
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIr(III)-catalyzed ortho C–H alkylations of (hetero)aromatic aldehydes using alkyl boron reagents†
Xiao-Yang
Chen
 * and
Erik J.
Sorensen
*
* and
Erik J.
Sorensen
*
Frick Chemistry Laboratory, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: xc3@princeton.edu; ejs@princeton.edu; Web: http://chemists.princeton.edu/sorensen/
First published on 3rd October 2018
Abstract
Transition-metal-catalyzed C–H alkylation reactions directed by aldehydes or ketones have been largely restricted to electronically activated alkenes. Herein, we report a general protocol for the Ir(III)-catalyzed ortho C–H alkylations of (hetero)aromatic aldehydes using alkyl boron reagents as the coupling partner. Featuring aniline as an inexpensive catalytic ligand, the method was compatible with a wide variety of benzaldehydes, heterocyclic aldehydes, potassium alkyltrifluoroborates as well as a few α,β-unsaturated aldehydes. An X-ray crystal structure of a benzaldehyde ortho C–H iridation intermediate was also successfully obtained.
Introduction
Aldehydes are among the most versatile functional groups in organic synthesis. However, their high reactivity and weak coordinating ability have rendered them recalcitrant directing groups in C–H functionalization reactions.1 In recent years, significant progress has been made to leverage this commonplace functional group as the directing element to promote the metalation and subsequent functionalizations of proximal C–H bonds.2 Nevertheless, missing from this repertoire is a general platform for the direct ortho C–H alkylations3 of (hetero)aromatic aldehydes. While related methods have been reported on the ortho C–H alkylations of aromatic ketones and aldehydes,4 β C–H alkylations of α,β-unsaturated ketones and aldehydes,5 as well as α C–H alkylations of aliphatic ketones,6 the coupling reagents were largely restricted to electronically activated alkenes in most cases, such as acrylates, styrenes and vinyl silanes.7 To further expand the utility of such transformations in synthesis, it would be ideal to develop aldehyde-directed C(sp2)–H alkylation reactions using versatile alkylating reagents.‡Our laboratory recently described a family of reactions to hydroxylate, methylate and fluorinate the ortho C–H bonds of benzaldehydes featuring Pd catalysis and the directivity of a structural element that arises in the course of the reaction through a reversible condensation.8 The ‘transient directing group (TDG)’ in these reactions is a putative Schiff base that is integral to catalyst binding and the subsequent palladation of a flanking arene C–H bond; this later chemical event initiates the process that replaces the ortho hydrogen with a new substituent group.9 Pioneered by the Yu group,10 this appealing concept for expanding the potential of group-directed C–H functionalizations in organic synthesis is amenable to a wide variety of aldehydes, ketones and amines.11 Our continued focus on C–H functionalizations mediated by TDGs prompted us to develop a new catalytic method for achieving direct ortho C–H alkylations of aromatic aldehydes. Unfortunately, in the evolution of this chemistry, we were unable to extend our successful, Pd-catalyzed ortho C–H methylation process to higher homologs (e.g. ethyl, propyl, etc.) due to the occurrence of undesired β-hydride eliminations (Scheme 1).8b
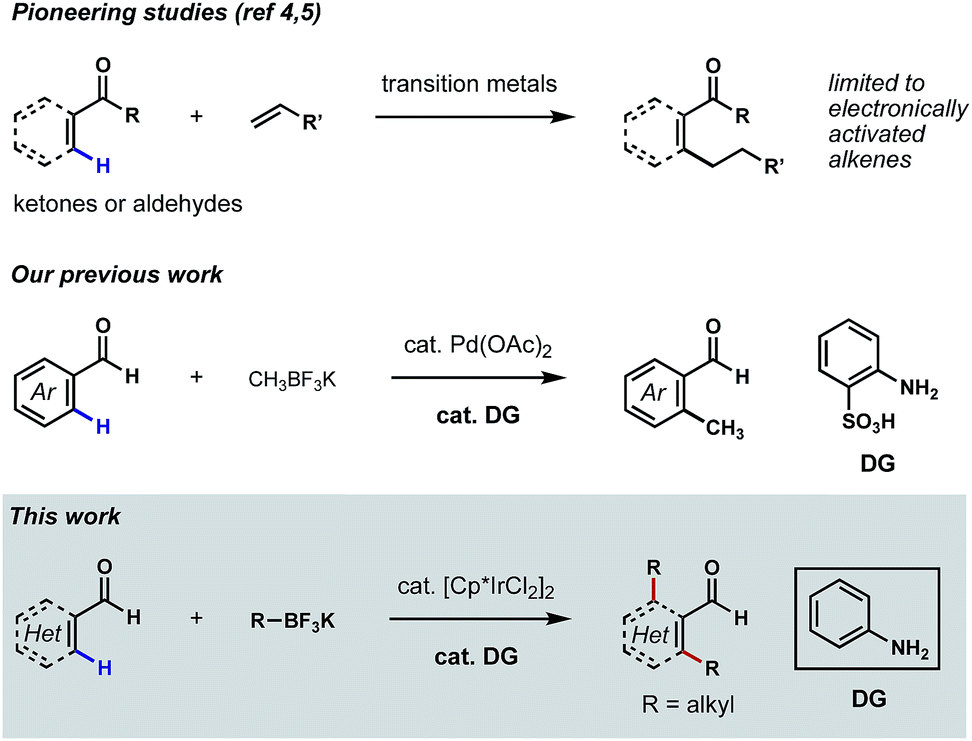 | ||
