
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold-catalyzed conversion of lignin to low molecular weight aromatics†
Yang
Song
 ab,
Justin K.
Mobley‡
c,
Ali Hussain
Motagamwala
cd,
Mark
Isaacs§¶
e,
James A.
Dumesic
cd,
John
Ralph
cf,
Adam F.
Lee
g,
Karen
Wilson
g and
Mark
Crocker
*ab
ab,
Justin K.
Mobley‡
c,
Ali Hussain
Motagamwala
cd,
Mark
Isaacs§¶
e,
James A.
Dumesic
cd,
John
Ralph
cf,
Adam F.
Lee
g,
Karen
Wilson
g and
Mark
Crocker
*ab
aDepartment of Chemistry, University of Kentucky, 505 Rose Street, Lexington, KY 40506, USA
bCenter for Applied Energy Research (CAER), University of Kentucky, 2540 Research Park Drive, Lexington, KY 40511, USA. E-mail: mark.crocker@uky.edu
cGreat Lakes Bioenergy Research Center, The Wisconsin Energy Institute, University of Wisconsin-Madison, 1552 University Avenue, Madison, WI 53726, USA
dDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53726, USA
eEuropean Bioenergy Research Institute, Aston University, Aston Triangle, Birmingham, B4 7ET, UK
fDepartment of Biochemistry, University of Wisconsin–Madison, 433 Babcock Drive, Madison, WI 53706, USA
gSchool of Science, RMIT University, 124 La Trobe Street, Melbourne, VIC 3000, Australia
First published on 6th September 2018
Abstract
A heterogeneous catalyst system, employing Au nanoparticles (NPs) and Li–Al (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) layered double hydroxide (LDH) as support, showed excellent activity in aerobic oxidation of the benzylic alcohol group in β-O-4 linked lignin model dimers to the corresponding carbonyl products using molecular oxygen under atmospheric pressure. The synergistic effect between Au NPs and the basic Li–Al LDH support induces further reaction of the oxidized model compounds, facilitating facile cleavage of the β-O-4 linkage. Extension to oxidation of γ-valerolactone (GVL) extracted lignin and kraft lignin using Au/Li–Al LDH under similar conditions produced a range of aromatic monomers in high yield. Hydrolysis of the Au/Li–Al LDH oxidized lignin was found to increase the degree of lignin depolymerization, with monomer yields reaching 40% for GVL extracted lignin. Based on these results, the Au/Li–Al LDH + O2 catalyst system shows potential to be an environmentally friendly means of depolymerizing lignin to low molecular weight aromatics under mild conditions.
:2) layered double hydroxide (LDH) as support, showed excellent activity in aerobic oxidation of the benzylic alcohol group in β-O-4 linked lignin model dimers to the corresponding carbonyl products using molecular oxygen under atmospheric pressure. The synergistic effect between Au NPs and the basic Li–Al LDH support induces further reaction of the oxidized model compounds, facilitating facile cleavage of the β-O-4 linkage. Extension to oxidation of γ-valerolactone (GVL) extracted lignin and kraft lignin using Au/Li–Al LDH under similar conditions produced a range of aromatic monomers in high yield. Hydrolysis of the Au/Li–Al LDH oxidized lignin was found to increase the degree of lignin depolymerization, with monomer yields reaching 40% for GVL extracted lignin. Based on these results, the Au/Li–Al LDH + O2 catalyst system shows potential to be an environmentally friendly means of depolymerizing lignin to low molecular weight aromatics under mild conditions.
Introduction
Due to rising political, economic, and environmental challenges associated with the extraction and use of fossil fuels, lignocellulosic biomass has come to prominence as a logical renewable alternative to petroleum-based resources for commercial carbon-based products.1,2 Although ethanol production from the cellulosic component of lignocellulosic biomass is being researched at a pilot scale as a replacement for grain ethanol,3 the utilization of lignin is limited,2 despite the fact that the cellulosic ethanol production cost is directly correlated to the valorization of lignin.4,5 Enzyme-induced combinatorial radical polymerization of monolignols leaves lignin with a complex, amorphous chemical structure.6–8 In view of its structural complexity, harnessing this resource requires chemical transformations that can depolymerize this recalcitrant material to useable monomers with high efficiency and selectivity.9 Although lignin possesses a heterogeneous structure, all lignins contain the alkyl aryl ether unit (i.e., contain the β-O-4 linkage) as the most abundant structural unit (up to 60% of all linkages)8 followed by β-5, β-β, and other minor units (Fig. 1).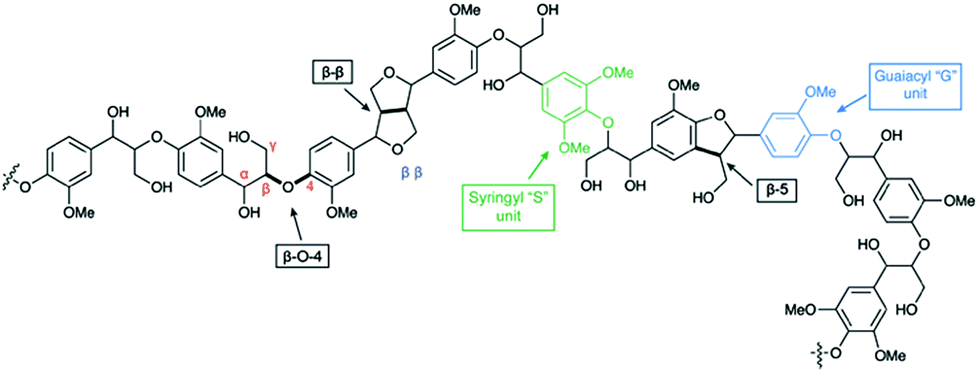 | ||
| Fig. 1 Structural representation of a lignin fragment illustrating selected linkages. | ||
