
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced annihilation electrochemiluminescence by nanofluidic confinement†
Hanan
Al-Kutubi‡
a,
Silvia
Voci‡
b,
Liza
Rassaei
cd,
Neso
Sojic
 *b and
Klaus
Mathwig
*a
*b and
Klaus
Mathwig
*a
aUniversity of Groningen, Groningen Research Institute of Pharmacy, Pharmaceutical Analysis, P.O. Box 196, 9700 AD Groningen, The Netherlands. E-mail: kmathwig@rug.nl
bUniversity of Bordeaux, Bordeaux INP, Institut des Sciences Moléculaires, UMR CNRS 5255, 33607 Pessac, France. E-mail: neso.sojic@enscbp.fr
cRotterdam School of Management, Erasmus University, Burgemeester Oudlaan 50, 3062 PA Rotterdam, The Netherlands
dDelft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 1st October 2018
Abstract
Microfabricated nanofluidic electrochemical devices offer a highly controlled nanochannel geometry; they confine the volume of chemical reactions to the nanoscale and enable greatly amplified electrochemical detection. Here, the generation of stable light emission by electrochemiluminescence (ECL) in transparent nanofluidic devices is demonstrated for the first time by exploiting nanogap amplification. Through continuous oxidation and reduction of [Ru(bpy)3]2+ luminophores at electrodes positioned at opposite walls of a 100 nm nanochannel, we compare classic redox cycling and ECL annihilation. Enhanced ECL light emission of attomole luminophore quantities is evidenced under ambient conditions due to the spatial confinement in a 10 femtoliter volume, resulting in a short diffusion timescale and highly efficient ECL reaction pathways at the nanoscale.
Introduction
Electrochemiluminescence (ECL) is the emission of light by electrochemical means: reactive species are generated at the electrode surface and undergo a highly exergonic electron-transfer reaction producing the excited state of the luminophore.1 It relaxes to the ground state, emitting a photon. The possibility to regenerate the reactive species in situ as well as not requiring an external light source for excitation, gives this method many advantages for (bio)sensing applications, single object imaging,2–4 and light-emitting devices.5 These advantages include a straightforward experimental setup, a very high sensitivity and a wide detection range. Thus, applications and fundamental aspects of ECL have been researched extensively.5Whereas the precise mechanism of light generation depends on the species involved, there are two dominant pathways: the annihilation and the co-reactant pathways.1 Here, we focus on the annihilation pathway in a nanochannel, in which light is generated through a reaction between the oxidized and reduced forms of the original compound. We selected tris(bipyridine)ruthenium(II), [Ru(bpy)3]2+, one of the most well-studied ECL compounds6,7 due to its relatively high quantum yield, water solubility and long-lived triplet excited state. Its annihilation pathway is:8
| [Ru(bpy)3]2+ + e− → [Ru(bpy)3]+ | (1) |
| [Ru(bpy)3]2+ − e− → [Ru(bpy)3]3+ | (2) |
| [Ru(bpy)3]+ + [Ru(bpy)3]3+ → [Ru(bpy)3]2+ + [Ru(bpy)3]2+* | (3) |
| [Ru(bpy)3]2+* → [Ru(bpy)3]2+ + hν. | (4) |
These reactions were initially investigated by rapidly pulsing a single electrode between the oxidizing and reducing potential.9,10 However, this technique suffers from drawbacks11 such as large charging currents. Using two electrodes, both [Ru(bpy)3]+ and [Ru(bpy)3]3+ can be generated in close vicinity under steady-state conditions. Various two-electrode setups have been employed, including ring and disk electrodes,12 thin layer cells,13 band electrodes,14–16 and scanning electrochemical microscopy.17,18 These studies have shown that annihilation occurs more efficiently at smaller inter-electrode distances16,18 as the diffusional distance between electrogenerated [Ru(bpy)3]+ and [Ru(bpy)3]3+ is decreased. Indeed, the stability of the reduced form, [Ru(bpy)3]+, is a key issue limiting the efficiency of the annihilation ECL. Yet, observation of light emission so far has been limited to a minimum inter-electrode distance of 2 μm.18,19 In addition, due to the sensitivity of annihilation ECL towards impurities such as dioxygen and water, experiments are performed in drastic conditions: dried and distilled solvents just before use, recrystallized and dried luminescent compounds and supporting electrolytes, under an inert atmosphere or in a dry box.15,18,20 This is especially true for platinum electrodes as the reduction potential for water/dioxygen competes with that of [Ru(bpy)3]2+.