| Scheme 1 Ir(III)-catalyzed, site-selective alkylations of (hetero)aromatic and α,β-unsaturated aldehydes using aniline as the catalytic ligand. | ||
We reasoned that it might be possible to suppress β-hydride elimination if the coordination sites on the transition metal were fully saturated during the C(sp3)–C(sp2) reductive elimination step. Inspired by the recent success of TDG-enabled ortho C–H amidation of benzaldehydes via Ir(III) catalysis,10d,11d,11e we envisioned that the well-established [Cp*IrCl2]2 pre-catalyst might be suitable for a successful C–H alkylation reaction,12 as coordinative saturation is enforced by the Cp* and the cyclometalated groups. The Cp*Ir(TDG-Ar)(alkyl) species could undergo selective C(sp2)–C(sp3) reductive elimination, followed by re-oxidation of Ir(I) to Ir(III) to complete the catalytic cycle. Herein, we describe the first examples of Ir(III)-catalyzed, mono- and bis-alkylations of the ortho C–H bonds of a range of substituted (hetero)aromatic aldehydes using potassium alkyltrifluoroborates as the source of the alkyl groups and aniline as the catalytic ligand (Scheme 1). The combination of reagents allowing these transformations also permit β C–H alkylations of α,β-unsaturated aldehydes, albeit in low yields.
Results and discussion
We began our studies by treating 4-bromobenzaldehyde with an excess of n-BuBF3K in the presence of [Cp*IrCl2]2, silver additives, anilines and oxidants in various solvents under an inert atmosphere at elevated temperatures. After an extensive screening and optimization effort, we were pleased to find that the desired ortho bis-alkylation reaction took place in a nearly quantitative yield after a 24 h reaction that employed 4 mol% of [Cp*IrCl2]2 as the catalyst, 16 mol% of AgNTf2 as the chloride scavenger, 20 mol% of aniline as the catalytic ligand, 3.0 equivalents of n-BuBF3K as the alkylating reagent, 4.0 equivalents of AgF as the oxidant and acetic acid (0.1 M) as the solvent at 100 °C (see ESI† for a complete list of optimization experiments).With an effective procedure in hand, we investigated the reactions of various substituted benzaldehydes to probe the scope of this C–H alkylation process. As depicted in Scheme 2, a number of electron-withdrawing groups, such as halogens, trifluoromethyl, ester and nitrile groups, were well tolerated at the para position, delivering the desired products mostly in excellent yields (1a–1f). Interestingly, with meta-trifluoromethyl and ester-substituted benzaldehydes as the substrates, the alkylation only took place at the less sterically crowded ortho position (Scheme 2, 1g, 1h, 1n and 1o). However, when smaller meta-substituents, such as halogens and methoxy groups were present, it was possible to introduce a second alkyl group even at the more crowded ortho position (Scheme 2, 1i–1m, 1p and 1q). This result also stands contrast to our previous Pd-catalyzed ortho C–H methylation reaction,8b which only afforded mono-methylated products with meta-substituents on the substrates. Mono-alkylation was also successful in the presence of an ortho substituent on the substrate, such as fluoro, methyl and methoxy groups. In most cases, the reactions proceeded without any erosions in performance (Scheme 2, 1r–1x). The current limitations of this method are (1) ortho-chlorinated or brominated substrates were incompatible with this process; and (2) mono-alkylations can be challenging to achieve on sterically unbiased substrates even with 1 equivalent of the coupling reagent.