Although much effort has been invested in hydrogenolytic approaches to lignin depolymerization, the chemistry can be non-selective;10 moreover, most phenolics produced via reductive pathways would face competition from the low cost phenolics produced from petroleum.11 Compared to hydrogenolytic methods, catalytic oxidative lignin depolymerization is desirable as the products exhibit increased functionalization and complexity,11–13 otherwise not readily available from petroleum resources.6 Many recent studies utilize homogenous catalytic systems for oxidative lignin depolymerization, and while promising results have been achieved for lignin model compounds, many systems suffer from a lack of selectivity,10,14 catalyst decomposition,12 and the requirement for harsh reaction conditions.15 In addition, problems inherent in the recycling of homogenous catalysts make the industrial application of these systems difficult. Heterogeneous catalyst systems are generally better suited for industrial applications,16 and recent reports have shown moderate to high yields in heterogeneously catalyzed oxidation of lignin model compounds and mono-aromatic substrates,17–23 albeit with few successful examples of lignin depolymerization having been reported. Aside from catalyst type, the oxidant is another key factor in oxidative lignin depolymerization processes, molecular oxygen being favored as the terminal oxidant for any large scale oxidative conversion process.17,24 In the past 20 years, Au nanoparticles (NPs) supported on metal oxide and layered double hydroxide (LDH) supports (Au/TiO2, Au/CeO2, Au/Mg–Al LDH, Au/Ni–Al LDH, etc.) have been shown to selectively catalyze aerobic oxidations of alcohols to the corresponding carbonyl compounds.25–29 Herein, we introduce a heterogeneous catalyst system for the aerobic oxidation and depolymerization of lignin by targeting the lignin β-aryl ether fragment, utilizing Au NPs supported on a basic Li–Al LDH30 under molecular oxygen at atmospheric pressure. We highlight the high oxidation activity of Au/Li–Al LDH with a variety of lignin model compounds, and its applicability towards lignin depolymerization.
Results and discussion
Our initial studies focused on the oxidation of simple benzylic alcohols (Tables 1 and 2). Encouragingly, we found that by employing a strongly basic Li–Al (1:2) LDH30 as support, the activity for benzylic alcohol oxidation increased significantly compared to less basic Au/LDH catalysts. When toluene was used as solvent for comparison with literature data, Au/Li–Al LDH exhibited the highest turnover frequency (TOF) compared to other supported Au NP catalyst systems that effectively oxidize 1-phenylethanol such as Au/Mg–Al LDH (3213 h−1),31 Au/Ni–Al LDH (5310 h−1),32 and Au/Al2O3 (825 h−1).27
| Catalyst | t [h] | Conv.b [%] | Sel.b [%] | TOFc [h−1] |
|---|---|---|---|---|
| a 1-Phenylethanol (1 mmol), catalyst (0.1 g, 1 wt% Au), diphenyl ether (10 mL), 80 °C, p = 1 atm. O2 (10 mL min−1). b Conversion and selectivity were determined by GC-MS using dodecane as internal standard. c TOF values are based on the number of surface Au atoms calculated for the mean Au particle size found via TEM analysis (see ESI Fig. S3).33,34 d TOF in parentheses was determined using toluene as solvent. Note: a control experiment conducted without catalyst showed no conversion. | ||||
| Au/Mg–Al LDH | 1 | 75 | >99 | 5926 |
| Au/Ni–Al LDH | 1 | 34 | >99 | 1317 |
| Au/Li–Al LDH | 1 | 98 | >99 | 11061 (29708)d |
| Entry | Substrate | t [h] | Conv. [%] | Product | Sel. [%] |
|---|---|---|---|---|---|
| a Substrate (1 mmol), Au/Li–Al LDH (50 mg, 1 wt% Au), diphenyl ether (DPE, 10 mL), 80 °C, p = 1 atm. O2 (10 mL min−1). Conversion and selectivity were determined by GC-MS using dodecane as internal standard. b Substrate (1 mmol), Au/Li–Al LDH (50 mg, 1 wt% Au), diphenyl ether (DPE, 10 mL), 100 °C, p = 1 atm. O2 (10 mL min−1). Conversion and selectivity were determined by GC-MS using dodecane as internal standard. Partial results shown, for full list of benzylic alcohols tested refer to ESI (see ESI Table S2). | |||||
| 1 |
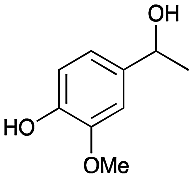
|
4 | 80 |
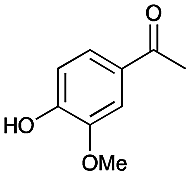
|
78 |
| 2 |

|
2 | 98 |
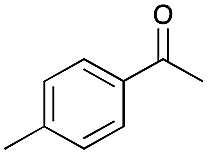
|
98 |
| 3 |

|
0.5 | >99 |
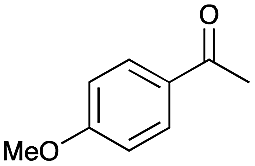
|
>99 |
| 4 |

|
2 | 67 |
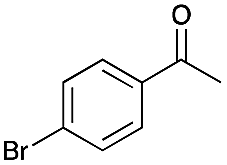
|
>99 |
| 5 |

|
4 | 80 |
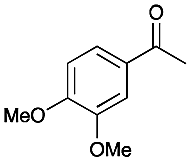
|
>99 |
| 6b |
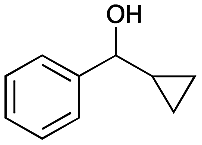
|
2 | >99 |
|
>99 |
| 7 |
|
0.5 | >99 |

|
>99 |
The high oxidation activity of Au/Li–Al LDH can be explained by the degree of charge transfer from the basic support to the Au NPs.30,35 According to CO2 pulse chemisorption measurements, the basic site concentration follows the order Mg–Al LDH (44.8 μmol gcat−1) < Ni–Al LDH (80.5 μmol gcat−1) < Li–Al LDH (102.7 μmol gcat−1), while evidence of charge transfer to the supported Au NPs was provided by X-ray photoelectron spectroscopy (XPS): as the basicity of the support increases, the binding energy of the Au 4f7/2 peak is shifted towards lower energy (see Fig. S1 and S2 in ESI†).
