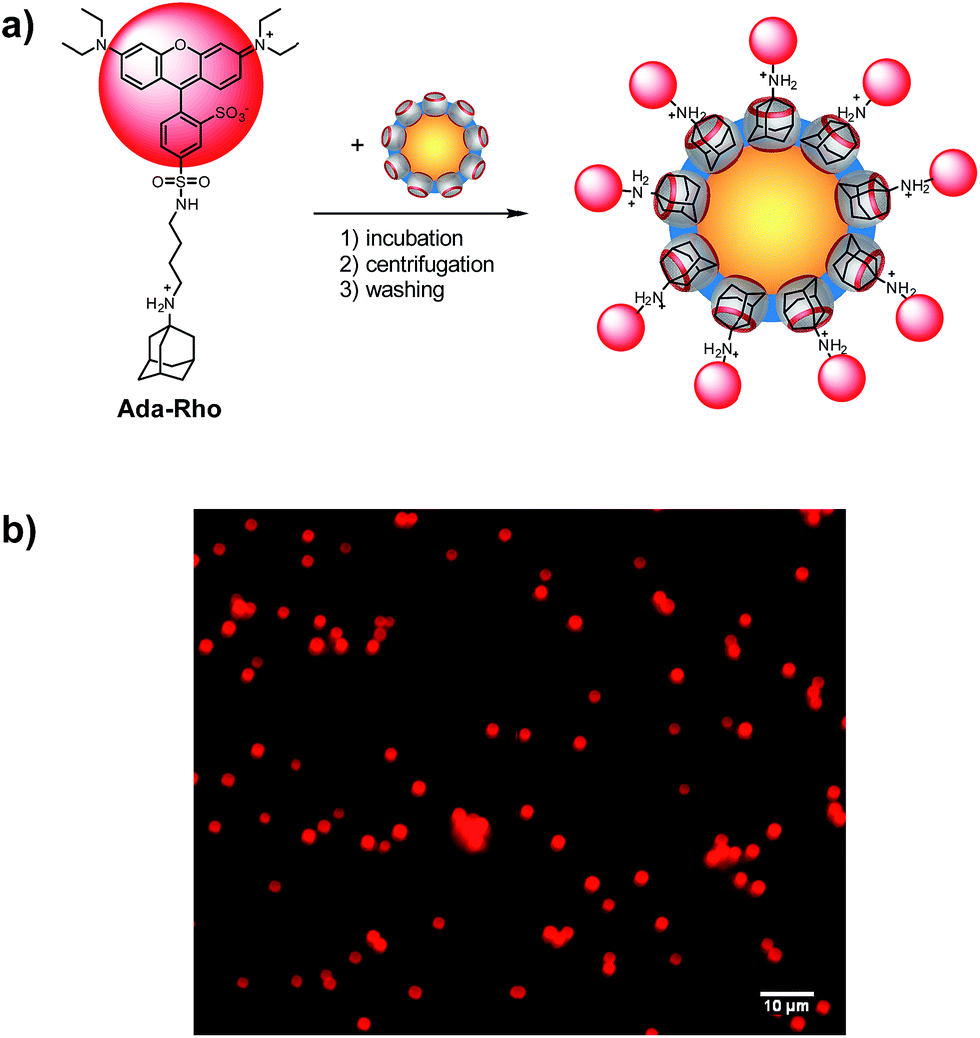 | ||
| Fig. 3 (a) Surface functionalization of CB7 particles with Ada-Rho. (b) Fluorescence microscopy images (λexc = 546 nm) of CB7-functionalized particles (5 mg mL−1) with surface-bound Ada-Rho. | ||
As in the case of AMADA (see above), the loading capacity of Ada-Rho could be determined from the intersection between the linear decrease and the plateau region of the titration plots (see ESI†), which revealed that the amount of Ada-Rho on the surface was significantly lower (3.0 μmol g−1) than the amount of CB7 (5.7 μmol g−1). Such a dependence of the surface coverage density on the size of the immobilized molecule is common, and may be due to steric repulsion between molecules at adjacent binding sites, a size-dependent diffusion through the grafted 3D PAA network, or a conformational rearrangement of the PAA chains in response to the presence of the hydrophobic dye, which may block otherwise available binding sites.