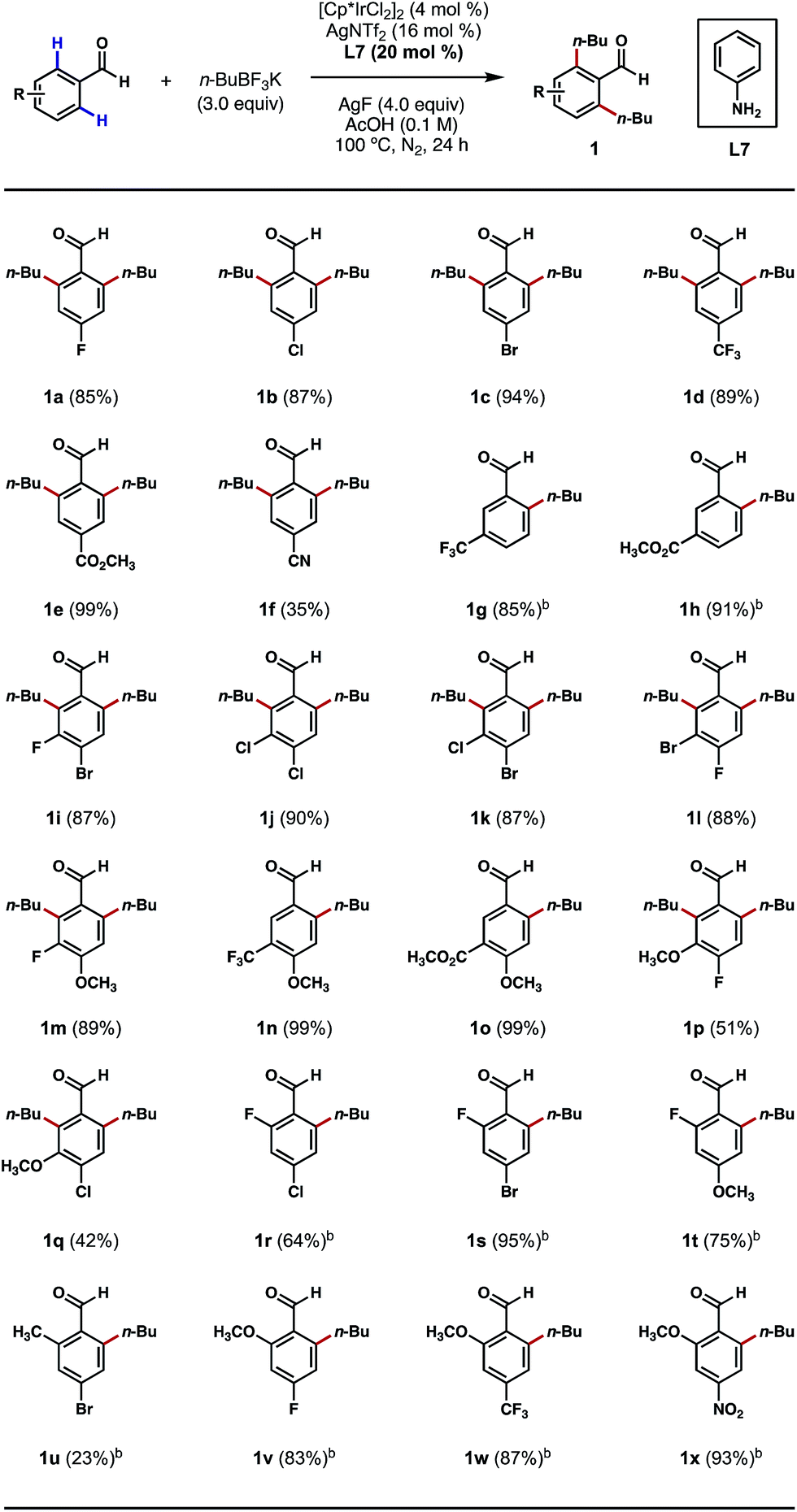 | ||
| Scheme 2 Scope of substituted benzaldehydes. aThe reactions were performed on a 0.3 mmol scale, and yields are reported as isolated yields. bn-BuBF3K (2 equiv.) and AgF (3 equiv.) were used. | ||
Encouraged by these outcomes, we next sought to apply this process to the ortho-alkylations of heterocyclic aldehydes. Traditionally, alkylations of N-heterocycles were realized through the venerable Friedel–Crafts and Minisci reactions.13 However, in these processes, alkylations typically occur at the most electron-rich and electron-poor sites of the heterocycles, respectively. We questioned if this electronic preference could be overridden by a TDG on the aldehyde component, so that the desired alkylation process would occur exclusively at the ortho positions of the substrates. Indeed, with nicotinaldehydes or isonicotinaldehydes as the substrates, the ortho C–H bis-alkylation process proceeded efficiently in most cases (Scheme 3, 2a–2f). While the reaction yields tended to vary with different substitution patterns, the regioselectivity was unaffected in all cases. Quinoline- and isoquinoline-carboxaldehydes were also amenable to this process, providing the ortho C–H alkylation products in greater than 80% yields (Scheme 3, 2g and 2h). Other five-membered heterocycles, such as indole, pyrrole, furan and thiophene were also viable substrates (Scheme 3, 2i–2l). Interestingly, alkylation only occurred at the 2′ position of the indole nucleus, a result that contrasts with the previous Pd-catalyzed ortho C–H arylation reaction (Scheme 3, 2i).10d
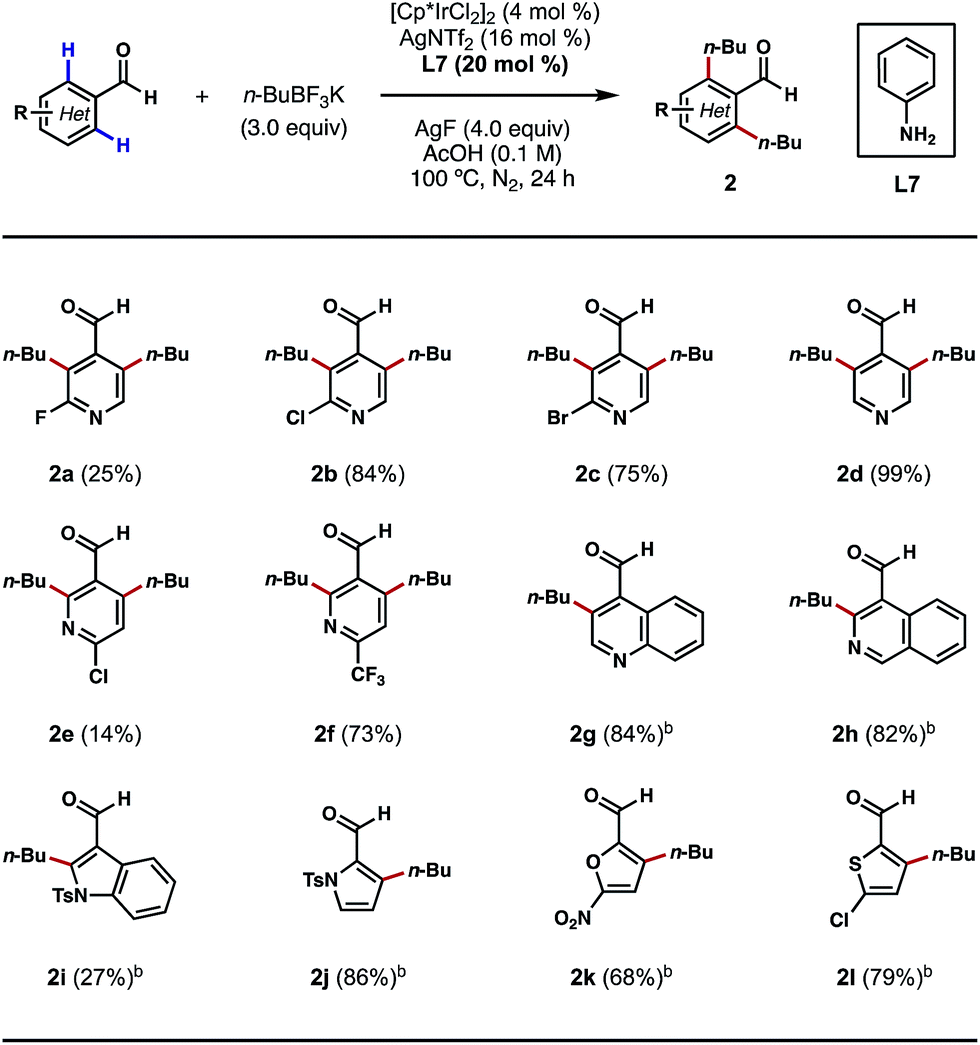 | ||
| Scheme 3 Scope of heterocyclic aldehydes. aThe reactions were performed on a 0.3 mmol scale, and yields are reported as isolated yields. bn-BuBF3K (2 equiv.) and AgF (3 equiv.) were used. | ||