Besides the basic nature of the Li–Al LDH, the Au nanoparticle size also plays an important role. It has been well established that reactivity towards alcohol oxidation using Au NPs decreases significantly when the Au particle size surpasses 5 nm;36–39 we observe via transmission electron microscopy (TEM) that most of the Au NPs in our catalyst are <5 nm, the median particle size being 2.1 nm (Fig. S3†), which should be optimal for alcohol oxidation. The Li–Al LDH used in this catalyst system possesses a crystalline structure (Fig. S4†) and exhibits a characteristic sand rose morphology (Fig. S5†) with a relatively high surface area of 85 m2 g−1 (Table S1†), which favors adsorption of the alcohol and facilitates the formation of small Au NPs on the catalyst surface.
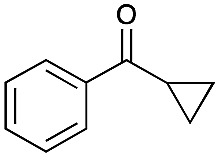
The difficulty of activating O2 represents one of the rate limiting factors in oxidation catalysis.40 It is contended that Au NPs adsorbed on the surface of a basic support contain coordinatively unsaturated Au atoms that are efficient in O2 dissociation, and the activity of adsorbed O2 on metallic Au follows Brønsted acid–base reaction patterns.38,40 Our proposed mechanism (Fig. 2) suggests that catalysis occurs at the Au–O–Li interface. Electron rich Au NPs activate molecular oxygen via electron donation to the LUMO (π*) of O2 (ref. 41) and simultaneously, the support deprotonates the benzylic hydroxyl group to allow adsorption to the catalyst surface. The oxidation process is then completed by a series of deprotonation, elimination, and catalyst regeneration steps. It should also be noted that the reaction proceeds via a two-electron pathway, as evidenced by the selective oxidation of α-cyclopropyl-benzenemethanol without ring opening (entry 6 in Table 2). Compared to radical pathways, a two-electron oxidation mechanism preserves desirable aromaticity in most products, and prevents repolymerization of intermediates that generate oligomers via irreversible bond formation.42–44
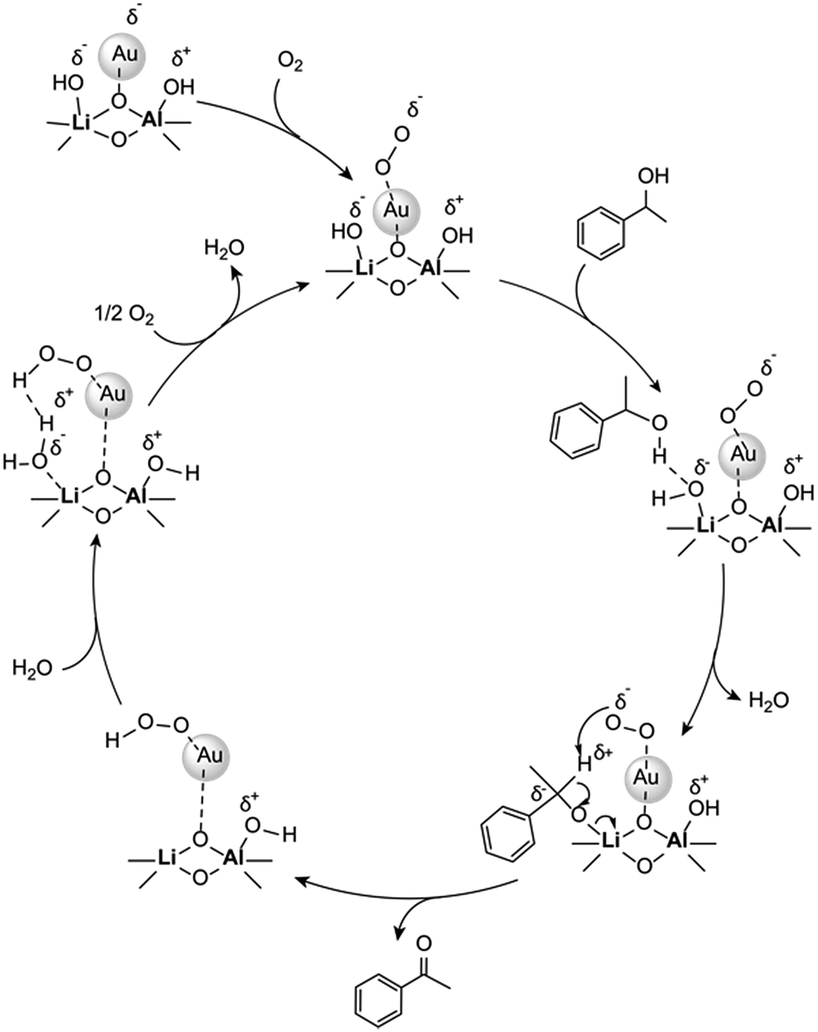 | ||
| Fig. 2 Proposed mechanism for aerobic oxidation of benzylic alcohols over Au/Li–Al LDH. | ||
The properties of Au/Li–Al LDH in aerobic oxidation were further evaluated using a lignin model dimer, 2a, containing the β-O-4 interunit linkage (Fig. 3). Our catalyst system showed selective oxidation of the secondary benzylic alcohol in 2a with a reaction rate of 10.8 mmol h−1 gcat−1, achieving a 37% yield of 2b in 5 h with 68% conversion of 2a (near complete conversion of 2a