The latter explanation can be ruled out, because particles, in which all binding sites were saturated with Ada-Rho could still extract additional 2.8 μmol g−1 AMADA from solution (Fig. S15†) and the sum of Ada-Rho (3.0 μmol g−1) and AMADA (2.8 μmol g−1) was in excellent agreement with the CB7 surface coverage density determined with AMADA only (5.7 μmol g−1). In other words, approximately 50% of all available CB7 binding sites were occupied when the particles were incubated with Ada-Rho first, and then, the remaining binding sites could be occupied in a second incubation step with AMADA.70 A different result was, however, obtained, when the CB7-functionalized particles were incubated with a mixture of both, Ada-Rho and AMADA. This reduced the amount of Ada-Rho that can be extracted with a specific amount of particles (Fig. S16†), which is consistent with a competitive occupation of the CB7 binding sites by AMADA, and suggests an elegant method to control the surface coverage density of CB7-functionalized particles.
Supramolecular control of surface coverage densities
In order to investigate in detail how the presence of AMADA during incubation with Ada-Rho influences the resulting surface coverage densities, varying amounts of CB7 particles were incubated with mixtures containing different mole fractions of AMADA and Ada-Rho. This indicated that the amount of particles, which are required to extract Ada-Rho from solution, remained approximately the same despite varying total Ada-Rho concentrations in the incubation solution (Fig. S17†). Consequently, the surface coverage densities of Ada-Rho as determined by our extraction-based surface quantification method depended linearly on the molar fraction of AMADA and Ada-Rho over its entire range (Fig. 4).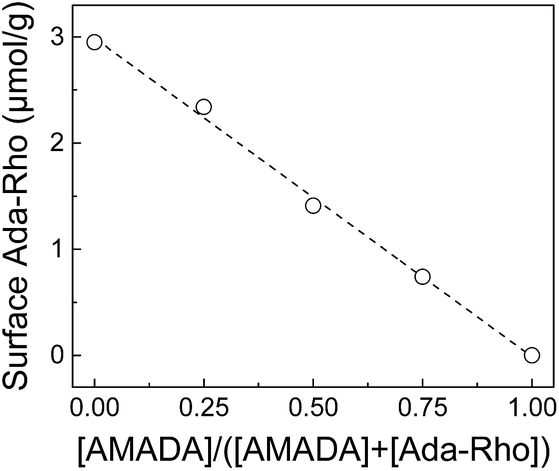 | ||
| Fig. 4 Dependence of resulting Ada-Rho surface coverage densities on the mole fraction of the competitor AMADA in mixtures of AMADA and Ada-Rho (6 μM total concentration). | ||
It is noteworthy that such a simple linear relationship came as a surprise, because first, the number of available binding sites is different for AMADA and Ada-Rho (see above), and second, the resulting surface concentrations of two competitors should, in a thermodynamically equilibrated mixture, also depend on their binding affinities.30,70 We conclude that the occupation of the CB7 binding cavities is diffusion-limited, which leads to a kinetically controlled competitive occupation of the CB7 binding sites with AMADA and Ada-Rho.
In addition to the mechanistic insights, our results clearly demonstrate that the surface coverage densities of application-relevant functional molecules on CB7-functionalized particles can be precisely adjusted by using two competitive cavity binders. This allowed us to prepare a series of particles with exactly known Ada-Rho surface coverage densities in a straightforward manner, which could then be analyzed by fluorescence spectroscopy and microscopy (Fig. 5). Therein, a linear increase in the fluorescence intensity was observed at low surface coverage densities (up to ca. 1 μmol g−1), whereas higher surface fluorophore coverage densities did not lead to a further increase in fluorescence intensity.