Having shown that direct ortho alkylations of diverse (hetero)aromatic aldehydes are possible, we evaluated the couplings of structurally diverse potassium alkyltrifluoroborates with 2-chloroisonicotinaldehyde. While the attempted ortho bis-methylations were disappointing (Scheme 4, 3a), to the best of our knowledge, this is the first example of directly transforming the ortho C–H bonds of a heterocyclic aldehyde into methyl groups, a challenging process that is unattainable using our previous Pd-catalyzed C–H methylation protocol.8b Gratifyingly, good reactivity was restored with other alkylation processes, such as ethylation, n-hexylation and n-octylation (Scheme 4, 3b–3d). The reaction can also affix branched alicyclic groups (e.g. cyclopentylmethyl and cyclohexylmethyl) to the pyridine nucleus (Scheme 4, 3e and 3f). Chains terminating in tert-butyl groups may also be introduced, albeit in a low yield (Scheme 4, 3g). We were also pleased to find that chains terminating in phenyl, acetoxy, and trifluoromethyl groups may also be introduced via this alkylation process (Scheme 4, 3h–3j). While reaction efficiency was not completely satisfactory in certain cases, these examples demonstrate the generality of this method in introducing structurally diverse alkyl groups to the ortho position of a heterocyclic aldehyde regardless of their lengths, bulkiness and the functionalities that they contain. For certain potassium alkyltrifluoroborates that are labile at high temperatures or under acidic conditions, dichloroethane (DCE) was used as the reaction solvent and a lowered reaction temperature was employed (Scheme 4, 3g and 3j). In its current state of development, this method does not permit the use of secondary potassium alkyltrifluoroborates.