was seen after 24 h). Under these conditions 2b reacted further to give the dehydration product 2c (rate = 3.6 mmol h−1 gcat−1) and the retro-aldol product 2d (rate = 36 mmol h−1 gcat−1). The high reaction rate of the retro-aldol product formation may be attributed to the basic property of the Li–Al support, such reactions having been reported in the literature.45,46 While 2c was not observed to react further (Fig. S6†), 2d reached a maximum yield of 40% after 24 h, in turn producing guaiacol (2e) and p-anisic acid (2f) at a rate of 10.8 mmol h−1 gcat−1. Formation of products 2e and 2fvia cleavage of the β-O-4 bond in 2d using molecular oxygen has also been observed in various literature reports.47–49 Indeed, the propensity of the oxidized (ketone) form of β-O-4 model compounds to undergo oxidative cleavage to monomers has been attributed to the significantly weaker C–O bond present in the ketone form compared to the initial benzylic alcohol compound, and a decrease in pKa of the proton located at Cβ.48,50 Schoenebeck and coworkers49 suggested that the first step would most likely involve the formation of an enolate intermediate after deprotonation of the Cβ–H, which can be further oxidized to give a hydroperoxide species. Thereafter, two possible routes would give products 2e and 2f: (1) an anionic pathway involving formation of a dioxetane intermediate, which could undergo subsequent fragmentation to a carboxylate and a phenyl ester; (2) a homolytic O–O scission step forms an O centred radical, which undergoes further C–C cleavage, as suggested by Wang and co-workers.47
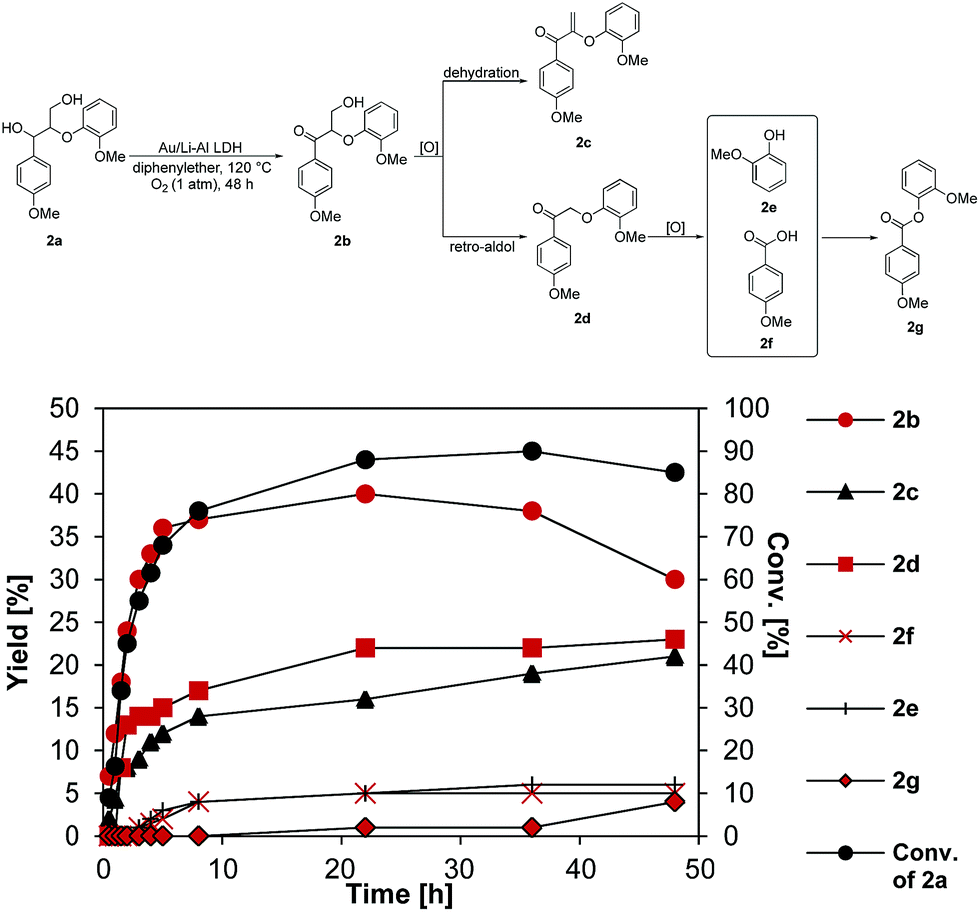 | ||
| Fig. 3 Aerobic oxidation of lignin model dimer 2a using Au/Li–Al LDH. Conditions: substrate (1 mmol), Au/Li–Al LDH (0.1 g, 1 wt% Au), DPE (10 mL), 120 °C, p = 1 atm. O2 (10 mL min−1). All reaction rates were obtained by oxidizing the individual intermediates using the same conditions. For full details refer to ESI (Fig. S5–S7 in ESI†). Conversion and yields were determined by GC-MS using dodecane as internal standard. | ||
In a subsequent esterification reaction, 2e and 2f reacted to form 2g with a rate of 5.4 mmol h−1 gcat−1. A similar rate of product formation was observed when only the Li–Al LDH support was used, consistent with base catalyzed esterification. Although esterification is an undesirable side-reaction, the ester linkage in 2g can be cleaved by means of simple hydrolysis, which, when translated to lignin, would result in net cleavage of the β-aryl ether linkage.