Here, ECL emission is observed under ambient conditions using platinum electrodes in electrochemical nanogap devices.21 We exploit the spatial confinement of such devices to enhance the annihilation ECL-reaction of [Ru(bpy)3]2+ and to reduce the effects of impurities. Microfabrication allows precise control of a nanofluidic channel geometry; two individually addressable electrodes positioned at the top and bottom of a nanochannel are separated by a distance of 100 nm or less (see Fig. 1).
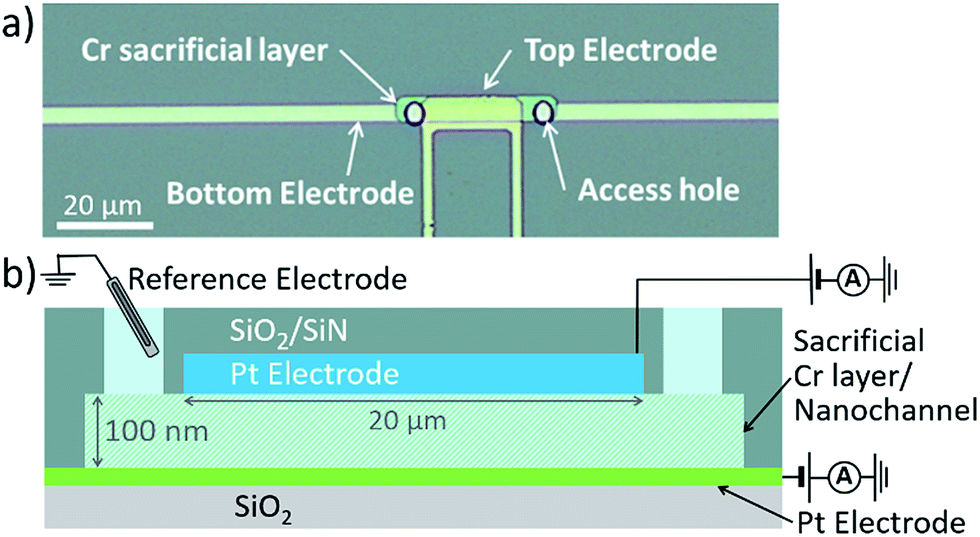 | ||
| Fig. 1 (a) Top view optical micrograph of an electrochemical nanogap device. (b) Schematic cross section of a device with access holes to reservoir and two individually addressable Pt electrodes. The sacrificial Cr layer is etched before the experiments to form the nanochannel. | ||
The nanoscale distance between electrodes results in short diffusion times between them and gives rise to highly amplified currents through electrochemical redox cycling. Reversibly electrochemically active analytes are repeatedly oxidized and reduced as they diffuse between both electrodes, shuttling electrons across the nanochannel. At diffusion times of ∼10 μs, each analyte molecule contributes thousands of electrons per seconds to the detected limiting current, which, thus, is highly amplified. Due to efficient redox cycling and a confined geometry, nanogap transducers and similar geometries have been employed for, e.g., studying adsorption,22 migration,23,24 biosensing,25 single-molecule electrochemistry,26 and spectroelectrochemistry.27
Herein, for the first time, annihilation ECL is examined in transparent nanofluidic electrochemical devices. We report enhanced light emission due to highly efficient reaction pathways at the nanoscale. Light emission is stable at ambient conditions due to reduced degradation by contaminants.