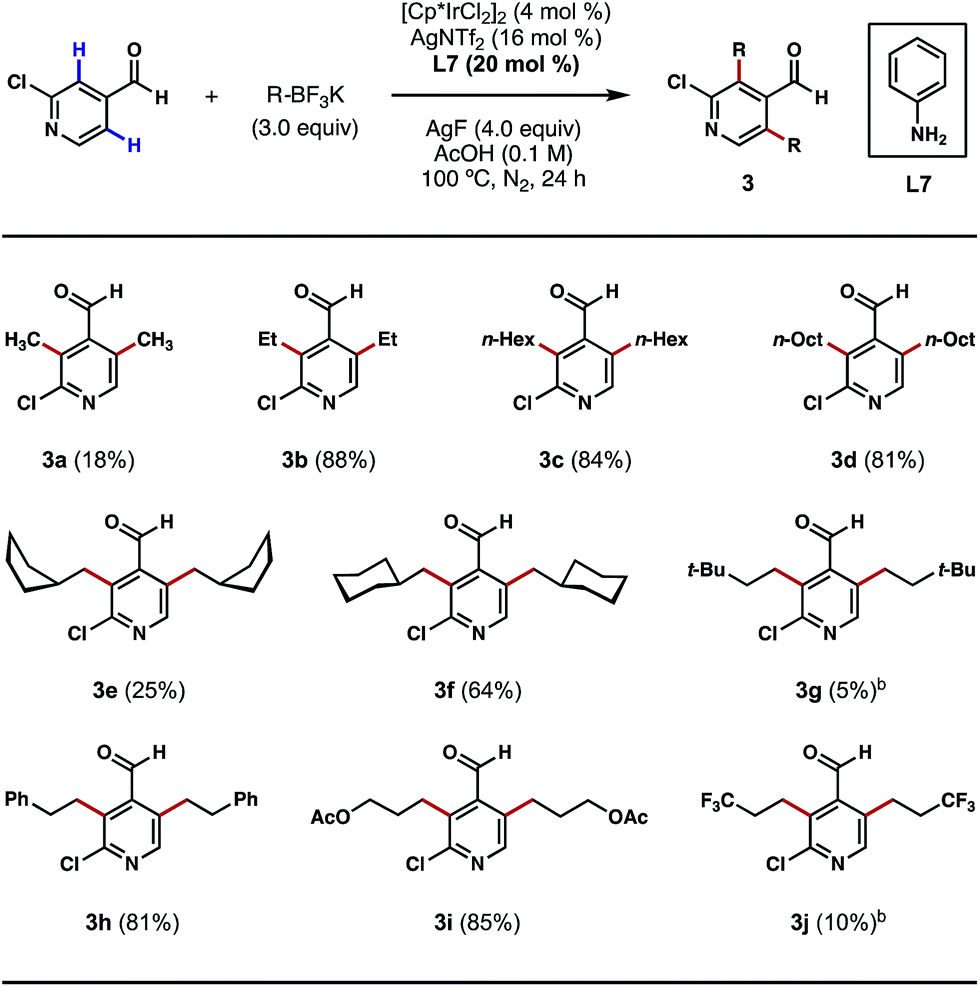 | ||
| Scheme 4 Scope of potassium alkyltrifluoroborates. aThe reactions were performed on a 0.3 mmol scale, and yields are reported as isolated yields. bDCE (0.1 M) was used as the reaction solvent and the reaction temperature was 80 °C. | ||
To gain mechanistic insights into this process, aldimine I (synthesized by a condensation reaction between 4-fluorobenzaldehyde and aniline; see ESI† for details) was exposed to a stochiometric amount of [Cp*IrCl2]2 and an excess of NaOAc in DCE at 60 °C (Scheme 5).11d In the wake of this reaction, we isolated a stable C–H iridacycle II; the structure of this complex was confirmed by X-ray crystallography (Scheme 5). This outcome provides a direct crystallographic evidence for an Ir(III) C–H insertion mechanistic pathway in this reaction instead of a simple conjugative addition of the Ir(III) catalyst to the electron-deficient arene. Further elaboration of complex II with AgNTf2 in acetonitrile results in the formation of cationic Ir complex III in a quantitative yield.11d To our delight, this air-stable cationic Ir species proved to be a competent intermediate in the bis-alkylation process and underwent conversion to compound 1a upon treatment with n-BuBF3K and AgF at 100 °C (Scheme 5).