In order to examine the reusability of Au/Li–Al LDH under these reaction conditions, recycle reactions were carried out using a slightly simpler model compound, 3a (Table S3 and S4†). After three runs, the conversion of 3a measured at 4 h showed no change (∼58%). However, for recycle experiments performed at longer reaction times (16 h), a gradual decrease in the yields of 2e and 2f, formed from 2d, was apparent. Yields of 2e, 2f, and 2g were largely restored after the spent catalyst was washed with water at room temperature (4th run in Table S4†), consistent with the removal of adsorbed 2e and 2d (identified in the water washings). This indicates that over time 2e and 2d accumulate on the catalyst surface, as would be expected for acidic compounds, slowing the further conversion of 2d. Leaching of the catalytically active species into the reaction medium is an underlying concern for heterogeneously catalyzed lignin depolymerization, as observed by Bolm and coworkers.17 In the present work, when Au/Li–Al LDH was removed via hot filtration after 2 h of oxidation of 2a, no further conversion was observed after an additional 10 h reaction time. Consistent with these findings, concentrations of Au, Li, and Al in solution were determined to be <0.1 ppm by ICP-OES.
Next, our focus shifted to the use of process lignins, namely, Indulin AT kraft lignin (softwood, from pine) and γ-valerolactone extracted lignin51 from maple wood (hereafter referred to as KL and GVL, respectively), as substrate. Reaction conditions similar to those used in the lignin model compound experiments were applied, albeit dimethylformamide (DMF) was used as solvent instead of DPE due to the higher solubility of lignin in DMF. Fig. 4 shows 2D HSQC NMR spectra comparing KL and GVL before and after oxidation with O2 in the presence of Au/Li–Al LDH (oxidized KL and GVL are abbreviated as KLox and GVLox, respectively).
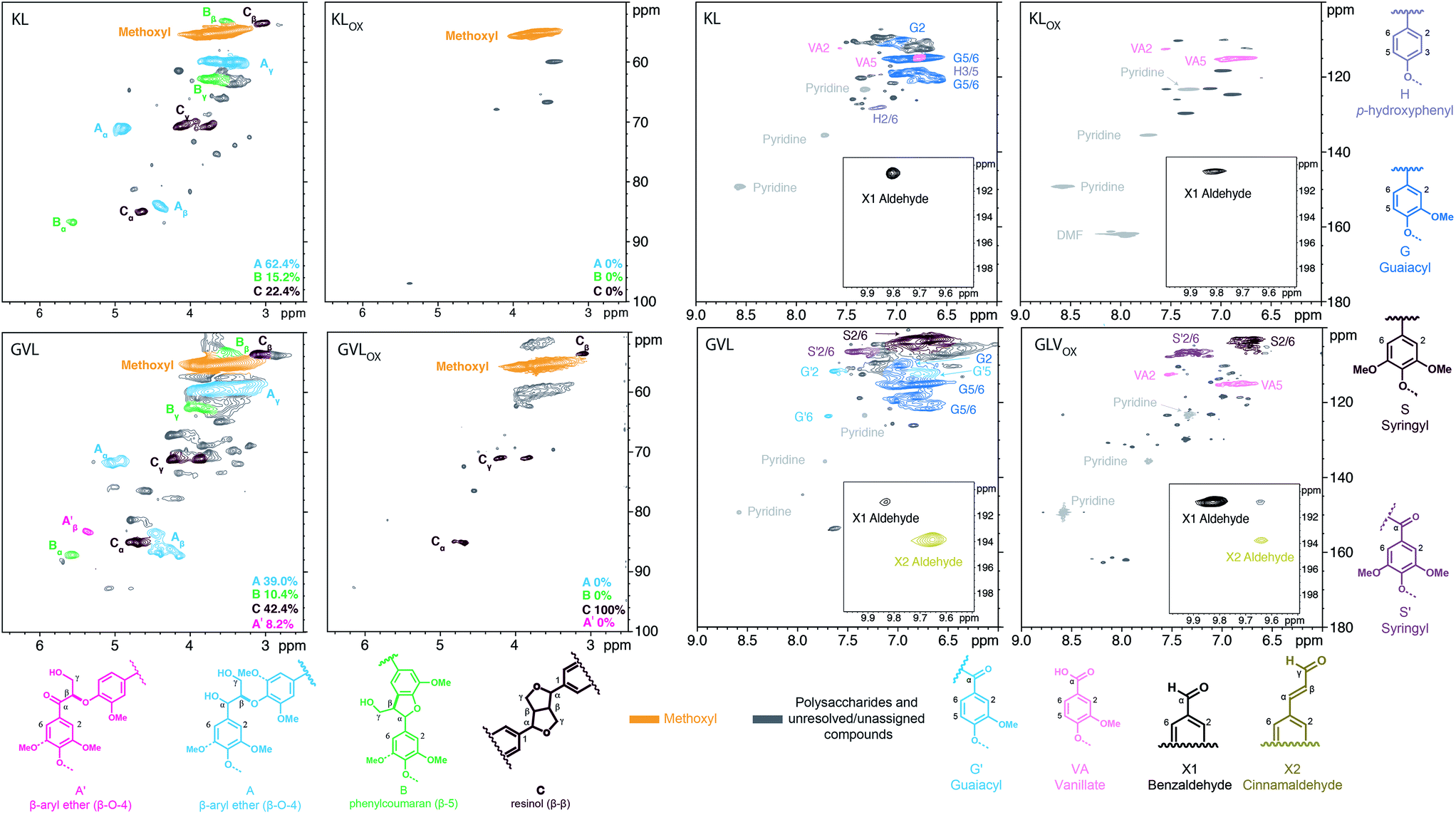 | ||
| Fig. 4 2D HSQC NMR spectra of KL, KLox, GVL, and GVLox lignin. All samples dissolved in 4:1 DMSO-d6/pyridine-d5; contours color-coded to structures responsible; percentages are determined from volume integrals based on αC–H with the exception of A′ which uses the βC–H signal. The assignment of peaks is based on known lignin spectra and available model compound data. | ||
Analysis of the oxygenated aliphatic region of GVLox and KLox revealed the absence of signals corresponding to β-aryl ether (A) and phenylcoumaran (B) units, albeit compared to KLox, some resinol (C) remains in GVLox. It is hypothesized that as for the β-aryl ether model dimer 2a, the β-aryl ether units in lignin may have gone through a similar oxidation-cleavage-re-coupling process forming the ester moiety, which has no sidechain C–H signals to observe via 2D HSQC NMR. Moreover, cross peaks correlating to the guaiacyl (G) units are no longer present in the HSQC spectrum after oxidation of KL and GVL (KLox and GVLox), and vanillate (VA) analogs are the only remaining signals for both. Additionally, the HSQC spectrum of GVLox displayed a significant decrease in syringyl (S) and an increase in oxidized syringyl units (S′).