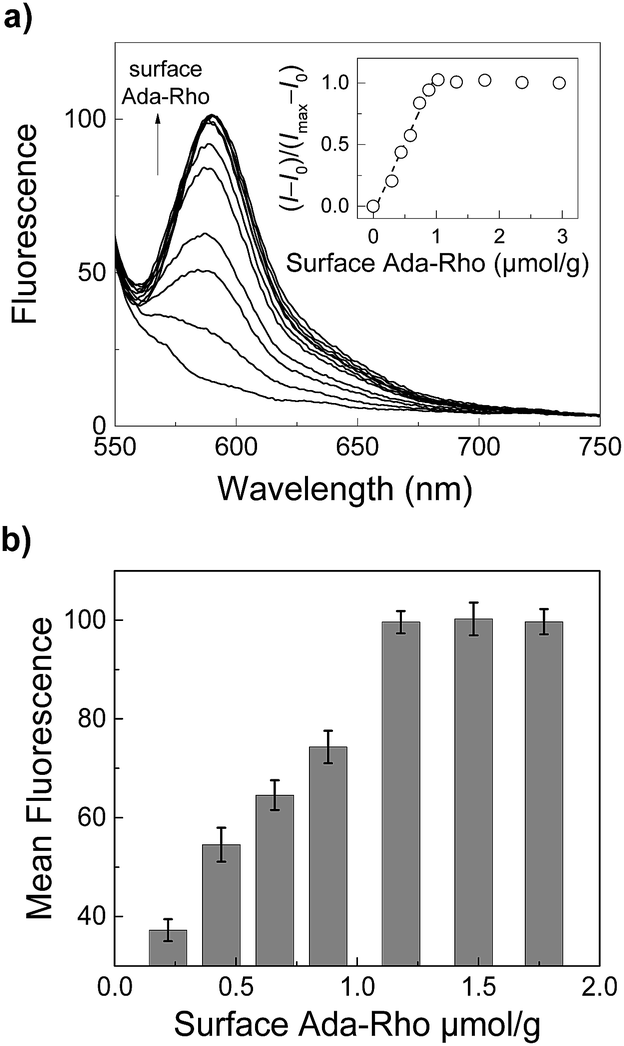 | ||
| Fig. 5 (a) Steady-state fluorescence spectra (λexc = 520 nm) of Ada-Rho-labeled particles (0.2 mg mL−1) in 10 mM (NH4)2HPO4, pH 7.2 with increasing surface coverage densities of Ada-Rho (adjusted by using mixtures of Ada-Rho and AMADA, see Fig. 4). The inset shows the dependence of the normalized fluorescence intensity (λem = 585 nm) on the surface coverage densities. (b) Dependence of the mean fluorescence intensity within the regions of interest (ROIs) in fluorescence microscopy images (see Fig. S18†). Error bars represent the standard deviation (n = 3). | ||
This result is consistent with our previous observations with covalently bound surface fluorophores and originates most likely from self-quenching due to an increased probability of non-fluorescent aggregate formation at high surface coverage densities.6,69 It is important to note that uncertainties arising from the covalent surface modification protocol needed to be previously eliminated by control measurements with absolute fluorometry involving an integrating sphere set-up,6 whereas in this report, we exploit supramolecular host–guest chemistry to unambiguously determine and control the fluorophore surface coverage densities. Another interesting consideration is that our competition-based surface functionalization protocol applies an excess of two competitors for a limited number of accessible binding cavities, which could lead to a more homogeneous distribution of surface coverage densities within a particle population than methods relying on substoichiometric amounts of reagent. The latter require a very efficient mixing to prevent a local depletion during reagent addition to a reaction mixture.
In contrast, when the particles were first surface-functionalized with Ada-Rho and subsequently incubated with high concentrations of AMADA for a longer period, a slow dissociation of Ada-Rho from the surface was observed (Fig. S19†). This clearly demonstrates the principal reversibility of the host–guest interaction despite the strong affinity. Furthermore, it enables the determination of exchange kinetics on particles surfaces and presents a complementary conceptual framework for kinetic studies of host–guest systems.71,72
Conclusions
In conclusion, we have introduced polymer particles surface-functionalized with the supramolecular host molecule CB7. Therefore, we synthesized monofunctionalized CB7 bearing a propargyl group, which can be covalently bound to azide-functionalized surfaces of polymer particles by click chemistry. The successful reaction and the resulting number of CB7 molecules on the particle surface was reliably quantified and the ease of subsequently introducing other molecular components was demonstrated with the fluorescent dye Ada-Rho. Compared with covalent conjugation strategies, simple mixing of the two components in water suffices and other additives such as coupling reagents are not required. Overall, this provides a reliable host–guest-based surface functionalization method in water with wide-ranging perspectives. We have shown that it allows to precisely control surface coverage densities, which unfolds numerous perspectives in varying areas. For example, it allows the investigation of fluorescence quenching mechanisms on surfaces and validation of absolute fluorometry,6,69 the systematic testing of hitherto unverified, theoretical quantification models for spherical substrates in X-ray photoelectron spectroscopy (XPS),68 as well as straightforward construction of multimodal probes for medical imaging,73 potentially for clinical applications, which require regulatory clearance and thus metrological traceability.67 Varying surface coverage densities can have a significant impact on the efficiency of nanoparticle-based diagnostics and therapy.1–3,74–77 We also demonstrated the controllable release of surface-bound Ada-Rho by a stronger cavity binder. This suggests the use of CB7-functionalized particles for the construction of stimuli-responsive release systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Deutsche Forschungsgemeinschaft (HE 5967/4-1 and NA 686/11-1), the China Scholarship Council (S. Z.), the Ministerio de Economía, Industria y Competitividad, Madrid (grant CTQ2014-54729-C2-1-P and predoctoral contract BES-2015-074458 for Z. D.), and the Ministerio de Ciencia, Innovación y Universidades, Madrid (grant CTQ2017-89832-P) is gratefully acknowledged.Notes and references
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef PubMed.