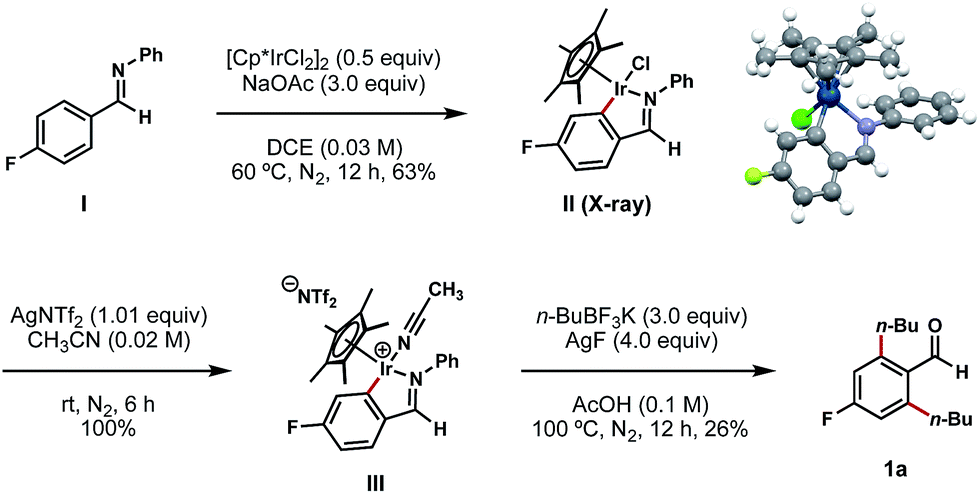 | ||
| Scheme 5 Isolation and X-ray characterization of iridacycle II and its subsequent transformations. aAll yields are reported as isolated yields. | ||
Motivated by the above studies, we next explored the possibility of alkylating vinylic C–H bonds in α,β-unsaturated aldehydes; this would be a unique and desirable transformation in synthesis. In comparison to the plethora of research on arene C–H functionalizations, activation of non-aromatic, vinylic C–H bonds using transition metals poses a steeper challenge and is still underdeveloped. While a number of C(sp2)–C(sp3) bond forming reactions were achieved primarily through C–H additions to carbonyls, imines and related, polarized π bonds,14 the direct coupling of vinylic C–H bonds with versatile alkyl boron reagents remains an unsolved problem in cross-coupling chemistry. With 4-trifluoromethylaniline (L1) as a key catalytic ligand and under the conditions shown in Scheme 6, a number of cyclic and acyclic α,β-unsaturated aldehydes were successfully alkylated at the β position (4a–4d). The exact role of crotonic acid is unclear at present, although it was found to be critical to reactivity in most cases. Surprisingly, both vinylic C–H bonds of acrolein substrates, either cis or trans to the formyl group, are alkylated under the conditions shown (Scheme 6, 4c and 4d); despite its modest efficiencies, this interesting and potentially important reaction type clearly merits further development.
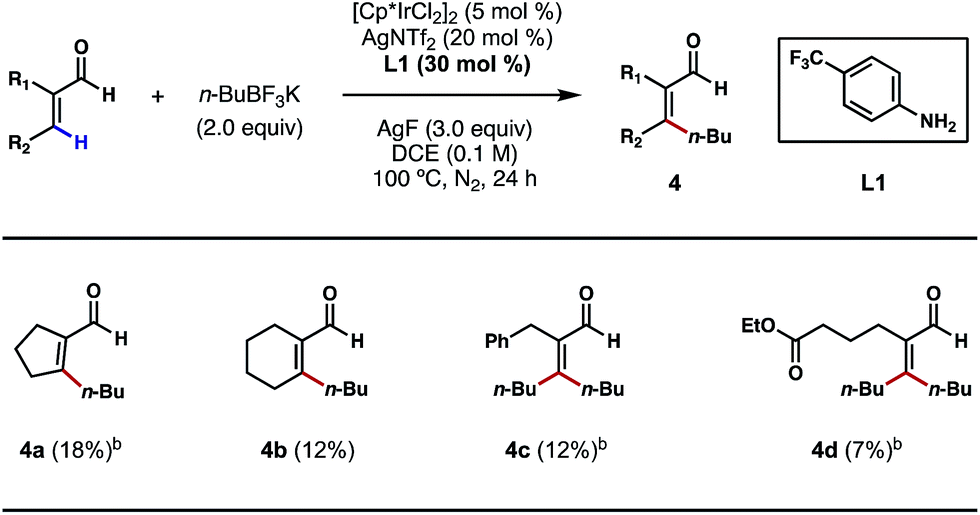 | ||
| Scheme 6 Preliminary studies on the β C–H alkylations of α,β-unsaturated aldehydes. aThe reactions were performed on a 0.3 mmol scale, and yields are reported as isolated yields. bCrotonic acid (30 mol%) was an additive. | ||
Conclusions
In summary, a convenient method was developed for the ortho C(sp2)–H alkylations of (hetero)aromatic aldehydes as well as β C–H alkylation of α,β-unsaturated aldehydes using potassium alkyltrifluoroborates as versatile alkylating reagents. The key to these transformations was the use of [Cp*IrCl2]2 as a precatalyst, which, in combination with aniline as the catalytic ligand, promotes the desired C(sp3)–C(sp2) reductive elimination without β-hydride elimination. This method offers an attractive, new approach to the late stage C(sp2)–H alkylations of aldehyde-containing substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Philip Provencher for generously providing the acrolein substrates, Dan Novoa for his suggestions on reaction engineering and other Sorensen group members for helpful discussions. We also thank the MacMillan and Chirik group at Princeton University for generously providing IrCl3 and the Cp* ligand. Dr Istvan Pelczer, Dr Ken Conover, Dr John Eng and Dr Phil Jeffrey at Princeton University are gratefully acknowledged for their assistance with our NMR, mass spectrometric and X-ray crystallographic analyses. This work was supported by the National Science Foundation (USA) under the CCI Center for Selective C–H Functionalization (CHE-1700982) and Princeton University.Notes and references
- (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (e) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- (a) N. Gürbüz, I. Özdemir and B. Çetinkaya, Tetrahedron Lett., 2005, 46, 2273–2277 CrossRef; (b) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134–1137 CrossRef CAS PubMed; (c) V. Lanke and K. Ramaiah Prabhu, Org. Lett., 2013, 15, 6262–6265 CrossRef CAS PubMed; (d) F. Z. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285–11288 CrossRef CAS PubMed; (e) R. Santhoshkumar, S. Mannathan and C.-H. Cheng, J. Am. Chem. Soc., 2015, 137, 16116–16120 CrossRef CAS PubMed.