The oxidized lignin samples were next subjected to hydrolysis with NaOH (0.1 M), after which the mixture was neutralized with HCl (1 M) and the soluble material was extracted with ethyl acetate (EtOAc). KLox yielded 20 wt% of EtOAc soluble material after hydrolysis (Fig. 5A), which is double the amount obtained from the initial oxidized lignin sample. Notably, GVLox after hydrolysis afforded 56 wt% EtOAc soluble material, versus 20 wt% without the additional hydrolysis step (Fig. 5A). The low percentage of soluble organic products formed from KLox is not surprising given that the kraft pulping process is known to produce a recalcitrant lignin that is C–C cross-linked, and hence difficult to depolymerize.8 However, the results obtained with GVLox are significant given that GVL more closely resembles the structure of native lignin than does KL.52 Gel permeation chromatography (GPC) revealed two peaks for both KLox and GVLox after hydrolysis (Fig. S9†). The first peak in both chromatograms represent fractions containing high molecular weight oligomers, and the second peak corresponds to low molecular weight components. The high molecular weight region shows a ∼3500 Da decrease in molecular weight for KLox post-hydrolysis while a decrease of ∼1000 Da was observed for GVLox post-hydrolysis. The low molecular weight range shows monomer production for both lignins, however, a significantly higher signal intensity was obtained from GVLox post-hydrolysis compared to KLox post-hydrolysis, consistent with the results previously mentioned in Fig. 5A.
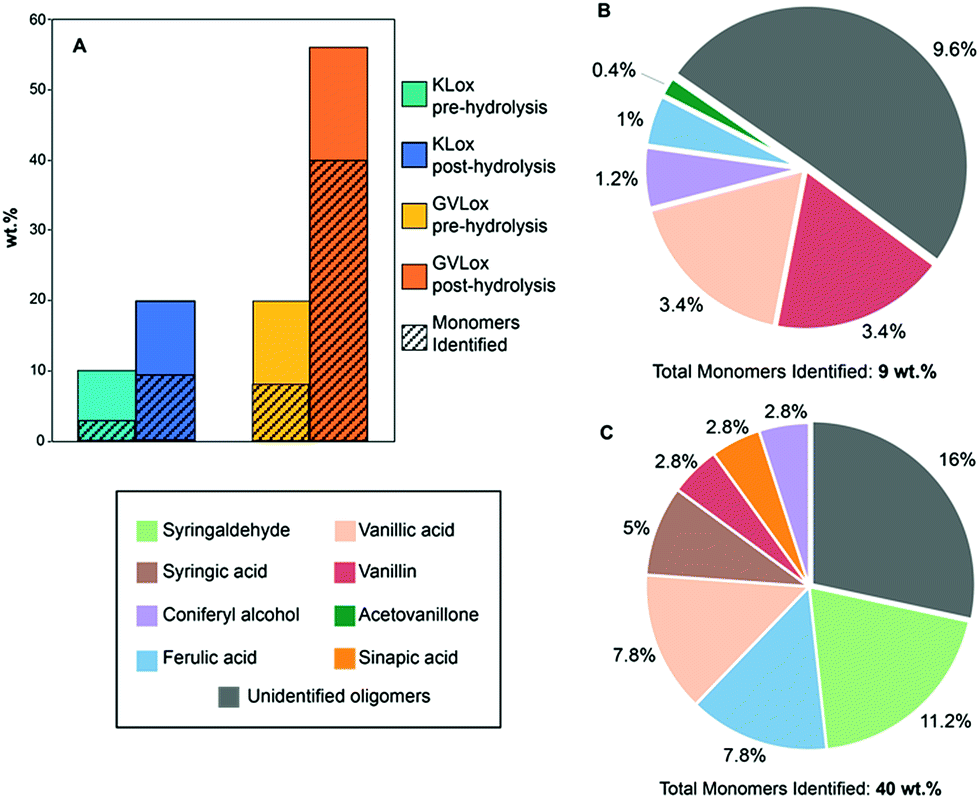 | ||
| Fig. 5 (A) Percent organic soluble fraction obtained from KLox and GVLox pre- and post-hydrolysis. Oxidized lignin samples (50 mg) were dissolved in 0.1 M NaOH (5 mL), followed by addition of 1 M HCl until pH 2 was reached, and were then extracted with EtOAc. For additional details refer to ESI. (B) Yields of monomer products from KLox and (C) GVLox after hydrolysis. Corresponding yields for control experiments (conducted without catalyst) are shown in the ESI.† | ||
Compound identification and quantification via GC-MS revealed that S- and G-derived carboxylic acids and aldehydes are the most prominent products (Fig. 5B and C). The yield of GC-MS identifiable monomers from GVL totaled 40 wt%, whereas a 10 wt% yield of GC-MS identifiable monomers was obtained from KL. Among the identified monomers, vanillin, vanillic acid, ferulic acid, and coniferyl alcohol were present from both lignins. Yields of these monomers, except for vanillin, were found to be higher from GVLox than from KLox. S-derived products were only obtained from GVL, as the KL used in this work was derived from pine, which consists of almost exclusively G units, a small amount of H units, and no S units.53 Notably, the monomers obtained, including vanillin, vanillic acid, and syringaldehyde, are of higher market value than those typically derived from hydrogenative methods of lignin depolymerization.54 These monomer yields are the highest reported to date for heterogeneously catalyzed oxidative lignin depolymerization55 and approach those reported for the most effective homogenous catalysts.56,57 Notably, the lignins employed in the latter case were either extracted enzymatically or via mild acidolysis, which preserves a high fraction of β-aryl ether linkages.57 The fact that we observe a 40 wt% monomer yield from a lignin that is extracted via a scalable method, utilizing a biomass-derived solvent, renders the Au/Li–Al LDH catalyst system particularly promising.