- I. Medintz, Nat. Mater., 2006, 5, 842 CrossRef PubMed.
- W. Liu, S. K. Samanta, B. D. Smith and L. Isaacs, Chem. Soc. Rev., 2017, 46, 2391–2403 RSC.
- C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef PubMed.
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Würth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef PubMed.
- T. Skorjanc, F. Benyettou, J.-C. Olsen and A. Trabolsi, Chem.–Eur. J., 2017, 23, 8333–8347 CrossRef PubMed.
- K. I. Assaf, A. Hennig, S. Peng, D.-S. Guo, D. Gabel and W. M. Nau, Chem. Commun., 2017, 53, 4616–4619 RSC.
- V. Montes-García, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzán, Chem.–Eur. J., 2014, 20, 10874–10883 CrossRef PubMed.
- H. Li, D.-X. Chen, Y.-L. Sun, Y. B. Zheng, L.-L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2013, 135, 1570–1576 CrossRef PubMed.
- S. S. Agasti, M. Liong, C. Tassa, H. J. Chung, S. Y. Shaw, H. Lee and R. Weissleder, Angew. Chem., Int. Ed., 2012, 51, 450–454 CrossRef PubMed.
- H. J. Kim, M. H. Lee, L. Mutihac, J. Vicens and J. S. Kim, Chem. Soc. Rev., 2012, 41, 1173–1190 RSC.
- Y. Yao, M. Xue, X. Chi, Y. Ma, J. He, Z. Abliz and F. Huang, Chem. Commun., 2012, 48, 6505–6507 RSC.
- R. de laRica, R. M. Fratila, A. Szarpak, J. Huskens and A. H. Velders, Angew. Chem., Int. Ed., 2011, 50, 5704–5707 CrossRef PubMed.
- R. Freeman, T. Finder, L. Bahshi and I. Willner, Nano Lett., 2009, 9, 2073–2076 CrossRef PubMed.
- R. Klajn, M. A. Olson, P. J. Wesson, L. Fang, A. Coskun, A. Trabolsi, S. Soh, J. F. Stoddart and B. A. Grzybowski, Nat. Chem., 2009, 1, 733–738 CrossRef PubMed.
- T. Jin, F. Fujii, E. Yamada, Y. Nodasaka and M. Kinjo, J. Am. Chem. Soc., 2006, 128, 9288–9289 CrossRef PubMed.
- A. Wei, Chem. Commun., 2006, 1581–1591 RSC.
- A. Arduini, D. Demuru, A. Pochini and A. Secchi, Chem. Commun., 2005, 645–647 RSC.
- T. Jin, F. Fujii, H. Sakata, M. Tamura and M. Kinjo, Chem. Commun., 2005, 2829–2831 RSC.
- B. Kim, S. L. Tripp and A. Wei, J. Am. Chem. Soc., 2001, 123, 7955–7956 CrossRef PubMed.
- J. Liu, S. Mendoza, E. Román, M. J. Lynn, R. Xu and A. E. Kaifer, J. Am. Chem. Soc., 1999, 121, 4304–4305 CrossRef.
- M. J. Ludden, D. N. Reinhoudt and J. Huskens, Chem. Soc. Rev., 2006, 35, 1122–1134 RSC.
- J. Voskuhl and B. J. Ravoo, Chem. Soc. Rev., 2009, 38, 495–505 RSC.
- D. Sigwalt, M. Šekutor, L. Cao, P. Y. Zavalij, J. Hostaš, H. Ajani, P. Hobza, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, J. Am. Chem. Soc., 2017, 139, 3249–3258 CrossRef PubMed.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef PubMed.
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984–12989 CrossRef PubMed.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef PubMed.
- D. Shetty, J. K. Khedkar, K. M. Park and K. Kim, Chem. Soc. Rev., 2015, 44, 8747–8761 RSC.