Results and discussion
Nanogap devices were fabricated as described previously,28,29 except that a transparent glass substrate was used. Due to the transparency and lower heat conductivity of glass, most fabrication steps had to be altered completely. In brief, structures comprising a thin 20 nm Pt bottom electrode, 100 nm Cr sacrificial layer (forming the nanochannel volume) and 120 nm Pt top electrode were patterned on a 10 cm borosilicate glass wafer using UV lithography followed by evaporation deposition and lift-off. A 500 nm SiO2/SiN passivation layer, deposited using chemical vapor deposition, encased the devices. Access holes were etched through the passivation layer by reactive ion etching. Finally, the wafer was diced into individual chips. Before experimentation, a polydimethylsiloxane fluid reservoir was placed on top of the device, and the sacrificial Cr layer was selectively wet-etched to form the channel. In Fig. 1a, a device is shown with a 5 μm × 32 μm × 100 nm channel volume, and an effective area of overlap between the electrodes of 3 μm × 20 μm.Electrochemical measurements were performed with a bipotentiostat (Autolab – PGSTAT30). Functioning simultaneously as both reference electrode and counter electrode, we used an Ag/AgCl/KCl (3 M) electrode in water, or an Ag wire in acetonitrile, respectively. For all experiments, freshly etched devices were cleaned by cycling the potential of both electrodes between −0.15 V and 1.2 V vs. Ag/AgCl in 50 mM H2SO4. Channels were thoroughly flushed with Milli-Q water, and subsequently with acetonitrile, which was then replaced by 10 mM Ru(bpy)3(PF6)2 and 0.1 M TBAPF6 in acetonitrile. This solution was used for all experiments.
We investigated two electrochemical ‘modes’ in the nanofluidic channel (see Fig. 2). In redox cycling, the top electrode is biased at an oxidizing potential generating [Ru(bpy)3]3+. Biasing the bottom electrode at 0 V leads to the one-electron reduction of [Ru(bpy)3]3+ and, thus, regeneration of [Ru(bpy)3]2+. In annihilation ECL, the same potential is imposed at the top electrode whereas the bottom electrode is placed at a sufficiently cathodic potential to reduce [Ru(bpy)3]2+ to [Ru(bpy)3]+. The annihilation reaction (3) takes place, and ECL light is emitted (4). Conceptually, this process is similar to redox cycling as it involves the constant consumption and regeneration of [Ru(bpy)3]2+. However, instead of taking place between the two electrode surfaces, regeneration (annihilation) occurs homogeneously in the channel bulk, where [Ru(bpy)3]+ and [Ru(bpy)3]3+ meet. In the top region, redox cycling between [Ru(bpy)3]2+ and its oxidized form occurs, whereas the bottom region shows cycling of [Ru(bpy)3]2+ with the reduced form. The channel is therefore effectively split, and anodic and cathodic redox cycling occur simultaneously (Fig. 2b). Therefore, as diffusion times scale inversely with the length of the diffusive path, switching from redox cycling to annihilation ECL mode should result in a higher faradaic current.
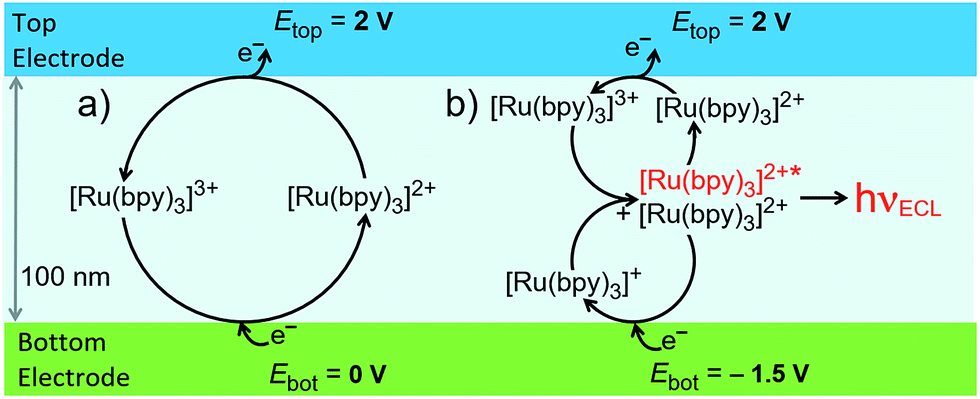 | ||
| Fig. 2 Schematic of two reaction pathways in the nanochannel: (a) redox cycling, and (b) annihilation leading to ECL emission. | ||
ECL-emission is produced in annihilation mode inside a nanochannel and was imaged and spatially resolved as shown in Fig. 3 using an epifluorescence microscope (DMI6000, Leica). Photon emission was collected by an inverted 40× microscope objective and detected by an Electron Multiplying Charge Coupled Device (EM-CCD) Camera (Hamamatsu, 9100-13). Light emission from a luminophore quantity of 60 attomoles present in the active area of the device was intense enough to be recorded through the 20 nm-thick semi-transparent Pt bottom electrode, due to nanogap amplification. The overlay image (Fig. 3a, right) shows that ECL intensity is uniform along the channel and decreases rapidly at the edges. No emission is seen from the bottom electrode area under the access holes. Furthermore, ECL emission is limited to a region narrower than the 5 μm wide channel. This indicates that emission is caused exclusively by reaction in the 3 μm × 20 μm effective area between the electrodes (as observed for purely electrochemical cycling30). Full-width-at-half-maxima of longitudinal and lateral intensity profiles amount to 20 μm and 4.3 μm, respectively. Presumably, the optical resolution of the 40× objective as well as inhomogeneous transparency of Pt and glass lead to a slightly wider profile than expected. Clearly, ECL emission is confined to the active area of the nanochannel, where reactions (1–4) (Fig. 2b) take place.