Conclusions
The heterogeneous catalyst Au/Li–Al LDH showed excellent activity and selectivity in the oxidation of benzylic alcohols using molecular oxygen as the terminal oxidant. XPS data suggest the high activity of this catalyst is facilitated by charge transfer from the basic Li–Al LDH support to the Au nanoparticles. As evidenced by the model β-O-4 linkage dimer oxidation results, our catalytic system was able to oxidatively cleave the linkage after selective benzylic alcohol oxidation, ultimately forming an ester moiety that can be depolymerized via simple hydrolysis. Similarly, NMR data obtained on oxidized kraft lignin (KLox) and GVL lignin (GVLox) showed the disappearance of benzylic alcohol groups, consistent with ester formation. After hydrolysis was performed on oxidized lignin samples, a 40 wt% yield of aromatic monomers was obtained from GVLox, while KLox afforded 10 wt% monomers. These monomer yields are the highest reported for heterogeneously catalyzed oxidative lignin depolymerization. Based on these results, Au/Li–Al LDH is identified as a promising catalyst system for lignin valorization to value-added low molecular weight aromatics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Science Foundation under Cooperative Agreement No. 1355438 and by the Global Bioenergy, Biofuels, and Biorefining network (GB3-Net) supported by the British Council and the Department for Business, Innovation and Skills (BIS). This work was also funded in part by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494 and DE-SC0018409). The authors thank Shelley Hopps for performing the XRD measurements and Dr Mark Meier for helpful discussions.References
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, et al. , Science, 2014, 344, 709 CrossRef PubMed.
- C. R. Brown and R. T. Brown, Why Are We Producing Biofuels?, Brownia LLC, 2012 Search PubMed.
- J. Kautto, M. J. Realff, A. J. Ragauskas and T. Kässi, BioResources, 2014, 9, 6041–6072 CrossRef.
- R. Davis, L. Tao, E. C. D. Tan, M. J. Biddy, G. T. Beckham, C. Scarlata, J. Jacobson, K. Cafferty, J. Ross, J. Lukas, et al., NREL/TP-5100-60223 2013.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef PubMed.
- C. Heitner, D. Dimmer and J. Schmidt, Lignin and Lignans: Advances in Chemistry, CRC Press, 2010 Search PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 2–54 Search PubMed.
- J.-P. Lange, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef PubMed.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef.
- L. Das, P. Kolar and R. Sharma-Shivappa, Biofuels, 2012, 3, 155–166 CrossRef.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef PubMed.
- O. Deutschmann, H. Knözinger, K. Kochloefl and T. Turek, in Ullmann's Encycl. Ind. Chem., Wiley-VCH Verlag GmbH & Co. KGaA, 2012, vol. 6, pp. 457–481 Search PubMed.
- J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2015, 8, 2106–2113 CrossRef PubMed.
- H. Deng, L. Lin and S. Liu, Energy Fuels, 2010, 24, 4797–4802 CrossRef.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, Z. Li and S. Liu, Catal. Lett., 2008, 126, 106–111 CrossRef.
- J. Zhang, H. Deng and L. Lin, Molecules, 2009, 14, 2747–2757 CrossRef PubMed.