- M. Nilam, P. Gribbon, J. Reinshagen, K. Cordts, E. Schwedhelm, W. M. Nau and A. Hennig, SLAS Discovery, 2017, 22, 906–914 Search PubMed.
- M. Schnurr, J. Sloniec-Myszk, J. Döpfert, L. Schröder and A. Hennig, Angew. Chem., Int. Ed., 2015, 54, 13444–13447 CrossRef PubMed.
- G. Ghale, A. G. Lanctôt, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762–2765 CrossRef PubMed.
- R. N. Dsouza, A. Hennig and W. M. Nau, Chem.–Eur. J., 2012, 18, 3444–3459 CrossRef PubMed.
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef PubMed.
- P. Neirynck, J. Brinkmann, Q. An, D. W. J. van der Schaft, L.-G. Milroy, P. Jonkheijm and L. Brunsveld, Chem. Commun., 2013, 49, 3679–3681 RSC.
- J. F. Young, H. D. Nguyen, L. Yang, J. Huskens, P. Jonkheijm and L. Brunsveld, ChemBioChem, 2010, 11, 180–183 CrossRef PubMed.
- I. Hwang, K. Baek, M. Jung, Y. Kim, K. M. Park, D.-W. Lee, N. Selvapalam and K. Kim, J. Am. Chem. Soc., 2007, 129, 4170–4171 CrossRef PubMed.
- J. Murray, J. Sim, K. Oh, G. Sung, A. Lee, A. Shrinidhi, A. Thirunarayanan, D. Shetty and K. Kim, Angew. Chem., Int. Ed., 2017, 56, 2395–2398 CrossRef PubMed.
- W. Li, A. T. Bockus, B. Vinciguerra, L. Isaacs and A. R. Urbach, Chem. Commun., 2016, 52, 8537–8540 RSC.
- D.-W. Lee, K. M. Park, M. Banerjee, S. H. Ha, T. Lee, K. Suh, S. Paul, H. Jung, J. Kim, N. Selvapalam, S. H. Ryu and K. Kim, Nat. Chem., 2011, 3, 154–159 CrossRef PubMed.
- M. J. Webber, E. A. Appel, B. Vinciguerra, A. B. Cortinas, L. S. Thapa, S. Jhunjhunwala, L. Isaacs, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14189–14194 CrossRef PubMed.
- S. K. Samanta, J. Quigley, B. Vinciguerra, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2017, 139, 9066–9074 CrossRef PubMed.
- K. L. Kim, G. Sung, J. Sim, J. Murray, M. Li, A. Lee, A. Shrinidhi, K. M. Park and K. Kim, Nat. Commun., 2018, 9, 1712 CrossRef PubMed.
- J. F. Young, H. D. Nguyen, L. Yang, J. Huskens, P. Jonkheijm and L. Brunsveld, ChemBioChem, 2010, 11, 180–183 CrossRef PubMed.
- P. Neirynck, J. Brinkmann, Q. An, D. W. van der Schaft, L.-G. Milroy, P. Jonkheijm and L. Brunsveld, Chem. Commun., 2013, 49, 3679–3681 RSC.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef PubMed.
- C.-A. Tao, Q. An, W. Zhu, H. Yang, W. Li, C. Lin, D. Xu and G. Li, Chem. Commun., 2011, 47, 9867–9869 RSC.
- T.-C. Lee and O. A. Scherman, Chem. Commun., 2010, 46, 2438–2440 RSC.
- F. Benyettou, I. Milosevic, Y. Lalatonne, F. Warmont, R. Assah, J.-C. Olsen, M. Jouaid, L. Motte, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. B, 2013, 1, 5076–5082 RSC.
- F. Benyettou, K. Nchimi-Nono, M. Jouiad, Y. Lalatonne, I. Milosevic, L. Motte, J. C. Olsen, N. i. Saleh and A. Trabolsi, Chem.–Eur. J., 2015, 21, 4607–4613 CrossRef PubMed.
- R. De La Rica and A. H. Velders, Small, 2011, 7, 66–69 CrossRef PubMed.
- I. Brouwer, G. Sitters, A. Candelli, S. J. Heerema, I. Heller, H. Zhang, D. Normanno, M. Modesti, E. J. Peterman and G. J. Wuite, Nature, 2016, 535, 566–569 CrossRef PubMed.