- (a) S. J. Tremont and H. U. Rahman, J. Am. Chem. Soc., 1984, 106, 5759–5760 CrossRef CAS; (b) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634–12635 CrossRef CAS PubMed; (c) X. Chen, J.-J. Li, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 78–79 CrossRef CAS PubMed; (d) L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS PubMed; (e) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed; (f) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (g) Q. Chen, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 428–429 CrossRef CAS PubMed; (h) Y. Zhao and G. Chen, Org. Lett., 2011, 13, 4850–4853 CrossRef CAS PubMed; (i) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS PubMed; (j) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906–3911 RSC; (k) S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302–2305 CrossRef CAS PubMed; (l) P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508–17513 CrossRef CAS PubMed; (m) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS PubMed; (n) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed; (o) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS PubMed; (p) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126–13129 CrossRef CAS PubMed; (q) R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194–13197 CrossRef CAS PubMed; (r) H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef CAS PubMed; (s) J. Wippich, I. Schnapperelle and T. Bach, Chem. Commun., 2015, 51, 3166–3168 RSC; (t) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531–539 CrossRef CAS PubMed; (u) K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Chem. Commun., 2016, 52, 1915–1918 RSC; (v) G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484–1488 CrossRef CAS PubMed; (w) P. Peng, J. Wang, H. Jiang and H. Liu, Org. Lett., 2016, 18, 5376–5379 CrossRef CAS PubMed; (x) J. M. Wiest, A. Pothig and T. Bach, Org. Lett., 2016, 18, 852–855 CrossRef CAS PubMed; (y) Z. Chen, L. a. Hu, F. Zeng, R. Zhu, S. Zheng, Q. Yu and J. Huang, Chem. Commun., 2017, 53, 4258–4261 RSC; (z) Q. Shao, J. He, Q.-F. Wu and J.-Q. Yu, ACS Catal., 2017, 7, 7777–7782 CrossRef CAS.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) F. Kakiuchi, M. Yamauchi, N. Chatani and S. Murai, Chem. Lett., 1996, 25, 111–112 CrossRef; (c) C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 6616–6623 CrossRef CAS; (d) C.-H. Jun, J.-B. Hong, Y.-H. Kim and K.-Y. Chung, Angew. Chem., Int. Ed., 2000, 39, 3440–3442 CrossRef CAS; (e) Y.-G. Lim, J.-S. Han, S.-S. Yang and J. H. Chun, Tetrahedron Lett., 2001, 42, 4853–4856 CrossRef CAS; (f) R. K. Thalji, K. A. Ahrendt, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 9692–9693 CrossRef CAS PubMed; (g) C.-H. Jun, C. W. Moon, J.-B. Hong, S.-G. Lim, K.-Y. Chung and Y.-H. Kim, Chem.–Eur. J., 2002, 8, 485–492 CrossRef CAS PubMed; (h) R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192–7193 CrossRef CAS PubMed; (i) R. Martinez, R. Chevalier, S. Darses and J.-P. Genet, Angew. Chem., Int. Ed., 2006, 45, 8232–8235 CrossRef CAS PubMed; (j) A. Watzke, R. M. Wilson, S. J. O'Malley, R. G. Bergman and J. A. Ellman, Synlett, 2007, 2007, 2383–2389 CrossRef; (k) K. Tsuchikama, M. Kasagawa, Y.-K. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939–3942 CrossRef CAS; (l) F. Kakiuchi, T. Kochi, E. Mizushima and S. Murai, J. Am. Chem. Soc., 2010, 132, 17741–17750 CrossRef CAS PubMed; (m) G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258–10261 CrossRef CAS PubMed; (n) N. Kimura, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2017, 139, 14849–14852 CrossRef CAS PubMed.
- (a) F. Kakiuchi, Y. Tanaka, T. Sato, N. Chatani and S. Murai, Chem. Lett., 1995, 24, 679–680 CrossRef; (b) B. M. Trost, K. Imi and I. W. Davies, J. Am. Chem. Soc., 1995, 117, 5371–5372 CrossRef CAS; (c) T. Sato, F. Kakiuchi, N. Chatani and S. Murai, Chem. Lett., 1998, 27, 893–894 CrossRef; (d) F. Kakiuchi, T. Sato, K. Igi, N. Chatani and S. Murai, Chem. Lett., 2001, 30, 386–387 CrossRef; (e) C.-H. Jun, C. W. Moon, Y.-M. Kim, H. Lee and J. H. Lee, Tetrahedron Lett., 2002, 43, 4233–4236 CrossRef CAS; (f) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 5604–5605 CrossRef CAS PubMed.