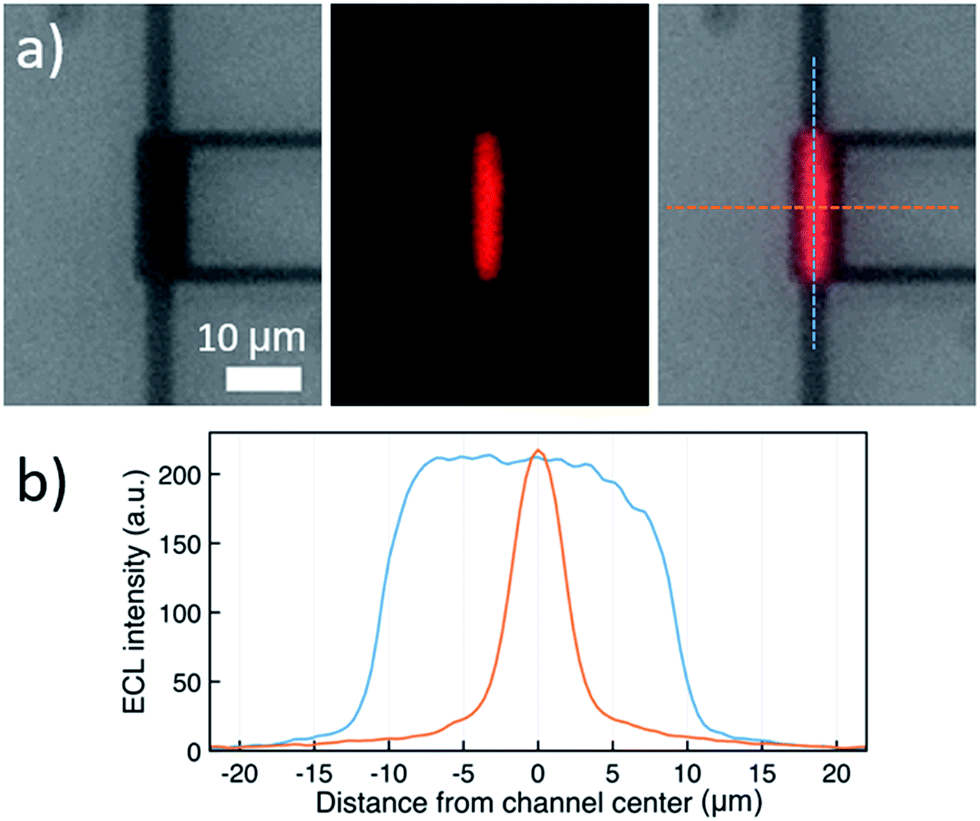 | ||
| Fig. 3 (a) From left to right: bright-field micrograph of the nanochannel, ECL emission recorded in the dark, and overlay of both images (dashed lines: direction of profiles in (b)). (b) ECL intensity profiles along (blue) and across (orange) the center of the channel, using a solution of 10 mM Ru(bpy)3(PF6)2 and 0.1 M TBAPF6 in acetonitrile. The top electrode was biased at 2 V, the bottom electrode at −1.5 V vs. Ag. | ||
The constant ECL intensity along the nanochannel (Fig. 3b, blue curve) indicates that only negligible degradation occurs. If [Ru(bpy)3]+ or [Ru(bpy)3]3+ would degrade, the intensity would be strongly attenuated in the center of the channel because the chance of encountering contaminants increases quadratically as luminophores diffuse towards the center.