- E. Mathiowitz, J. S. Jacob, Y. S. Jong and G. P. Carino, Nature, 1997, 386, 410 CrossRef PubMed.
- D. Zhang, G. Chen, D. Manwani, A. Mortha, C. Xu, J. J. Faith, R. D. Burk, Y. Kunisaki, J.-E. Jang and C. Scheiermann, Nature, 2015, 525, 528–532 CrossRef PubMed.
- J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef PubMed.
- M. Pierobon, J. Wulfkuhle, L. Liotta and E. Petricoin, Oncogene, 2015, 34, 805–814 CrossRef PubMed.
- Y. Lu, J. Lu, J. Zhao, J. Cusido, F. M. Raymo, J. Yuan, S. Yang, R. C. Leif, Y. Huo, J. A. Piper, J. Paul Robinson, E. M. Goldys and D. Jin, Nat. Commun., 2014, 5, 3741 CrossRef PubMed.
- M. Liong, A. N. Hoang, J. Chung, N. Gural, C. B. Ford, C. Min, R. R. Shah, R. Ahmad, M. Fernandez-Suarez, S. M. Fortune, M. Toner, H. Lee and R. Weissleder, Nat. Commun., 2013, 4, 1752 CrossRef PubMed.
- D. Quesada-Gonzalez and A. Merkoci, Biosens. Bioelectron., 2015, 73, 47–63 CrossRef PubMed.
- Q. Y. Xie, Y. H. Wu, Q. R. Xiong, H. Y. Xu, Y. H. Xiong, K. Liu, Y. Jin and W. H. Lai, Biosens. Bioelectron., 2014, 54, 262–265 CrossRef PubMed.
- H. Chen, Z. Huang, H. Wu, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 129, 16802–16805 CrossRef.
- M. M. Ayhan, H. Karoui, M. Hardy, A. Rockenbauer, L. Charles, R. Rosas, K. Udachin, P. Tordo, D. Bardelang and O. Ouari, J. Am. Chem. Soc., 2015, 137, 10238–10245 CrossRef PubMed.
- A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Chem. Commun., 2011, 47, 7842–7844 RSC.
- A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Anal. Chem., 2011, 83, 4970–4974 CrossRef PubMed.
- A. Hennig, P. M. Dietrich, F. Hemmann, T. Thiele, H. Borcherding, A. Hoffmann, U. Schedler, C. Jager, U. Resch-Genger and W. E. S. Unger, Analyst, 2015, 140, 1804–1808 RSC.
- P. M. Dietrich, A. Hennig, M. Holzweber, T. Thiele, H. Borcherding, A. Lippitz, U. Schedler, U. Resch-Genger and W. E. S. Unger, J. Phys. Chem. C, 2014, 118, 20393–20404 CrossRef.
- A. Hennig, S. Hatami, M. Spieles and U. Resch-Genger, Photochem. Photobiol. Sci., 2013, 12, 729–737 RSC.
- The reported binding constants for the cationic ammonium forms of 1-aminoadamantane and 1-aminomethyladamantane are 1.7 × 1014 M−1 and 7.7 × 1014 M−1, respectively (ref. 27). As a consequence of this strong affinity, the resulting host–guest complex with CB7 has also an appreciable kinetic stability. Assuming a maximally diffusion-controlled association reaction (kass = 7.4 × 109 M−1 s−1), the dissociation half-life times are >4.4 h and >20 h for 1-aminoadamantane and 1-aminomethyladamantane.
- E. Masson, M. Raeisi and K. Kotturi, Isr. J. Chem., 2018, 58, 3–4 CrossRef.
- C. Bohne, Chem. Soc. Rev., 2014, 43, 4037–4050 RSC.
- S. Kim, G. Yun, S. Khan, J. Kim, J. Murray, Y. M. Lee, W. J. Kim, G. Lee, S. Kim, D. Shetty, J. H. Kang, J. Y. Kim, K. M. Park and K. Kim, Mater. Horiz., 2017, 4, 450–455 RSC.
- M. Colombo, L. Fiandra, G. Alessio, S. Mazzucchelli, M. Nebuloni, C. De Palma, K. Kantner, B. Pelaz, R. Rotem, F. Corsi, W. J. Parak and D. Prosperi, Nat. Commun., 2016, 7, 13818 CrossRef PubMed.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42–47 CrossRef PubMed.
- M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef PubMed.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterisation data. See DOI: 10.1039/c8sc03150a |
| This journal is © The Royal Society of Chemistry 2018 |