- (a) Z. Wang, B. J. Reinus and G. Dong, J. Am. Chem. Soc., 2012, 134, 13954–13957 CrossRef CAS PubMed; (b) F. Mo and G. Dong, Science, 2014, 345, 68–72 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- (a) X.-Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 6280–6283 CrossRef CAS PubMed; (b) X.-Y. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS PubMed.
- (a) Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 49, 4808–4826 CrossRef CAS; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 1–24 CrossRef; (c) S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC.
- (a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed; (b) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (c) K. Hong, H. Park and J.-Q. Yu, ACS Catal., 2017, 7, 6938–6941 CrossRef CAS PubMed; (d) X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed; (e) H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755–762 CrossRef CAS PubMed.
- (a) F. Ma, M. Lei and L. Hu, Org. Lett., 2016, 18, 2708–2711 CrossRef CAS PubMed; (b) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 CrossRef CAS PubMed; (c) R. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef PubMed; (d) Y.-F. Zhang, B. Wu and Z.-J. Shi, Chem.–Eur. J., 2016, 22, 17808–17812 CrossRef CAS PubMed; (e) D. L. Mu, X. M. Wang, G. Chen and G. He, J. Org. Chem., 2017, 82, 4497–4503 CrossRef CAS PubMed; (f) X.-Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 1140–1143 CrossRef CAS PubMed; (g) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CAS; (h) S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC; (i) J. Xu, Y. Liu, Y. Wang, Y. Li, X. Xu and Z. Jin, Org. Lett., 2017, 19, 1562–1565 CrossRef CAS PubMed; (j) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073–1076 CrossRef CAS PubMed; (k) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS PubMed; (l) B. Li, K. Seth, B. Niu, L. Pan, H. Yang and H. Ge, Angew. Chem., Int. Ed., 2018, 57, 3401–3405 CrossRef CAS; (m) D. Mu, G. He and G. Chen, Chem.–Asian J., 2018, 13, 2423–2426 CrossRef CAS PubMed; (n) F. Li, Y. Zhou, H. Yang, D. Liu, B. Sun and F.-L. Zhang, Org. Lett., 2018, 20, 146–149 CrossRef CAS PubMed; (o) G. Liao, Q. J. Yao, Z. Z. Zhang, Y. J. Wu, D. Y. Huang and B. F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 CrossRef CAS PubMed; (p) X. Liu, Z. Wang, Q. Chen, M.-y. He and L. Wang, Appl. Organomet. Chem., 2018, 32, e4039 CrossRef; (q) L. Pan, K. Yang, G. Li and H. Ge, Chem. Commun., 2018, 54, 2759–2762 RSC; (r) J. Xu, Y. Liu, J. Zhang, X. Xu and Z. Jin, Chem. Commun., 2018, 54, 689–692 RSC; (s) D.-Y. Wang, S.-H. Guo, G.-F. Pan, X.-Q. Zhu, Y.-R. Gao and Y.-Q. Wang, Org. Lett., 2018, 20, 1794–1797 CrossRef CAS PubMed; (t) O. K. Rasheed, Synlett, 2018, 29, 1033–1036 CrossRef CAS; (u) X. Wang, S. Song and N. Jiao, Chin. J. Chem., 2018, 36, 213–216 CrossRef CAS.
- (a) J. Wencel-Delord, F. W. Patureau and F. Glorius, in C-H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 1–27 Search PubMed; (b) J. G. Kim, K. Shin and S. Chang, in C–H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 29–51 Search PubMed.
- (a) M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 CrossRef PubMed; (b) M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, and crystallographic data (CIF). CCDC 1861852. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03606c |
| ‡ General experimental procedure for the ortho C–H alkylations of (hetero)aromatic aldehydes: a 20 mL oven-dried scintillation vial equipped with a cross-shaped stir bar was charged with an aromatic aldehyde (0.30 mmol), [Cp*IrCl2]2 (9.6 mg, 0.012 mmol), AgF (0.152 g, 1.20 mmol) and n-BuBF3K (0.147 g, 0.9 mmol). The vial was transferred to a glovebox filled with N2, wherein AgNTf2 (18.6 mg, 0.048 mmol) and aniline (5.6 μL, 0.060 mmol) were added. The vial was capped tightly with a PTFE-lined green cap and taken out of the glovebox. Degassed AcOH (3 mL) was added through a syringe, and the vial was heated in a pie-block at 100 °C under vigorous stirring for 24 h. After cooling to r.t., the reaction mixture was filtered through a pad of Celite® and washed with EtOAc (15 mL). The filtrate was then concentrated in vacuo and the resulting residue was purified by flash silica gel column chromatography. |
| This journal is © The Royal Society of Chemistry 2018 |