Fig. 4a displays the chronoamperometric currents at both electrodes when stepping between redox cycling and annihilation ECL modes. In redox cycling mode, the oxidation and reduction currents are symmetric and constant. The observed currents of ∼0.65 μA are in agreement with an expected analytical estimate31 of 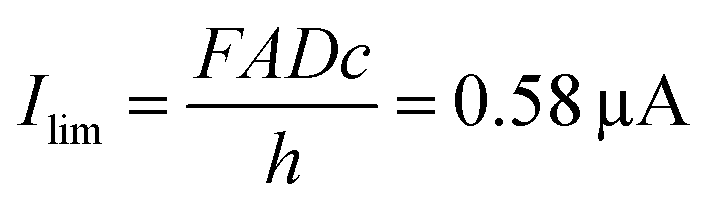 (F: faraday constant; A: active area of 3 μm by 20 μm; D = 1 × 10−9 m2 s−1 diffusion coefficient, c: 10 mM [Ru(bpy)3]2+ concentration, h: 100 nm channel height).
(F: faraday constant; A: active area of 3 μm by 20 μm; D = 1 × 10−9 m2 s−1 diffusion coefficient, c: 10 mM [Ru(bpy)3]2+ concentration, h: 100 nm channel height).
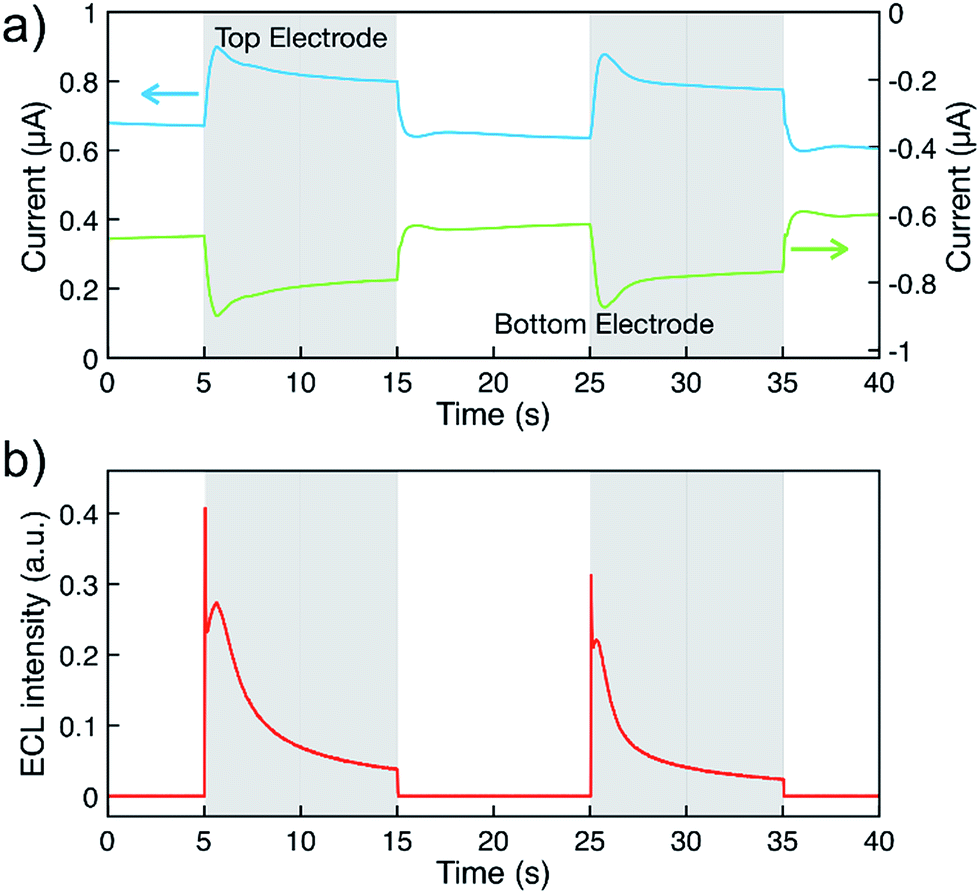 | ||
| Fig. 4 (a) Measured chronoamperometric currents at the top (blue) and bottom (green) electrodes, and (b) corresponding ECL intensity. The top electrode was maintained at 2.3 V while the bottom electrode was pulsed between 0 V and −1.5 V vs. Ag, highlighted in white (redox cycling mode) and grey (annihilation ECL mode), respectively. Solution consisted of 10 mM Ru(bpy)3(PF6)2 and 0.1 M TBAPF6 in acetonitrile. | ||
ECL intensity was measured simultaneously by using a photomultiplier tube (Hamamatsu R4632). The signal was amplified by a Keithley Picoammeter before acquisition with the second input signal of a μAutolab type II potentiostat. Emission was not observed in redox cycling mode (Fig. 4b). Upon switching to annihilation mode, an initial transient current increase can be ascribed to adsorption of analytes onto the electrode surfaces due to the high surface-to-volume ratio of the nanogap devices.22,29,32 Oxidation and reduction currents are symmetric as expected for reactions (1–4). In annihilation mode, the light intensity (Fig. 4b) exhibits a strong transient behavior before approaching a steady state. Understanding this transient behavior will require further studies, we partially attribute the complex transient to desorption when stepping the potential as well as accumulating [Ru(bpy)3]3+ during redox cycling.