- M. R. Sturgeon, M. H. O'Brien, P. N. Ciesielski, R. Katahira, J. S. Kruger, S. C. Chmely, J. Hamlin, K. Lawrence, G. B. Hunsinger and T. D. Foust, et al. , Green Chem., 2014, 16, 824–835 RSC.
- J. S. Kruger, N. S. Cleveland, S. Zhang, R. Katahira, B. A. Black, G. M. Chupka, T. Lammens, P. G. Hamilton, M. J. Biddy and G. T. Beckham, ACS Catal., 2016, 6, 1316–1328 CrossRef.
- X. Wu, S. Guo and J. Zhang, Chem. Commun., 2015, 51, 6318–6321 RSC.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef PubMed.
- J. K. Mobley and M. Crocker, RSC Adv., 2015, 5, 65780–65797 RSC.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890–1896 CrossRef.
- L. M. D. R. S. Martins, S. A. C. Carabineiro, J. Wang, B. G. M. Rocha, F. J. Maldonado-Hodar and A. J. L. Pombeiro, ChemCatChem, 2016, 9, 1211–1221 CrossRef.
- A. Abad, C. Almela, A. Corma and H. García, Tetrahedron, 2006, 62, 6666–6672 CrossRef.
- L. C. Wang, Y. M. Liu, M. Chen, Y. Cao, H. Y. He and K. N. Fan, J. Phys. Chem. C, 2008, 112, 6981–6987 CrossRef.
- J. L. Shumaker, C. Crofcheck, S. A. Tackett, E. Santillan-Jimenez, T. Morgan, Y. Ji, M. Crocker and T. J. Toops, Appl. Catal., B, 2008, 82, 120–130 CrossRef.
- P. Zhang, Y. Sui, G. Xiao, Y. Wang, C. Wang, B. Liu, G. Zou and B. Zou, J. Mater. Chem. A, 2013, 1, 1632–1638 RSC.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203–1207 CrossRef PubMed.
- L. Li, L. Dou and H. Zhang, Nanoscale, 2014, 6, 3753–3763 RSC.
- S. Wang, S. Yin, G. Chen, L. Li and H. Zhang, Catal. Sci. Technol., 2016, 6, 4090–4104 RSC.
- L. Wang, J. Zhang, X. Meng, D. Zheng and F. S. Xiao, Catal. Today, 2011, 175, 404–410 CrossRef.
- W. Fang, J. Chen, Q. Zhang, W. Deng and Y. Wang, Chem.–Eur. J., 2011, 17, 1247–1256 CrossRef PubMed.
- W. Fang, Q. Zhang, J. Chen, W. Deng and Y. Wang, Chem. Commun., 2010, 46, 1547–1549 RSC.
- G. J. Hutchings, in Nanotechnol. Catal., ed. B. Zhou, S. Han, R.Raja and G. A. Somorjai, Springer, New York, NY, 2007, pp. 39–54 Search PubMed.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef.
- T. A. Baker, X. Liu and C. M. Friend, Phys. Chem. Chem. Phys., 2011, 13, 34–46 RSC.
- S. Nishimura, Y. Yakita, M. Katayama, K. Higashimine and K. Ebitani, Catal. Sci. Technol., 2013, 3, 351–359 RSC.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef PubMed.
- J. Long, Q. Zhang, T. Wang, X. Zhang, Y. Xu and L. Ma, Bioresour. Technol., 2014, 154, 10–17 CrossRef PubMed.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- L. Hora, V. Kelbichová, O. Kikhtyanin, O. Bortnovskiy and D. Kubička, Catal. Today, 2014, 223, 138–147 CrossRef.
- J. C. A. A. Roelofs, D. J. Lensveld, A. J. Van Dillen and K. P. De Jong, J. Catal., 2001, 203, 184–191 CrossRef.
- H. Liu, M. Wang, H. Li, N. Luo, S. Xu and F. Wang, J. Catal., 2017, 346, 170–179 CrossRef.
- T. Rinesch, J. Mottweiler, M. Puche, P. Concepción, A. Corma and C. Bolm, ACS Sustainable Chem. Eng., 2017, 5, 9818–9825 CrossRef.
- A. S. K. Tsang, A. Kapat and F. Schoenebeck, J. Am. Chem. Soc., 2016, 138, 518–526 CrossRef PubMed.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef.
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Energy Environ. Sci., 2015, 8, 2657–2663 RSC.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen and R. J. A. Gosselink, et al. , Green Chem., 2016, 18, 2651–2665 RSC.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals From Biomass, Pacific Northwest Natl. Lab., 2007, vol. 2, p. 87 Search PubMed.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef PubMed.
- A. Das, A. Rahimi, A. Ulbrich, M. Alherech, A. Hussain Motagamwala, A. Bhalla, L. da Costa Sousa, V. Balan, J. A. Dumesic and E. L. Hegg, et al. , ACS Sustainable Chem. Eng., 2018, 6, 3367–3374 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03208d |
| ‡ Present affiliation: Department of Chemistry, University of Kentucky, 505 Rose Street, Lexington, KY 40506, USA. |
| § Present affiliation: Harwell XPS, Research Complex at Harwell (RCaH) Rutherford Appleton Laboratory, Harwell Oxford, Didcot, OX11 0FA (UK). |
| ¶ Present affiliation: Department of Chemistry, University College London, 20 Gordon St, Kings Cross, London WC1H 0AJ (UK). |
| This journal is © The Royal Society of Chemistry 2018 |