Upon switching from redox cycling to annihilation mode, an expected increase in the current is observed as the diffusive path for cycling molecules halves to 50 nm. Concentration profiles of all [Ru(bpy)3] species are illustrated in Fig. 5 (COMSOL Multiphysics, see ESI†). Considering the symmetry of the nanofluidic channel and the annihilation reaction (i.e., double cycling redox process), ECL is emitted in the middle of the nanogap where both [Ru(bpy)3]3+ and [Ru(bpy)3]+ react to form [Ru(bpy)3]2+*, assuming similar diffusion coefficients for [Ru(bpy)3]3+ and [Ru(bpy)3]+. The ECL emission should occur in the vertical symmetry plane of the device and be confined to the electroactive area of the nanochannel (see Fig. 5d). However, it is not possible to resolve this with optical microscopy due do the nanometric dimension of the inter-electrode gap.
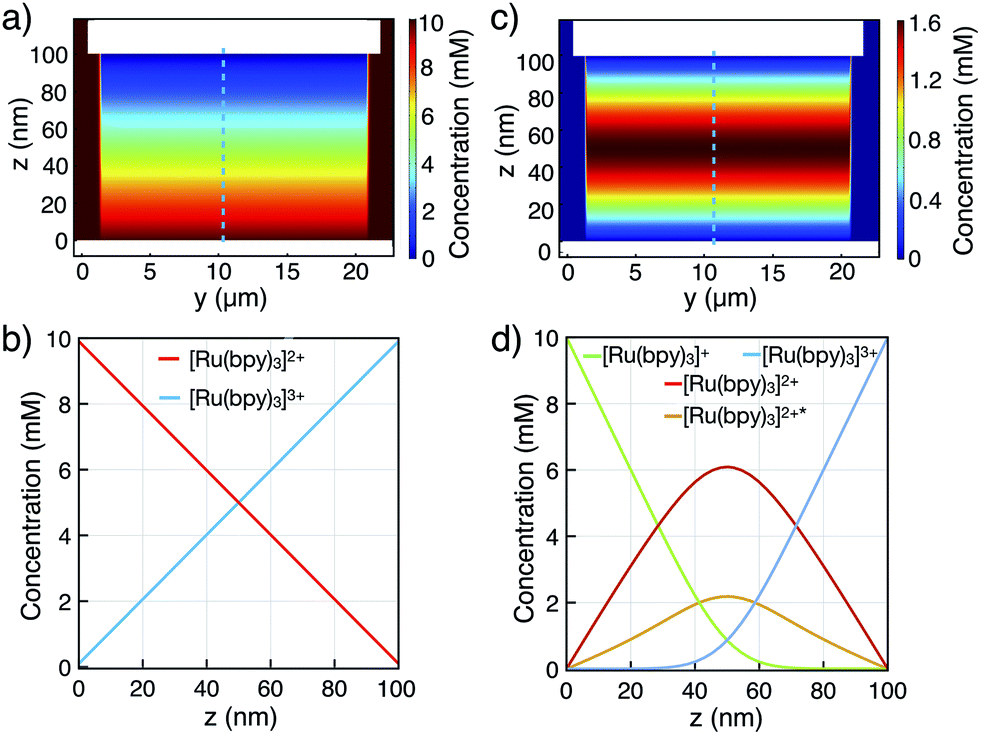 | ||
| Fig. 5 Finite element simulations of concentration profiles for a 10 mM [Ru(bpy)3]2+ bulk concentration in the nanochannel. Two-dimensional [Ru(bpy)3]2+ profiles in (a) redox cycling mode, and (c) annihilation mode. (b and d) Corresponding cross-sectional profiles in the channel center (indicated by the dashed line in both modes, respectively). | ||
The simulated diffusive fluxes (i.e., slopes of concentration profiles) at electrode surfaces double when switching from redox cycling to annihilation. However, a ratio of only 1.2 for the steady-state redox cycling and annihilation currents is observed (Fig. 4a). Understanding this low ratio requires further studies. As origin we rule out degradation (see above) and a limited annihilation rate (as a two-electron electrode reaction of [Ru(bpy)3]+ and [Ru(bpy)3]3+ would exactly compensate for every annihilation not taking place18,33,34). Furthermore, any influences on the currents by unequal diffusivities as well as by adsorption are balanced by coupling to the bulk reservoir.29,31 We speculate that more complex concentration profiles are caused by contribution of electrical migration (at an overall potential drop of 3.5 V over 100 nm).
Classically, annihilation ECL experiments are performed using highly inert conditions (vide supra).15,18,20 In the present work, we imaged an intense ECL signal under ambient conditions using platinum electrodes. In the nanochannel, neither strong degradation of [Ru(bpy)3]+ nor greatly reduced annihilation occurs (previously reduction of the annihilation rate from >107 M−1 s−1 to ∼103 M−1 s−1 was observed under ambient conditions18,35). Negligible degradation is caused by the short diffusive path, which greatly reduces the chance to encounter contaminants such as dioxygen, and/or by depletion of contaminants in the nanochannel by electrochemical deactivation at the electrodes (in the ESI† we further explore the effect of channel height, lifetime of the excited state and possible degradation on concentration profile by finite element calculations).
Conclusions
ECL-emission was investigated in transparent nanofluidic devices for the first time. Light emission was shown to occur through an annihilation reaction pathway when both electrodes were biased at appropriate potentials. Light emission is enhanced in two ways: first, the close vicinity of the two electrodes leads to efficient annihilation reactions due to very fast diffusion. Second, stable light emission is achieved as the very short diffusive distances protect from degradation by contaminants, and our results indicate that measurements in nanodevices are possible under ambient conditions, which cannot be achieved using classical setups. Microfabricated nanodevices combine the properties of exact control of an inter-electrode distance, a highly reproducible device geometry and strong confinement of analytes in the channel volume. Therefore, we believe that nanogap transducers are powerful tools for investigating pathways in ECL and also to exploit their unique nanometric properties to develop multicolor ECL emission systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Agence Nationale de la Recherche (MOLY, ANR-15-CE19-0005-01; NEOCLASSIC ANR-15-CE09-0015-03). We thank Dr Giovanni Valenti (University of Bologna) for helpful discussions.References
- A. Bard, Electrogenerated chemiluminescence, Marcel Dekker, New York, 2004 Search PubMed.
- A. J. Wilson, K. Marchuk and K. A. Willets, Nano Lett., 2015, 15, 6110–6115 CrossRef CAS PubMed.
- J. E. Dick, C. Renault, B. K. Kim and A. J. Bard, Angew. Chem., Int. Ed., 2014, 53, 11859–11862 CrossRef CAS PubMed.
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed.
- L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275–3304 RSC.
- B. A. Gorman, P. S. Francis and N. W. Barnett, Analyst, 2006, 131, 616–639 RSC.
- X. B. Yin, S. Dong and E. Wang, TrAC, Trends Anal. Chem., 2004, 23, 432–441 CrossRef CAS.
- R. J. Forster, P. Bertoncello and T. E. Keyes, Annu. Rev. Anal. Chem., 2009, 2, 359–385 CrossRef CAS PubMed.
- R. Breslow, R. Hill and E. Wasserman, J. Am. Chem. Soc., 1964, 83, 5350–5351 Search PubMed.
- K. S. V. Santhanam and A. J. Bard, J. Am. Chem. Soc., 1965, 87, 139–140 CrossRef CAS.
- F. F. Fan, in Electrogenerated Chemiluminescence, ed. A. J. Bard, CRC Press, New York, 1st edn, 2004, pp. 23–99 Search PubMed.
- J. T. Maloy, K. B. Prater and A. J. Bard, J. Am. Chem. Soc., 1971, 93, 5959–5968 CrossRef CAS.
- G. H. Brilmyer and A. J. Bard, J. Electrochem. Soc., 1980, 127, 104–110 CrossRef CAS.
- J. E. Bartelt, S. M. Drew and R. M. Wightman, J. Electrochem. Soc., 1992, 139, 70–74 CrossRef CAS.
- C. Amatore, C. Pebay, L. Servant, N. Sojic, S. Szunerits and L. Thouin, ChemPhysChem, 2006, 7, 1322–1327 CrossRef CAS PubMed.
- G. C. Fiaccabrino, M. Koudelka-Hep, Y. T. Hsueh, S. D. Collins and R. L. Smith, Anal. Chem., 1998, 70, 4157–4161 CrossRef CAS PubMed.
- F.-R. F. Fan, D. Cliffel and A. J. Bard, Anal. Chem., 1998, 70, 2941–2948 CrossRef CAS.
- J. Rodríguez-López, M. Shen, A. B. Nepomnyashchii and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 9240–9250 CrossRef PubMed.
- M. Shen, N. Arroyo-Currás and A. J. Bard, Anal. Chem., 2011, 83, 9082–9085 CrossRef CAS PubMed.
- A. Kapturkiewicz and G. Angulo, Dalton Trans., 2003, 3907–3913 RSC.
- M. A. G. Zevenbergen, D. Krapf, M. R. Zuiddam and S. G. Lemay, Nano Lett., 2007, 7, 384–388 CrossRef CAS PubMed.
- D. Mampallil, K. Mathwig, S. Kang and S. G. Lemay, J. Phys. Chem. Lett., 2014, 5, 636–640 CrossRef CAS PubMed.
- Q. Chen, K. McKelvey, M. A. Edwards and H. S. White, J. Phys. Chem. C, 2016, 120, 17251–17260 CrossRef CAS.
- C. Ma, W. Xu, W. R. A. Wichert and P. W. Bohn, ACS Nano, 2016, 10, 3658–3664 CrossRef CAS PubMed.
- L. Rassaei, K. Mathwig, S. Kang, H. A. Heering and S. G. Lemay, ACS Nano, 2014, 8, 8278–8284 CrossRef CAS PubMed.
- S. Kang, A. F. Nieuwenhuis, K. Mathwig, D. Mampallil, Z. Kostiuchenko and S. G. Lemay, Faraday Discuss., 2016, 193, 41–50 RSC.
- D. Han, G. M. Crouch, K. Fu, L. P. Zaino III and P. W. Bohn, Chem. Sci., 2017, 8, 5345–5355 RSC.
- K. Mathwig and S. G. Lemay, Micromachines, 2013, 4, 138–148 CrossRef.
- S. Kang, K. Mathwig and S. G. Lemay, Lab Chip, 2012, 12, 1262–1267 RSC.
- S. Kang, A. F. Nieuwenhuis, K. Mathwig, D. Mampallil and S. G. Lemay, ACS Nano, 2013, 7, 10931–10937 CrossRef CAS PubMed.
- D. Mampallil, K. Mathwig, S. Kang and S. G. Lemay, Anal. Chem., 2013, 85, 6053–6058 CrossRef CAS PubMed.
- E. Kätelhön, K. J. Krause, K. Mathwig, S. G. Lemay and B. Wolfrum, ACS Nano, 2014, 8, 4924–4930 CrossRef PubMed.
- Q. Wang, J. Rodríguez-López and A. J. Bard, ChemPhysChem, 2010, 11, 2969–2978 CrossRef CAS PubMed.
- C. Amatore, F. BonhSomme, J. L. Bruneel, L. Servant and L. Thouin, J. Electroanal. Chem., 2000, 484, 1–17 CrossRef CAS.
- M. M. Collinson, R. M. Wightman and P. Pastore, J. Phys. Chem., 1994, 98, 11942–11947 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemical reagents, device fabrication, background measurement, and finite element modeling, numerical concentration profiles. See DOI: 10.1039/c8sc03209b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |